Wojciech Piątkowski Inżynieria Chemiczna i Procesowa Wykład VII II stopień ADSORPCJA I CHROMATOGRAFIA
|
|
|
- Aneta Sadowska
- 8 lat temu
- Przeglądów:
Transkrypt


1 W y d z i a ł C h e m i c z n y P o l i t e c h n i k a R z e s z o w s k a i m. I g n a c e g o Ł u k a s i e w i c z a Wojciech Piątkowski Inżynieria Chemiczna i Procesowa Wykład VII II stopień ADSORPCJA I CHROMATOGRAFIA Katedra Inżynierii Chemicznej i Procesowej Wydział Chemiczny, Politechnika Rzeszowska
2 LITERATURA D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin, wyd.2 zmienione, WNT W-wa, K. Kaczmarski, W. Piątkowski Podstawy przenoszenia masy, Of. Wyd. PRz, Rzeszów R. Petrus, G. Aksielrud, J. Gumnicki, W. Piątkowski, Wymiana masy w układzie ciało stałe-ciecz, monografia, Of. Wyd. PRz., Rzeszów Z. Witkiewicz Podstawy chromatografii, WNT W-wa, G. Guiochon, A. Felinger, D. G. Shirazi, A.M. Katti, Fundamentals of Preparative and Nonlinear Chromatography, Elsevier, NY, Carta, G., Jungbauer, A., Protein Chromatography: Process Development and Scale Up, Wiley-VCH, Weinheim, T. Bredsznajder, Własności gazów I cieczy, WNT W-wa, Praca zbiorowa pod red. J. Bandrowskiego Materiały pomocnicze do ćwiczeń i projektów z inżynierii chemicznej skrypt Pol. Śląskiej 2

3 ADSORPCJA Naturalna - obecne zastosowania Przykłady adsorpcji klasycznej naturalnej, to np.: pochłanianie zapachów przez filtr węglowy w lodówce lub samochodzie, pochłanianie toksyn przez węgiel medyczny (carbo medicinalis) z przewodu pokarmowego pacjenta procesy sorpcji/desorpcji w migracji zanieczyszczeń organicznych i nieorganicznych w środowisku glebowym oraz inne patrz strona Google po linkach Zeolity skuteczne minerały Zeolity stosowane są w ośrodkach leczniczych w wielu krajach. Zeolity są znanym adsorbentem mikotoksyn, metali ciężkich oraz wirusów, zwalczają drobnoustroje, są bronią przeciwko wolnym rodnikom. 3
4 ADSORPCJA Podstawowe pojęcia Powierzchnia adsorbentu znajduje się w innym stanie energetycznym niż jego wnętrze, a siły działające na tej powierzchni są niezrównoważone. Wyeksponowanie ciała stałego w płynie spowoduje zjawisko gromadzenia się cząstek płynu na powierzchni tego ciała. Efekt ten będzie tym lepszy, im większa będzie powierzchnia właściwa (przypadająca na jednostkę masy ciała stałego). Zdolność pokrywania powierzchni ciała stałego cząstkami tego samego rodzaju substancji jest charakterystyczna dla danego układu płyn-ciało stałe. Zatem cząstki płynu będącego mieszaniną dwu- lub więcej składników mogą mieć różne powinowactwo do powierzchni ciała stałego. Spowoduje to zmianę stężenia każdej substancji biorącej udział w ruchu masy: stężenia objętościowego tej substancji w płynie oraz jej stężenia powierzchniowego, w warstwie osadzonej na powierzchni ciała stałego. Opisane zjawisko nosi nazwę adsorpcji. Równowaga termodynamiczna układu dwufazowego płyn-ciało stałe jest zazwyczaj silnie przesunięta na korzyść stężenia substancji zaadsorbowanej w warstwie powierzchniowej i przez to adsorpcja jest szczególnie efektywną metodą rozdzielania mieszanin, pozwalającą na zmniejszenie stężenia danej substancji w płynie, nawet do wartości stężenia rzędu ppm. 4
5 ADSORPCJA Podstawowe pojęcia Charakter sił wiążących adsorbat z powierzchnią adsorbentu dzieli adsorpcję na dwa rodzaje: (*) adsorpcję typu fizycznego oraz (*) adsorpcję typu chemicznego. Adsorpcja fizyczna jest spowodowana działaniem przyciągających sił międzycząsteczkowych, w większości przypadków sił typu van der Waalsa, spotęgowanych niekiedy siłami elektrostatycznymi lub siłami wiązania wodorowego. Adsorpcja chemiczna - chemisorpcja, zwana także adsorpcją aktywowaną, jest związana z wytworzeniem wiązania typu (siły, energii) chemicznego między adsorbatem a adsorbentem. Następujące kryteria pozwalają rozróżnić adsorpcję fizyczną od chemisorpcji: 1. ciepło adsorpcji - małe wartości ciepła adsorpcji, rzędu 220 [kj/mol] dla adsorpcji fizycznej, duże dla - zmiana równowagi procesu jest łatwa w przypadku adsorpcji fizycznej, spowodowanie chemisorpcji, rzędu [kj/mol], porównywalne z ciepłem reakcji; 2. odwracalność procesu takiej zmiany dla chemisorpcji wiąże się z koniecznością zastosowania bardziej drastycznych warunków, na przykład zwiększenia temperatury desorpcji, 3. grubość warstw adsorpcyjnych - w przypadku adsorpcji fizycznej tworzą się na powierzchni warstewki adsorbatu, których grubość odpowiada kilku średnicom cząstek tego ostatniego; w przypadku chemisorpcji powstają warstewki jednocząsteczkowe warstewki adsorbatu. Podczas adsorpcji w układzie ciecz-ciało stałe możliwe jest często wystąpienie obu typów adsorpcji - fizycznej oraz chemicznej jednocześnie już w temperaturze otoczenia. 5
6 ADSORPCJA Podstawowe pojęcia Z termodynamicznego punktu widzenia, w adsorpcji, która jest procesem odwracalnym - jak w każdym procesie równowagowym, obowiązuje równanie Gibbsa: G = H r - TS Ze względu na to, że adsorpcja jest procesem samorzutnym to energia swobodna Gibbsa G < 0. Przejście adsorptywu A z płynu w adsorbat zaadsorbowany na powierzchni adsorbentu A S k k 1 A( S) -1 jest związane z utratą co najmniej jednego stopnia swobody (zahamowany ruch translacyjny), a więc wystąpi także zmiana entropii układu S < 0. Wtedy H < 0, ADSORPCJA JEST WIĘC PROCESEM EGZOTERMICZNYM! W związku z tym - stężenie adsorbatu na powierzchni adsorbentu maleje ze wzrostem temperatury procesu a rośnie ze wzrostem stężenia adsorptywu w płynie. 6
7 ADSORPCJA i Chromatografia Nazewnictwo Dobrany i zoptymalizowany pod względem selektywności rozdzielania danej mieszaniny układ, w skład którego wchodzą: rozdzielana mieszanina składników, a w tym: składnik kluczowy A - inaczej adsorptyw (w płynie); adsorbat (na powierzchni) adsorbowany w adsorpcji, lub chromatografowany w chromatografii: analitycznej określa się jako analit,, preparatywnej jako składnik chromatografowany,, rozpuszczona w fazie ruchomej (eluencie) nazywana jest próbką w chromatografii okresowej oraz cyklicznej lub strumieniem zasilającym w adsorpcji oraz w chromatografii ciągłej, + faza ruchoma w chromatografii cieczowej to rozpuszczalnik lub mieszanina rozpuszczalników nazywana eluentem + adsorbent (złoże), nazywa się układem. 7
8 Typy oddziaływań w chromatografii Jak wywołać inny stan energetyczny powierzchni adsorbentu? Siły: Adsorpcyjne Faza stacjonarna wyższa polarność od próbki oraz fazy ruchomej (od analitów i rozpuszczalników) Biospecyficzne Powinowactwa (np. Proteina A) Podziałowe Odwrócone Fazy (RPC) Faza stacjonarna niższa polarność od próbki oraz fazy ruchomej (od analitów i rozpuszczalników) Hydrofobowe Chromatografia Oddziaływań Hydrofobowych (HIC) Elektrostatyczne Kompleksowanie Wymiana Jonowa (IEC) Oddziaływanie z Metalami (MIC) Steryczne Wykluczanie (SEC) 8
9 ADSORPCJA i Chromatografia Mechanizmy rozdzielania Chromatografia adsorpcyjna Chromatografia jonowymienna Chromatografia powinowactwa (affinity chromatography) Chromatografia ciecz ciecz Chromatografia żelowa Chromatografia adsorpcyjna 9


10 Chromatografia podziałowa (ciecz- ciecz) Chromatografia jonowymienna 10
")

11 Chromatografia żelowa (SEC) Chromatografia powinowactwa (affinity chromatography) 11
12 Dynamika adsorpcji i chromatografii Proces w kolumnie adsorpcyjnej lub chromatograficznej z nieruchomym złożem adsorbentu jest procesem okresowym procesem nieustalonym! Różnice pracy kolumny okresowej: -- adsorpcyjnej Krzywa wyjścia (przebicia) oraz -- chromatograficznej Pik chromatograficzny. R. Petrus; G. Aksielrud; J. Gumnicki; W. Piątkowski Wymiana masy w układzie ciało stałe ciecz, rozdz.7. 12

13 Schemat aparatury chromatograficznej zbiornik eluentu, 2 pompa, 3 dozownik, 4 piec, 5 kolumna, 6 detektor, 13
14 Chromatografia cieczowa kolumnowa LC Chromatografia w układzie ciecz-ciało stałe LSC Chromatografia nadkrytyczna SFC podziałowa adsorpcyjna podziałowa adsorpcyjna wykluczania jonowymienna hydrofobowa powinowactwa a) Techniki chromatograficzne okresowa cykliczna ciągła b) elucja izokratyczna elucja gradientowa rugowanie Schemat klasyfikacji technik chromatograficznych LSC w chromatografii preparatywnej D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.1. 14
15 ADSORPCJA Podstawowe pojęcia adsorbenty naturalne Podział adsorbentów ze względu na porowatość: Adsorbenty nieporowate - mają one niewielką powierzchnię właściwą, rzadko przewyższającą 10 m 2 /g. Najczęściej powierzchnia ta wynosi od 0,1 do 1 m 2 /g. Należą do nich: sadza grafitowana, BaSO 4, aerożele krzemionkowe. Adsorbenty porowate - ciała stałe o powierzchniach właściwych od setek do tysiąca m 2 /g. Adsorbenty takie stosuje się w postaci ziarnistej (tabletki, granulki, kulki) w celu nadania im odpowiedniej wytrzymałości i zmniejszenia oporu w stosunku do strumienia gazu lub cieczy. Rozmiary ziaren wynoszą najczęściej od 0,1 do 15 m. Wyróżniamy wśród nich: żele krzemionkowe, uwodniony Al 2 O 3, węgle aktywne, sita molekularne (zeolity), szkła porowate. 15
 pory o promieniach większych od 2 nm a mniejszych od 200 nm, makropory pory o promieniach większych od 200 nm. 16")
16 ADSORPCJA Adsorbenty porowate Podstawowe pojęcia Klasyfikacja porów wg Dubinina: mikropory pory o promieniach mniejszych od 2 nm, pory pośrednie (mezopory) pory o promieniach większych od 2 nm a mniejszych od 200 nm, makropory pory o promieniach większych od 200 nm. 16
 opór dyfuzji powierzchniowej dąży do 0. Iberer, G., Hahn, R. and Jungbauer, A.")
17 Flow Monolity oraz złoża Membrane Flow Flow Monolith Bead (*) budowa przestrzenna wypełnienia kolumny monolitycznej składa się z cylindrycznych porów połączonych ze sobą: (*) opór dyfuzji zewnętrznej dąży do 0, (*) opór dyfuzji powierzchniowej dąży do 0. Iberer, G., Hahn, R. and Jungbauer, A. (1999) Monoliths as stationary phases for separation of biopolymers: the fourth generation of chromatography sorbents LC-GC, 11, Svec, F., Tennikova, T.B. & Deyl, Z. Monolithic material: preparation, properties and applications, Journal of chromatography library, Vol. 67. (Elsevier), Amsterdam; (2003). 17
18 Monolity oraz złoża c.d. Historia materiałów zol-żelowych rozpoczęła się w w połowie XIX w., kiedy Ebelman jako pierwszy doniósł o tworzeniu się przezroczystego materiału wskutek powolnej hydrolizy estru kwasu krzemowego. Zauważył on, że tetraetoksysilan (TEOS) tworzy roztwory, które w procesie żelowania zastygają w różne bryły, w zależności od użytej formy. Powyższe spostrzeżenia ponad sto lat później wykorzystał Dislich, syntezując szkło borowo-krzemianowe poprzez ogrzewanie w łaźni wodnej sproszkowanych tlenków, otrzymanych za pomocą niskotemperaturowego procesu zol-żel. Istnieje cały szereg procedur preparatywnych należących do grupy procesów zol-żel. Jedna z prostszych i zarazem najpopularniejszych metod polega na przygotowaniu roztworu koloidalnego (zolu) i przeprowadzeniu go w żel. Otrzymany (pół)stały materiał poddaje się dojrzewaniu, podczas którego następuje wyrzucanie cieczy z jego wnętrza, określane mianem synerezy. W ten sposób powstają żele mokre (alko- i hydrożele), które następnie suszy się, co prowadzi do otrzymania szklistych i stabilnych kserożeli. Różnorodność form i kształtów materiału otrzymanego w metodzie zol-żel jest praktycznie nieograniczona. Dzięki temu, że zol jest jednorodną cieczą o względnie małej lepkości, może być odlewany w dowolnych kształtach, zależnie od jego przeznaczenia. W efekcie uzyskuje się więc zarówno transparentne monolity, jak i cienkie filmy, włókna, proszki czy też granulaty. Ważny jest jednak wybór odpowiedniej formy, do której wlewa się zol. Musi ona mieć tak przygotowaną powierzchnię, aby nie dochodziło do adhezji żelu lub powstawania pęcherzyków na granicy forma/żel. 18
19 Matryca ciała stałego Materiały nieorganiczne Polimery naturalne Polimery organiczne Chromatografia podziałowa Chromatografia jonowymienna Chromatografia powinowactwa Chromatografia chiralna i inne 19
20 Chromatografia Różnice między chromatografią analityczną a preparatywną 0.5 stężenie czas [s] Kształt pików chromatograficznych: - w chromatografii analitycznej. - w chromatografii preparatywnej: c F c F V F c m szok fala prosta c m ogon piku Przeładowanie stężeniowe t r b) t r Przeładowanie objętościowe D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.1. 20
21 Chromatografia preparatywna Impurities Concentration Threshold I II Threshold t s 350 t I 400 t II 450 t 500 e 550 Time 21
22 Chromatografia Różnice między chromatografią analityczną a preparatywną analityczna preparatywna CEL analiza rozdzielanie WYNIK informacja produkty STĘŻENIE niskie różne OBCIĄZENIE MASOWE KOLUMNY niskie wysokie IZOTERMA liniowa nieliniowa KONKURENCJA między adsorbatami SPRAWNOŚĆ KOLUMNY MODEL DYNAMIKI OPTYMALIZACJA nie znacząca wysoka Szybkie rozwiązanie, wymaga tylko znajomości sprawności kolumny oraz selektywności rozdzielania znacząca niska Trudne rozwiązanie. Funkcją celu jest produktywność kolumny. Wymaga założenia tzw. ograniczeń optymalizacji 22
23 TECHNIKI CHROMATOGRAFICZNE 23
24 Idea okresowej chromatografii izokratycznej Zasilanie próbką Eluent Eluent Eluent Kolumna chromatograficzna upakowana złożem adsorbentu Czas: 0 Czas: 10 Czas: 20 Dorota Antos, Krzysztof Kaczmarski, Wojciech Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.1. Czas: 30 24
25 Idea procesu chromatografii izokratycznej c f1, c f2 t imp x = 0 x = L 25

26 Idea procesu chromatografii izokratycznej 26
27 Eluent (E) Chromatografia okresowa, E = R C Concentration Elucja izokratyczna H (A+E) Eluat (ang. Efluent) (B+E) E = R +ME Time [s] Elucja gradientowa; gradient gradient stężenia ME 27
28 Gradient liniowy vs gradient schodkowy % salt or temp 0 C % salt c [g/l] * Lys simulation exp. Chym CV c [g/l] * % sal Lys Chym simulation Temp CV tv V CV 0 Muca R., Piątkowski W., Antos D., Altering efficiency of hydrophobic interaction chromatography by combined salt and temperature effects, J. Chromatogr. A, 1216 (2009)

29 Okresowość a półciągłość procesu chromatografii Chromatografia okresowa Chromatografia cykliczna 29
30 Chromatografia cykliczna 1.4 I fraction II fraction 1.2 interfraction Concentration [g/l] C treshold t si t ei t sii t eii Time [s] t end Chromatografia cykliczna Czas trwania t c dla dwóch kolejnych cykli: t c = t end t si ; Czas odbioru i-tej frakcji: t frac = t ei - t si 30
31 Przeciwprądowa Chromatografia ciągła (TMB) - IDEA A + B Zone IV A A+B Zone III B A Zone II B Zone I Zone II Zone III Desorbent 31
32 ADSORPCJA I CHROMATOGRAFIA Dynamika procesu Proces jednostkowy w inżynierii produkcji jest to elementarna część składowa procesu wytwórczego, w którym surowce są przetwarzane w produkty. Proces (produkcyjny) jest uporządkowanym zbiorem procesów jednostkowych, w tym: reakcji chemicznych, procesów fizycznych. Procesy jednostkowe są analizowane z użyciem podobnych technik badawczych; ilustrują to przykłady metod modelowania kinetyki reakcji chemicznych w reaktorach chemicznych albo sposoby prezentacji bilansu energetycznego (zobacz: zasada zachowania energii) lub bilansu masy (zobacz: prawo zachowania masy). Projektowanie i prowadzenie procesów produkcji wymaga stosowania złożonych modeli matematycznych zwanych modelami dynamiki procesu oraz specjalistycznych programów komputerowych. 32
33 ADSORPCJA Podstawy procesu 33
34 Proces cząstkowy kontrolujący szybkość procesu ogólnego nazywanego adsorpcją 1. wnikanie masy adsorptywu i od płynu do zewnętrznej powierzchni adsorbentu (dyfuzja zewnętrzna); 2. dyfuzja adsorptywu i poprzez porowatą strukturę adsorbentu do wnętrza ziarna (dyfuzja wewnętrzna); 3-4. adsorpcja składnika i na powierzchni adsorbentu (proces powierzchniowy) - równowaga termodynamiczna procesu powierzchniowego równoległa dyfuzja powierzchniowa adsorbatu A 1 2,5 A B A 6 Płyn 1 2 A Konwekcja, dyspersja A Dyfuzja zewnętrzna przenoszenie masy z płynu do powierzchni ziarna 2. Dyfuzja wewnętrzna (w porach) 3. Adsorpcja Dyfuzja powierzchniowa 4. Desorpcja 5. Dyfuzja wewnętrzna odwrotna 6. Dyfuzja zewnętrzna odwrotna Powierzchnia ziarna (zewnętrzna) Powierzchnia porów (wewnętrzna) 34
35 ADSORPCJA Statyka (termodynamika) procesu (Równowaga procesu powierzchniowego adsorpcja-desorpcja) (Proces cząstkowy 3-4) 35
36 ADSORPCJA Statyka procesu O ilości parametrów, jakie możemy przyjmować dowolnie (zmienne niezależne) w warunkach równowagi adsorpcyjnej w układzie gaz(lub ciecz) płyn-ciało stałe informuje nas reguła faz. W przypadku najprostszego układu 3-składnikowego: s = i f - gdzie: i - ilość składników; f - ilość faz; s - ilość stopni swobody. i = 3; f = 2; s = = 3 stopnie swobody. Dwa z nich zajmujemy: T oraz p. Jeżeli 3-ci stopień swobody zajmiemy jednym ze stężeń np. C A - stężeniem składnika kluczowego (adsorptywu) A w fazie płynu, to stężenie składnika A na powierzchni adsorbentu (adsorbat) * q A A* po osiągnięciu stanu równowagi będzie funkcją: = f(c A ; T; p). Jeżeli ustalimy * T; p, wówczas q A = f(c A ). 36 * q A
37 ADSORPCJA Statyka procesu c.d. Ze zmianą dwóch, głównych parametrów T oraz p (dla układu gaz-ciało stałe) adsorpcja przebiega następująco. Ze wzrostem temperatury stężenie równowagowe adsorbatu się zmniejsza, a ze wzrostem ciśnienia rośnie (układ gaz-ciało stałe!). Z tego powodu adsorpcję należy prowadzić w jak najniższej temperaturze i jak najwyższym ciśnieniu (zakresy zmian tych parametrów są charakterystyczne dla danego układu adsorpcyjnego) ponieważ maksymalizujemy wówczas siłę napędową procesu. Każdorazowy stan równowagi powiązany jest zależnością: K q * A T, p const CA gdzie: K - stała równowagi adsorpcyjnej. stezenie rownowagoweskladnikaa na powierzchni adsorbentu stezenie rownowagoweskladnikaa w fazie plynu Prawidłowy, naturalny (szczegółowy) rodzaj stężenia użytego w modelu adsorpcji wynika z wymiaru bilansu masy w sześcianie jednostkowym, w którym każdy człon winien mieć wymiar [kmola/m 3 s]. Mimo tego, że adsorpcja to przypadek dyfuzji 1) lub 3) rodzaju to najbardziej dogodnym i podstawowym używanym w opisie stężeniem jest koncentracja molowa lub masowa. 37
38 ADSORPCJA Statyka procesu c.d. Adsorpcja idealna: faza ruchoma to idealny roztwór, idealny adsorbent ciało stałe o idealnej (homogenicznej) powierzchni. Adsorpcja nieidealna - rzeczywista: 1. faza ruchoma to idealny roztwór, nieidealny adsorbent ciało stałe o rzeczywistej (heterogenicznej) powierzchni. 2. faza ruchoma to nieidealny (rzeczywisty) roztwór, idealny adsorbent ciało stałe o idealnej (homogenicznej) powierzchni. 3. faza ruchoma to nieidealny (rzeczywisty) roztwór, adsorbent nieidealny ciało stałe o rzeczywistej (heterogenicznej) powierzchni. D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.2. 38
39 ADSORPCJA Statyka procesu c.d. Izoterma Gibbsa Opis procesu adsorpcji gazów ma swoją podbudowę teoretyczną, w tym termodynamiczną w teorii kinetycznej budowy gazów Maxwella-Stefana: Ogólna, klasyczna izoterma Gibbsa dla idealnego układu gaz ciało stałe: A p T const RT p n s gdzie: p to ciśnienie adsorptywu; to ciśnienie powierzchniowe monowarstwy adsorbatu; n s jest liczbą moli adsorbatu; A jest powierzchnią adsorpcji. Z izotermy Gibbsa oraz równania stanu dla gazów może zostać wyprowadzonych szereg szczegółowych modeli izotermy. 39
40 ADSORPCJA Izotermy pojedyncze Statyka procesu c.d. Modele izotermy dla idealnej adsorpcji na homogenicznej powierzchni adsorbentu Izoterma Langmuira: A S k k 1 A( S) -1 Izoterma Henry ego: q H C * i i i qi 1 1 (1 ) * k i Cpi q qi k i KriCpi q qi 0 i i t H i q K ri q * i q K ri C i H i C i 1K C 1K C ri i ri i q* I II III q * /c m nachylenie cięciwa 0 0 a) c m b) c m 40
41 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. Modele izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu Istnieje wiele klasycznych modeli izoterm uwzględniających nieidealność zachowania się adsorbatów w fazie zaadsorbowanej. Modele te nie uwzględniają odstępstw od ideału w fazie ciekłej. Model izotermy Fowlera Jednym z takich modeli jest izoterma Fowlera, która została zaproponowana przez Fowlera i Guggenheima aby uwzględnić odchylenia tzw. 1-go rodzaju od modelu izotermy Langmuira. Model ten zakłada idealną adsorpcję na homogenicznej powierzchni adsorbentu z bardzo słabymi oddziaływaniami pomiędzy cząsteczkami adsorbatu na sąsiadujących miejscach aktywnych. Energia tych interakcji jest tak mała, że nie zmienia ona przypadkowego charakteru rozkładu cząstek adsorbatu na powierzchni. Przy wyżej wymienionych założeniach izoterma Fowlera dla układu gaz-ciało stałe przyjmuje postać: 2 bp exp 1 RT gdzie: 2 jest energia oddziaływań pomiędzy sąsiadującymi adsorbatami. 41
42 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. odele izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Modele izoterm S-kształtnych oraz izotermy kwadratowej Modele izoterm S-kształtnych opisuje przypadek termodynamiki adsorpcji, który może być wynikiem silnych oddziaływań między molekułami adsorbatu oddziaływań bocznych między adsorbatami w monowarstwie lub oddziaływań prowadzących do powstania adsorpcji wielowarstwowej. Tego rodzaju modele izotermy mogą być przedstawione przez izotermę wielomianową, która może opisywać izotermy wklęsłe w niskim zakresie stężenia. Ze wzrostem wartości stężenia, w dalszej części izotermy S-kształtnej występuje punkt przegięcia i izoterma zmienia się na wypukłą. Równanie dla tej izotermy jest zwykle zapisywane w postaci stosunku dwóch wielomianów: q * m m 2... m n b C b C nb C q 1 b1 C b2 C... b C 2 m m m n Izoterma 2-go stopnia otrzymana dla n = 2 nazywana jest izotermą kwadratową. n n 42
43 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. odele izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Rys. Izotermy opisane równaniem izotermy kwadratowej dla n = 1, 2, 3, 4 (od dołu go góry) q = 10, b 1 = 1, b 2 = b 3 = b 4 = 0.8, w porównaniu z przebiegiem izotermy anty-langmuira (linia wklęsła czerwona) q* [g/cm 3 ] c m [g/cm 3 ] Izoterma antylangmuira: q antylangmuir * q b ' ri C i H ' i C i qi 1b C 1b C ri i ri i q * C 43
44 Breakthrough curves ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. Modele izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Izoterma BET dla układu gaz-ciało-stałe MeOH/H 2 O (65/35) 24 Packed Izoterma BET w układzie ciecz-ciało stałe: q * i qi Kri Ci 1 K C 1 K C K C L i L i ri i q*/c minimum slope of chord isotherm Monolith Izoterma BET dla układu ciecz-ciało stałe. Kształt zależności wartości q*/c i - w funkcji stężenia C i oraz charakterystyczny kształt krzywych wyjścia naprowadza na prawidłowy model izotermy adsorpcji tutaj BET c [g/dm 3 ] Gritti, F., Piątkowski, W., Guiochon, G., Study of the mass transfer kinetics in a monolithic column, J. Chromatogr. A, 983 (2003)
45 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. Izoterma bilangmuira: q * A Klasyczne modele izotermy dla idealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu q K C q K C 1K C 1K C AI I i AII II i Izoterma Freundlicha: I i II i q * 1/ n i k F Ci gdzie: k F i n parametry równania Izoterma Langmuira-Freundlicha: q * A 1/ n ra ma ( A) 1/ n ra A K A C 1 K ( C ) 45
46 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. Modele izotermy dla idealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. Niejednorodność energetyczna powierzchni adsorbentów Wpływ na termodynamikę adsorpcji wywierają nie tylko takie czynniki, jak np. charakter wiązania adsorbatu z adsorbentem, ale też tzw. heterogeniczność energetyczna powierzchni adsorbentu, np. zróżnicowany skład chemiczny powierzchni, tzw. grupy powierzchniowe funkcyjne, defekty krystaliczne powierzchni, itp. Oznacza to istnienie na powierzchni adsorbentu obszarów o różnej zdolności adsorpcyjnej (różnej sile wiązania adsorpcyjnego) powierzchnia jest powierzchnią heterogeniczną (energetycznie niejednorodną). Obecność energetycznej niejednorodności powierzchni oznacza najczęściej różną, silniejszą adsorpcję dla niskich i bardzo niskich wartości stężenia adsorbatu. Praktycznie nie istnieje idealna jednorodna powierzchnia adsorbentu, powierzchnia adsorpcyjna jest zawsze rzeczywista, czyli heterogeniczna. 46
47 ADSORPCJA Izotermy pojedyncze c.d. Statyka procesu c.d. Modele izotermy dla idealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. Rzeczywistą powierzchnię adsorbentu można traktować jako powierzchnię o ciągłym rozkładzie energii adsorpcji. Celem opisania adsorpcji na takiej powierzchni stosuje się całkowe równanie adsorpcji: * * E max q p f E q p,e de lub p f E p,e de t E min gdzie: t (p) to izoterma globalna - izoterma adsorpcji otrzymana przez uśrednienie izotermy lokalnej dla różnych typów miejsc aktywnych (średnia dla całej powierzchni); (p,e) to izoterma lokalna, lokalne pokrycie powierzchni, które jest zależne od energii adsorpcji danego miejsca adsorpcyjnego E; f(e) - znormalizowana do jedności funkcja rozkładu energii adsorpcji (gęstość prawdopodobieństwa) charakterystyczna dla adsorbentu i adsorbatu. 47
48 ADSORPCJA Izotermy pojedyncze c.d. * q K C q A 1 i exp A ln 2 h 1 K Ci exp h h Izoterma UNILAN Do uwzględnienia heterogeniczności powierzchni adsorbentu w literaturze proponuje się m.in. model izotermy UNILAN. Do wyznaczenia izotermy globalnej w tym przypadku stosuje się następujący rozkład energii E: Statyka procesu c.d. Modele izotermy dla idealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. f E 1 dla Emin E Emax Emax Emin 0 dla E Emin lub E E Równanie izotermy UNILAN, zapisanej dla i-tego składnika, otrzymuje się przez całkowanie powyższej funkcji zakładając izotermę Langmuira jako izotermę lokalną. * q K r C q f E de m 1 K C r m max Dla h = 0 równanie redukuje się do modelu izotermy Langmuira. 48
49 ADSORPCJA CHROMATOGRAFIA Metody pomiaru równowagi adsorpcyjnej Izoterma pojedyncza Termodynamika procesu adsorpcji jest dominującym lub jednym z najważniejszych etapów cząstkowych decydujących o przebiegu oraz szybkości procesu adsorpcyjnego. 1. Aby wyznaczyć równowagę adsorpcyjną należy stworzyć zbiór doświadczalnych danych równowagowych. Sposób otrzymania tych danych oraz ich interpretacji zależny jest od metody pomiaru równowagi adsorpcyjnej, jaką się zastosuje. 2. Podstawą każdego sposobu interpretacji jest założenie mechanizmu odwracalnego procesu powierzchniowego adsorpcja-desorpcja, na bazie którego dobiera się adekwatny model izotermy adsorpcji oraz wyznacza jego parametry modelu (parametry izotermy). 3. Procedurę kończy porównanie wyników symulacji z danymi doświadczalnymi i określenie błędu opisu walidacja modelu izotermy. D. Antos, K.Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.5. 49

50 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izotermy pojedynczej Metoda statyczna Metoda statyczna jest najprostszą, ale zarazem najbardziej czasochłonną metodą pomiaru izoterm. Polega na zmieszaniu określonej ilości adsorbentu o objętości V a, z roztworem o objętości V 0 i stężeniu C 0 badanego analitu. Po dostatecznie długim czasie kontaktu ustali się równowaga między stężeniem analitu w roztworze, C i na powierzchni sorbentu, q. q V ( C C) V * 0 0 a W ten sposób znajduje się jedną parę stężeń równowagowych, czyli jeden punkt na linii równowagi. Następnie procedurę powtarza się dla innych wartości stężenia początkowego. W kolejnym kroku, należy zaproponować adekwatne do założonego mechanizmu procesu powierzchniowego równanie izotermy adsorpcyjnej q* = f(c m,p 1,,p n ) oraz wyestymować jego parametry p 1,,p n, dopasowując krzywą uzyskaną z symulacji komputerowych do danych doświadczalnych. 50
51 ADSORPCJA Metody wyznaczania izoterm metody dynamiczne Metoda analizy frontalnej krzywej wyjścia CHROMATOGRAFIA W metodzie tej, roztwór adsorptywu o znanym stężeniu wlotowym może być wprowadzony na wlot kolumny w postaci odpowiednio szerokich impulsów, tak, aby profil stężenia otrzymany jako odpowiedź układu w efluencie na wylocie z kolumny posiadał plateau odpowiadające wartości stężenia wlotowego adsorptywu. Oznacza to podanie w impulsie takiej masy adsorptywu, aby cała objętość złoża w kolumnie zapełniła swą powierzchnię adsorpcyjną aż do warunków równowagi termodynamicznej (przeładowanie objętościowe kolumny) Metoda krzywej wyjścia zaliczana jest do najdokładniejszych metod badań równowagi adsorpcyjnej. Stężenie równowagowe, q*, na powierzchni adsorbentu do stężenia, C, w płynie, oblicza się w oparciu o krzywe wyjścia wykonane dla szeregu stężeń analizowanego składnika. c 6 c 6 c 5 c 5 c 5 c 4 stężenie c 4 c 3 stężenie c 3 c 4 stężenie c 2 c 3 c 2 c 2 c 1 c 1 c 1 a) czas b) czas Rys. Krzywe wyjścia dla różnych wartości stężenia wlotowego analizowanego adsorptywu: c F6 > c F3 > c F1. a) izoterma Langmuira (wypukła) b) izoterma anty-langmuira (wklęsła). czas D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.5. 51
52 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne Metoda analizy frontalnej krzywej wyjścia c.d. C C ( V V ) C ( V V ) t t t t q C f C * F r 0 F r 0 t r 0 r 0 F Va ( Vcol V0 ) 1 t t0 F t0 V r, t r plateau V lub t stężenie P P1 P2 t r, V r F W metodzie analizy frontalnej należy przez optymalizację porównać założony model izotermy adsorpcji z danymi doświadczalnymi - walidacja modelu izotermy F 1 t t Rys. Ilustracja metody równych pól 52
53 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne c.d. Metoda impulsowa - perturbacji W metodzie roztwór danego adsorptywu, rozpuszczonego w eluencie (fazie ruchomej) o znanym stężeniu pompowany jest przez kolumnę aż do pojawiania się plateau krzywej wyjścia na wylocie kolumny, tzn. do osiągnięcia warunków równowagi adsorpcyjnej pomiędzy fazą ruchomą a całym złożem zawartym w kolumnie. Złoże zapełnione jest adsorbatem do wartości równowagi. Następnie na wlot kolumny wprowadza się niewielki, krótkotrwały, impuls próbki, o wartości stężenia badanej substancji niewiele różniącej się, w górę lub w dół, od jej stężenia w eluencie. Doświadczenie powtarza się dla szeregu rosnących wartości początkowych stężenia adsorptywu. C t 10 2 r Metoda impulsowa zilustrowana jest dla izotermy jednoskładnikowej. Metoda ta, jak poprzednia wymaga założenia modelu izotermy adsorpcji i estymacji jej parametrów. q t * i r t t tr t F t q C q K r t ( C) t 1 F q K r C H C 1K C 1K C 1 K C q r * 0 C C C F r Ogólnie F r 1 t t 53
54 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne c.d. Metoda impulsowa - perturbacji 450 q t ( C) t 1 F r * 0 C C C F t r [s] c m F [mol/cm3 ] Otrzymane dane doświadczalne należy przedstawić jak na wykresie Aby wyznaczyć izotermę adsorpcji dla otrzymanych doświadczalnie wartości czasów retencji impulsów w funkcji stężenia plateau, można równanie przekształcić obliczając: q C * F a następnie scałkować je względem f t ( C ) r F C F Następnie, dla uzyskanych w ten sposób danych doświadczalnych w formie zbioru par stężeń równowagowych dobiera się model izotermy i estymuje jego parametry. 54
55 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne c.d. Metoda impulsowa perturbacji c.d. PRZYKŁADY dla metody perturbacji PM t ( C) t 1 F r q * 0 C C C F q C * F f t ( C ) r F Izoterma Henry ego: Izoterma Langmuira: q q * H C i i i * i H i C i 1 K C ri i t t F H r 1 * 0 q H f t ( ) r CF tr t 1F H KC r q C * F C F H 1 KC r f t ( ) 2 r CF 55
56 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne c.d. Metoda elucji punktów charakterystycznych (EPC) W metodzie FACP stężenie adsorbatu, q*, w równowadze ze stężeniem w fazie ruchomej c m, oblicza się ze zbocza o opadającej wartości stężenia od wartości plateau krzywej wyjścia do zera Każdy punkt opadającego zbocza daje jeden punkt izotermy. Klasyczna metoda ECP daje dobre wyniki dla kolumn o liczbie półek teoretycznych większej od około (To WADA! To jest równoznaczne z założeniem nieskończenie dużej sprawności kolumny). Izotermę q* = f(c), oblicza się według wzoru: c * r inj 0 q C dc m m 0 V ( C) V V V a m ( t t ) u L 1 t ( C) t c c r inj t m r inj dc (1 ) 0 t L F t dc stężenie czas c m =c F m c m (t) opadające zbocze c m =0 56
57 ADSORPCJA CHROMATOGRAFIA Metody wyznaczania izoterm metody dynamiczne c.d. Metoda dopasowania do piku (IM) Metoda nazywana inaczej metodą odwrotną, wymaga znacznie mniejszej ilości odczynników i mniejszego nakładu pracy. Metoda ta polega na rejestrowaniu przeładowanych profili stężenia (pików). Następnie proponowany do opisu termodynamiki adsorpcji model izotermy adsorpcyjnej wprowadza się do wybranego modelu matematycznego dynamiki kolumny chromatograficznej, który rozwiązuje się z jednoczesną estymacją parametrów modelu, tak, aby symulowany profil stężenia był zgodny z profilem doświadczalnym. Wstępnie dobiera się wartości startowe parametrów izotermy dla procedury optymalizacyjnej tak, aby pik symulowany leżał, przynajmniej w pewnym zakresie, w obszarze piku doświadczalnego patrz rys. - pik 1. Następnie parametry izotermy są zmieniane zgodnie z algorytmem optymalizacyjnym, tak, aby, zminimalizować różnice kwadratów między doświadczalnym i obliczonym profilem stężenia. Przebieg poszczególnych faz estymacji ilustrują krzywe 2-5 na rys. 1. Wadą metody IM jest trudność w prawidłowym oszacowaniu pojemności chłonnej złoża. Ze 2 względu na silne rozcieńczenie profili stężenia 1 3 podczas chromatografii, stężenie maksymalne w piku opuszczającym kolumnę może być 4 znacznie niższe od stężenia odpowiadającego silnej nieliniowości izotermy i całkowitemu pokryciu miejsc aktywnych Metoda IM jest natomiast bardzo szybka i dlatego często stosowana do celów czas praktycznych. Stężenie 57
58 ADSORPCJA gdzie: * q i, j IZOTERMY KONKURENCYJNE - WIELOSKŁADNIKOWE Współczynnik rozdzielenia adsorpcyjnego i,j w przypadku adsorpcji z płynu więcej niż jednego adsorptywu, jest mierzony analogicznie jak w przypadku innych metod rozdzielania przez stosunek stałych równowagi adsorpcyjnej (analog np. lotności względnej w destylacji), definiowany jako: CHROMATOGRAFIA Statyka procesu c.d. k q / C H * j i i ri ij * ki q j / C j Hrj ilość zaadsorbowanej substancji (adsorbatu i/j) w jednostce masy adsorbentu (stężenie powierzchniowe adsorbatu) w warunkach równowagi termodynamicznej; C i,j - stężenie adsorptywu w płynie; Rozdzielanie mieszaniny przez adsorpcję jest możliwy, jeśli równowagowa selektywność substancji i od j jest rzędu i,j = lub większa. Izotermy klasyczne - Izoterma konkurencyjna Langmuira: Izoterma Langmuira: q * i H i C i 1 K C ri i q * i qi K i C i 2 1 K C j1 j j założenie: q i q tot const 58
59 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. jeśli: N q tot q * i i1 wówczas izoterma konkurencyjna Langmuira zamienia się w izotermę stechiometryczną Izoterma stechiometryczna: k 1 A B( S) A( S) k q -1 q K C B q K C * A ra A A ra A A 1 C A KrA CA 1KrA 1CA 59
60 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. W układzie adsorpcyjnym ciecz-ciało stałe, w fazie ruchomej nie ma praktycznie inertów, a wszystkie składniki próbki: składniki chromatografowane oraz rozpuszczalniki biorą udział w adsorpcji w procesie transportu masy (czyli wszystkie są adsorptywami). Tylko niektóre składniki eluentu, będącego mieszaniną rozpuszczalników, w konkretnych warunkach procesu, mogą jedynie zbliżać w się swym zachowaniu do zachowania się inertu. Z tego powodu termodynamika układu ciecz-ciało stałe dotyczy zawsze układów wieloskładnikowych - co najmniej dwuskładnikowych (pseudo-jednoskładnikowych, tzn. adsorbat eluent). Klasyczna zależność Duhema-Margelusa w tym układzie dla fazy zaadsorbowanej oraz analogiczna zależność dla fazy ruchomej (ciekłej) mają postacie analogiczne jak dla układu gazciało stałe: n n s s s s i i i i i i i i1 i1 n i1 Ad Ad n d n d Ad 0 n m i m i d 0 Modele izotermy dla nieidealnej adsorpcji w płynie na heterogenicznej lub homogenicznej powierzchni adsorbentu c.d. gdzie: ciśnienie powierzchniowe (ang. Spreading Pressure) i jest dla układu ciecz-ciało stałe definiowane jako różnica napięcia powierzchniowego. 60
61 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Chemiczne potencjały składnika i w rozpatrywanych fazach wynoszą: m 0 m i i RT ln a i s 0 s i i RT ln a i Modele izotermy dla nieidealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. Porównując i podstawiając równania otrzymuje się: n n s s m m ni i A i ni i i1 i1 d d d 0 gdzie; to odpowiednio aktywności molowe w fazie ruchomej oraz zaadsorbowanej: a m i a s i = γ m m i x i = γ s s i x i Powyższy ogólny układ równań może zostać uproszczony do kilku typowych przypadków szczególnych. 61
62 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Model izotermy dla nieidealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. Równowaga w układzie pseudo-jednoskładnikowym, gdy wartości stężenia obydwu składników są porównywalne x A m x B m Przykładem jest równowaga adsorpcyjna konkurencyjna składników fazy ruchomej, będącej dwuskładnikową mieszaniną rozpuszczalników. Jeden z rozpuszczalników, silniej adsorbujący się, pełni rolę modyfikatora eluentu. Drugi z rozpuszczalników, słabiej adsorbujący się, może zbliżać się w swym zachowaniu do inertu. x A m + x B m = 1 x A s + x B s = 1 Podział adsorpcyjny i-tego składnika (i = A, B) w wyżej wymienionym układzie między fazy ruchomą oraz zaadsorbowaną może być zapisany następującym układem równań równowagi z podstawionymi wartościami potencjałów chemicznych: a A s exp A π A π A 0 q A R T a B s exp A π B π B 0 q B R T = a A m = a B m 62
63 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Model izotermy dla nieidealnej adsorpcji w płynie na heterogenicznej powierzchni adsorbentu c.d. Równowaga w układzie pseudo-jednoskładnikowym, gdy wartości stężenia obydwu składników są porównywalne W dyskusji równań można poczynić następujące założenia upraszczające: Zał.1) Pojemność chłonna dla obu składników fazy ruchomej ma tę samą wartość: Zał.2) Powierzchnia adsorbentu jest całkowicie pokryta przez cząsteczki adsorbatów obydwu rozpuszczalników, tzn. występuje całkowity brak wolnych, nieobsadzonych przez adsorbaty miejsc aktywnych. Wówczas można zapisać, że całkowita pojemność chłonna złoża jest sumą: Otrzymuje się następującą zleżność: x x s m s m B xa x A B K s m A s m A xb B A q i q q q * q * A B gdzie: wszystkie parametry równań równowagi są uwzględnione w parametrze izotermy czyli w wartości stałej równowagi adsorpcyjnej, odpowiadającej układowi wymiarowemu stężenia wyrażonego za pomocą ułamka molowego. x K A 63
64 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Model izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Równowaga w układzie pseudo-jednoskładnikowym, gdy wartości stężenia obydwu składników są porównywalne Otrzymuje się postać izotermy, opisującą nieidealną adsorpcję dla składnika A w obecności składnika B na homogenicznej powierzchni adsorbentu: q A = q K A x x A m γ A m γ B s γ A s 1 x A m γ B m + K A x x A m γ A m γ B s Rys. Rzeczywista równowaga adsorpcyjna rozpuszczalników organicznych na żelu krzemionkowym i wypełnieniu CSP (teicoplanina) w układzie woda-rozpuszczalnik organiczny; x s, x m to odpowiednio ułamki molowe rozpuszczalnika w fazie ruchomej oraz zaadsorbowanej. x s alk MeOH EtOH Propan-2-ol chiral silikażel x m alk. W tym przypadku najczęściej proste postaci równania izotermy jednoskładnikowej, jak np. izoterma Langmuira, nie opisują poprawnie danych eksperymentalnych, przez co nie mogą zostać użyte. 64

65 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Modele izotermy dla idealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Równowaga w układzie pseudo-jednoskładnikowym, gdy wartości stężenia obydwu składników są porównywalne Jeżeli powyższy pseudo-jednoskładnikowy układ zbliża się w swoim zachowaniu do zachowania mieszaniny idealnej cieczy (roztwory rozpuszczalników o analogicznych własnościach np. związków homologicznych), to współczynnik rozdzielania adsorpcyjnego takiego dwuskładnikowego układu adsorpcyjnego dany jest przez stosunek stałych równowagi adsorpcyjnej: lub: x A s = α AB = x A s x B m x B s x A m m α AB x A 1 + α AB 1 m x A Równanie izotermy adsorpcji z roztworów dwuskładnikowych. x A s = m K 1 x A 1 + K 1 1 m x A Dla homogenicznej powierzchni adsorbentu α AB = K 1 Izoterma Everetta 65
66 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Równowaga w układzie pseudo-dwuskładnikowym przy dużym nadmiarze jednego ze składników adsorbentu c.d. x m A + x m B + x m C = 1 Model izotermy dla nieidealnej adsorpcji w płynie na homogenicznej powierzchni Przykładem jest równowaga adsorpcyjna konkurencyjna dwóch adsorptywów: składnika chromatografowanego A oraz modyfikatora eluentu B. Stężenie składnika chromatografowanego A jest wielokrotnie niższe od stężenia modyfikatora eluentu B, x m A x m B. Trzecim składnikiem układu jest drugi z rozpuszczalników C, znajdujący się w fazie ruchomej, słabiej adsorbujący się, o zachowaniu zbliżającym się do inertu. Wówczas, w równaniu: można przyjąć, że: q A = q K A x x A m γ A m γ B s γ A s 1 x A m γ B m + K A x x A m γ A m γ B s 1 x A m 1 x A m x B m + x C m = 1 W rozpatrywanym przypadku zachowanie się fazy zaadsorbowanej niewiele odbiega od ideału, wartości współczynników aktywności są praktycznie równe współczynnikom aktywności przy nieskończonym rozcieńczeniu roztworu i są stałe, niezależne od stężenia adsorptywu. Upraszcza to zapis równania do postaci: q A = q K A x x A m γ A m γ B s γ A s γ B m + K A x x A m γ A m γ B s 66
67 ADSORPCJA CHROMATOGRAFIA IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Model izotermy dla pseudoidealnej adsorpcji w płynie na homogenicznej powierzchni adsorbentu c.d. Równowaga w układzie pseudo-dwuskładnikowym przy dużym nadmiarze jednego ze składników Jeżeli wartości współczynników aktywności są stałe to mogą być ujęte w wartości stałej x równowagi ഥK A, po przekształceniach otrzymuje się klasyczne równanie izotermy Langmuira:: q A = q ഥK x m A x A 1 + ഥK x m A x = q ഥK c m A C A A 1 + ഥK c m A C = ഥH c m A C A A 1 + ഥK c m A C A Model izotermy Langmuira, należy więc do klasy izoterm pseudo-jednoskładnikowych, opisujących idealną adsorpcję składnika A na homogenicznej powierzchni adsorbentu. 67
68 ADSORPCJA NADMIAR ADSORPCYJNY CHROMATOGRAFIA Gibbs, posługując się teorią termodynamiki gazów, w roku 1878 rozpatrzył po raz pierwszy proces adsorpcji na granicy międzyfazowej układu gaz-ciecz. Rozważania Gibbsa są słuszne dla wszystkich dwufazowych układów adsorpcyjnych i należą do podstaw fizykochemii zjawisk powierzchniowych. Z natury używanych metod pomiarowych równowagi adsorpcyjnej mierzony jest nadmiar powierzchniowy - a nie stężenie równowagowe q* na powierzchni adsorbentu. a) warstwa międzyfazowa b) c y i c f i c m i m x powierzchnia Gibbsa f s c) c i i m c i I q * i f m warstwa międzyfazowa y f x powierzchnia Gibbsa m c i s i f m III q * i y x powierzchnia Gibbsa f s i II q * i 68
69 ADSORPCJA NADMIAR ADSORPCYJNY c.d. CHROMATOGRAFIA Dla dwuskładnikowej mieszaniny tzw. powierzchniowy nadmiar stężenia substancji i-tej - zwany adsorpcją nadmiarową, w układzie ciecz-ciało stałe, może być wyrażony zależnością: Γ i A = n i A gdzie: A to pole powierzchni granicznej (czyli powierzchnia adsorpcyjna); n i to nadmiar liczby moli i-tej substancji w warstwie powierzchniowej z powodu adsorpcji w porównaniu z liczbą moli jaka istniałaby w tej warstwie, gdyby nie było adsorpcji. Nadmiar powierzchniowy, dla mieszaniny pseudo-jednoskładnikowej, złożonej ze składnika czynnego w ruchu masy (adsorptywu) oraz inertu, wyraża następujące równanie: Γ i q i q i x i m q i 1 x i m Dla adsorpcji wieloskładnikowej w układzie ciecz-ciało stałe przyjmuje się, że wszystkie miejsca aktywne na powierzchni adsorbentu są zajęte. Prowadzi to do następującej postaci równania wyrażającego nadmiar powierzchniowy : Γ i = q i q x i m gdzie: N q = i=1 q i 69
70 ADSORPCJA NADMIAR ADSORPCYJNY c.d. CHROMATOGRAFIA Dla najprostszego przypadku idealnego układu dwuskładnikowego A, B, dla którego stężenie w fazie zaadsorbowanej jest opisane równaniem równowagi w gdzie: układzie pseudo-jednoskładnikowym, gdy wartości stężenia obydwu składników są porównywalne: Równanie przekształca się do następującej postaci: Γ A = q x m m A α AB 1 1 x A m 1 + α AB 1 x A x A s = q A m q = α AB x A m A 1 + α AB 1 x A q A = q B = q ; q A + q B = q ; α AB = x A s x B m x B s x A m 1. Jeśli x m A 0 to wartość mianownika dąży do 1, x S m A α AB x A, wartość Γ A 0 2. Jeśli α to dla Γ AB = const. α AB > 0 Γ A A > 0 0, dla α AB < 0 Γ A < 0 3. Jeśli α AB 0 x A m α AB 1 α AB x A m 70
71 ADSORPCJA x A s NADMIAR ADSORPCYJNY c.d. α AB x A m 1 + α AB x A m CHROMATOGRAFIA Ad. 3. Ogólne równanie izotermy adsorpcji przyjmuje wtedy postać: A równanie izotermy nadmiarowej: α AB 0 α AB x A m 0 x A S 1 Γ A q 1 x A m 4. Jeśli to także iloczyn,, a Otrzymana izoterma nadmiarowa jest krzywą wypukłą. mod * [mole/cm 3 ] *10 3 a) propan-2-ol - dane dośw. izoterma bil izoterma Unilan octan etylu - dane dośw x mod A, [mol/cm 3 ] b) dane dośw. symulacja RP18 RP18e RP8e x A Γ i = q x m m A α AB 1 x A m 1 + α AB x A Przedstawione krzywe określają odpowiednio: dodatnią i ujemną adsorpcję składnika i w całym zakresie wartości ułamka molowego, lub niewielką adsorpcję obu składników roztworu. c) * rozp. org. [mol/dm 3 ]* metanol etanol propan-2-ol acetonitryl x rozp. org. Rys. a) Izoterma nadmiarowa dla układów: (a) NP: octan etylu - n-heksan; propan-2-ol n-heksan; b) dla układu: metanol woda na RP-18, RP-18e i RP-8e; c) CSP (teicoplanina): woda-rozpuszczalnik organiczny. 71
72 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Podstawy teoretyczne Zasadniczym celem chromatografii preparatywnej nie jest chromatografia jednoskładnikowa czy pseudo-jednoskładnikowa. Celem jest rozdzielanie mieszanin wieloskładnikowych. Do modelowania i optymalizacji procesu rozdzielania chromatograficznego niezbędna jest znajomość izoterm opisujących termodynamikę adsorpcji takich mieszanin. Izotermy te muszą uwzględniać rywalizację różnych molekuł o centra aktywne, a także wzajemne wypieranie się molekuł. Ponadto, często należy również brać pod uwagę oddziaływania między różnymi zaadsorbowanymi molekułami. Dalszą komplikację opisu adsorpcji wieloskładnikowej powoduje konieczność uwzględnienia heterogeniczności powierzchni. Z powyższego wynika, że określenie mechanizmu adsorpcji z roztworu wieloskładnikowego jest znacznie trudniejsze niż dla jednoskładnikowego lub pseudojednoskładnikowego. 72
73 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. TEORIA AST Model AST został wyprowadzony z równania adsorpcji Gibbsa w warunkach izotermicznych, dla i-tego składnika oraz dla rozpuszczalnika ( solv ): NC A dπ i = i q i dμ s s i + q solv dμ solv Analogiczna zależność dla fazy ruchomej jest zapisana jako: NC i m x i dμ m m i + x solv dμ m solv = 0 W równowadze termodynamicznej potencjał chemiczny fazy zaadsorbowanej oraz ruchomej jest taki sam, a więc dla każdego i-tego adsorptywu i/lub modyfikatora eluentu izotermiczne równanie Gibbsa-Duhema może być otrzymane przez podstawienie wartości potencjałów chemicznych do ww. równań. Otrzymuje się: NC A dπ i = i തq i dμ i s gdzie tzw. wartość adsorpcji składnika i w rozpuszczalniku: തq i = q i x i m m x solv q solv (ang. Adsorbed Solution Theory) 73
74 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. NC TEORIA AST c.d. Z równania można wyznaczyć ciśnienie powierzchniowe: π i a i 0 = RT A 0 a i 0 തq i a i m a i m da i m A dπ i = i തq i dμ i s gdzie: a i 0 to aktywność dla czystych składników, dla której każdy z tych składników może osiągnąć tę samą wartość ciśnienia powierzchniowego π mix samodzielnie lub w mieszaninie: π mix = π i a i 0 തq i q i തq mod q mod Równanie przekształci się do postaci: Π i a i 0 = 0 a i 0 q i a i m a i m da i m gdzie: Π i = π i Τ A R T q i jest izotermą jednoskładnikową i-tego składnika w obecności rozpuszczalnika solv (INERTU), wyrażoną jako funkcja aktywności fazy ruchomej. 74
75 ADSORPCJA (2.97) Chcąc obliczyć wartości oraz, układ równań ( ) musi być rozwiązywany numerycznie. TEORIA AST c.d. Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. Model AST dodatkowo zakłada, że zależność równowagowa wynikająca z prawa Raoulta, lecz zapisana dla układu rzeczywistego, gdzie stężenie zastąpione jest aktywnością molową, jest dana równaniem: a m i = x s 0 n i a i m przy: a i 0 = 1 1 a i i=1 Chcąc obliczyć wartości a i 0 oraz x i s, układ równań od równania na Π i oraz podanych powyżej musi być rozwiązywany numerycznie. 75
76 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. TEORIA AST c.d. Model AST bazuje na wcześniej znalezionych doświadczalnie izotermach jednoskładnikowych dla każdego i-tego składnika w obecności rozpuszczalnika solv (INERTU) adsorbowanej mieszaniny, opisujących rzeczywiste (nieidealne) zachowanie się fazy ruchomej. * * m i i i i i q a q x Uwzględnia się także rzeczywiste zachowanie fazy zaadsorbowanej. Zawarte jest ono w wartościach współczynników aktywności dla fazy zaadsorbowanej, których wartości należy estymować. Wieloskładnikowa izoterma konkurencyjna q q a NCMN s NCMN s 1 xi s lnγ i x * * 0 i tot i1 i i i1 i gdzie: s γi jest współczynnikiem aktywności w fazie zaadsorbowanej 76
77 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. TEORIA RAST Model RAST należy do modeli prostszych. Zakłada się w nim, że nieidealność dotyczy tylko fazy zaadsorbowanej: q q x NCMN s NCMN s 1 x ln i s γi x * * 0 i tot i1 i1 i i i TEORIA IAST Postępowanie analogiczne do modelu AST W przypadku układu idealnego wszystkie modele upraszczają się do modelu IAST: q q x NCMN s NCMN s 1 x ln i s γi x * * 0 i tot i1 i1 i i i (ang. Real Adsorbed Solution Theory) (ang. Ideal Adsorbed Solution Theory) 77
78 ADSORPCJA Statyka procesu c.d. IZOTERMY KONKURENCYJNE WIELOSKŁADNIKOWE c.d. TEORIE IAST i HIAST W modelu IAST można: - uwzględnić heterogeniczność powierzchni (nieidealność w fazie zaadsorbowanej poprzez wprowadzenie do modelu pojedynczej izotermy heterogenicznej: NCMN s 1 xi * * 0 tot i1 i i konkurencyjna Langmuira * 0 q i xi q q x Przykład z układu klasycznego - izoterma q K x m γ m i i i i 2 1 K x m γ m j j j j 1 - lub też zastosować model HIAST, w którym wstawia się izotermę konkurencyjną. Model HIAST składa się z modelu IAST do wyrażenia izotermy lokalnej dla danego miejsca aktywnego oraz ciągłego rozkładu energii adsorpcji do obliczenia izotermy globalnej: NCMN s 1 xi * * tot i i * i1 * i i d q q x q x f E q x,e E (ang. Heterogeneuos Ideal Adsorbed Solution Theory) 78
79 ADSORPCJA I CHROMATOGRAFIA Dynamika procesu D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.3. 79
80 Dynamika adsorpcji bilans(bilanse) ciepla kinetyka transportu ciepla bilans(bilanse)masy kinetyka transportu masy statyka (termodynamika) procesu kinetykareakcji równanie przenoszenia pedu Matematyczne zależności między istotnymi dla danego procesu wielkościami nazywa się modelem dynamiki układu (procesu; aparatu) Proces może być ustalony (1) lub nieustalony (2). W przypadkach: (2) procesu nieustalonego w czasie; zmiennej powierzchni procesu (powierzchnia procesu powierzchnia izokoncentryczna, to miejsce geometryczne punktów o jednakowym stężeniu w danej chwili czasu) Model dynamiki procesu transportu masy to układ równań różniczkowych wyrażonych jako bilanse masy dla sześcianu jednostkowego Proces w kolumnie adsorpcyjnej lub chromatograficznej z nieruchomym złożem adsorbentu jest procesem okresowym procesem nieustalonym! 80

81 Dynamika adsorpcji natezenie natezenie akumulacja ubytek skl. A doplywu skl. A odplywu skl. A skl. A w wyniku reakcji Stężeniem najczęściej używanym w modelu adsorpcji jest koncentracja molowa C na / V A [kmola/m 3 ] Definicja gęstości strumienia masy miary prędkości procesu: N C u ' A A A [kmola/m 2 s] N Ax dy dz N Ax N x Ax dx d ydz N d y dz dm ' A ' A [kmola/s] 1. R. Petrus; G. Aksielrud; J. Gumnicki; W. Piątkowski Wymiana masy w układzie ciało stałe ciecz, 2. D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin, 3. Z. Kembłowski, St. Michałowski, Cz. Strumiłło, R. Zarzycki Podstawy teoretyczne Inżynierii Chemicznej i Procesowej 81
82 Dynamika adsorpcji Model ogólny GR (tylko dla Chromatografii) ruch masy c if (t) x = 0 x = L Solid phase Bondary Layer c pi R p q * i c i Założenia upraszczające: (*) proces jest izotermiczny, (*) szybkość fazy ruchomej jest stała, jej ściśliwość jest do pominięcia, (*) złoże jest upakowane porowatymi ziarnami adsorbentu o sferycznym kształcie i ujednoliconej wielkości, (*) gradient stężenia w kierunku promieniowym w aparacie jest do pominięcia, (*) dla każdego składnika systemu istnieje równowaga lokalna pomiędzy wartością stężenia na powierzchni adsorbentu a wartością stężenia w nieruchomym filmie płynu, (*) wartości współczynników: dyspersji wzdłużnej oraz przenikania masy w aparacie są stałe. 82
83 Model ogólny GR (tylko dla Chromatografii) c.d. akumulacja natezenie natezenie ubytek skl. A skl. A doplywu skl. A odplywu skl. A w wyniku procesu czastkowego Bilans masy dla i-tego składnika w fazie ruchomej: C u C C ' ( ) 2 t x x 2 i i i D L F kext, iap Ci Cpi r Rp e dla t > 0; r = 0 oraz Człon hydrodynamiczny dyspersja wzdłużna c pi ( t, r) r 0 C pi ( t, r) D k ext, i C C ( t, r) eff r i pi Bilans masy w nieruchomym filmie płynu w porach: Cpi q D i effi 1 C 2 pi F" r 2 t t p r r r gdzie: Bondary Layer D effi F' 1 ; p e Solid phase e C i F'' 1 p 83
84 Dynamika adsorpcji Model ogólny GR (tylko dla Chromatografii) c.d. D eff jest efektywnym współczynnikiem dyfuzji w porach: Dyfuzyjność wewnętrzna obliczana jest następująco: dq D D D dc * 1 i effi pi p p si pi C 2 p Dmi p Dpi 2 p D mi 2 Zależność między porowatościami układu t, e, p : ( 1 ) gdzie: t, e, p są odpowiednio porowatością całkowitą, zewnętrzną oraz wewnętrzną układu; Do równań bilansów masy w modelu GR należy dołączyć równania: - zależności pomiędzy stężeniem powierzchniowym a stężeniem w płynie w postaci: * równania kinetycznego qi f C ; równania izotermy adsorpcjiq p i f Cp ; w przypadku gdy kinetyka jest szybka tylko izotermę, - warunki początkowe oraz warunki brzegowe całkowania dla chromatografii. t e e p 84
85 Dynamika adsorpcji Zaletą modelu GR jest możliwość analizowania na jego podstawie wpływu prawie wszystkich oporów transportu masy występujących w dynamice adsorpcji lub chromatografii. Jednocześnie model GR jest relatywnie skomplikowany a rozwiązanie numeryczne wymaga stosunkowo długiego czasu komputerowego obliczeń. W praktyce, w budowie modelu dynamiki adsorpcji lub chromatografii bardzo często stosuje się uproszczenia modelu GR, które mają w określonych warunkach podobną dokładność opisu. Rozwiązania tych uproszczonych modeli są wystarczające do opisu rzeczywistości adsorpcji lub chromatografii, ale otrzymuje się je znacznie szybciej. 85
86 Dynamika adsorpcji Model ogólny GR (dla adsorpcji) uzupełnienie Bilans ciepła w fazie ruchomej: Tc u (Tc ) * T D c 4 Lc t x 2 x d c e 2 k w p,c c T T a T T c sc c F' p,c c p c c srr * D Lc gdzie: - współczynnik dyspersji wzdłużnej w aparacie dla transportu ciepła; c p,c - ciepło właściwe płynu; c - gęstość płynu; c - współczynnik wnikania ciepła od płynu do powierzchni adsorbentu; w - współczynnik wnikania ciepła od płynu do ściany aparatu; T c - temperatura cieczy; T s - temperatura zewnętrznej powierzchni adsorbentu; T sc - temperatura ściany; d k - średnica aparatu. Bilans ciepła w ziarnie adsorbentu: Ts es, 2 0 Ts 2 T s H q t c p,s 2 s r r r c p,s s t gdzie: es0 - efektywny współczynnik przewodzenia ciepła w ziarnie; c p,s - ciepło właściwe ciała stałego; s - gęstość ziarna; H - ciepło adsorpcji. + warunki początkowe oraz warunki brzegowe całkowania dla adsorpcji. 86
87 Dynamika adsorpcji Rozwiązania analityczne modelu dynamiki adsorpcji W rozdz. 7 podręcznika 1) - podano rozwiązania analityczne modelu dynamiki adsorpcji dla wybranego aparatu i odpowiednich, założonych uproszczeń modelu, W rozdz. 3 podręcznika 2) - podano rozwiązania analityczne modelu dynamiki chromatografii dla odpowiednich, założonych uproszczeń modelu. Analityczne rozwiązania modelu pozwalają poznać jak zachowa się proces w odpowiedzi na konkretny sygnał wlotowy oraz ustalone warunki prowadzenia procesu mimo tego, że są to w większości przypadków rozwiązania mało dokładne! 1) R. Petrus; G. Aksielrud; J. Gumnicki; W. Piątkowski Wymiana masy w układzie ciało stałe ciecz, podrozdz ) D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.3; 4. 87
88 Dynamika adsorpcji Proces nazywany adsorpcją jest procesem ogólnym, złożonym z szeregowo równoległych procesów cząstkowych. Należy szukać i stawiać hipotezę mechanizmu tego procesu dla każdego, badanego przypadku. Jednym z punktów tej hipotezy jest założenie, który z procesów cząstkowych jest natychmiastowy, szybki, a który wolny, bardzo wolny czyli który z procesów jest mechanizmem kontrolującym szybkość procesu ogólnego. W adsorpcji oraz chromatografii wszystkie kombinacje, który z procesów cząstkowych kontroluje szybkość ogólną procesu są możliwe. Za pomocą modelu GR oraz różnych zakładanych mechanizmów sorpcji można interpretować dane doświadczalne, weryfikować hipotezę mechanizmu i oceniać dokładność modelu. Na ogólną szybkość procesu adsorpcji mogą mieć wpływ szybkości następujących etapów: 1. wnikania masy adsorptywu A od płynu do zewnętrznej powierzchni adsorbentu (dyfuzji zewnętrznej), 2. dyfuzji adsorptywu poprzez porowatą strukturę adsorbentu do wnętrza ziarna (dyfuzji wewnętrznej), 3. adsorpcji składnika A na powierzchni adsorbentu (procesu powierzchniowego adsorpcja-desorpcja). 88
89 Proces cząstkowy kontrolujący szybkość procesu ogólnego nazywanego adsorpcją 1. wnikanie masy adsorptywu i od płynu do zewnętrznej powierzchni adsorbentu (dyfuzja zewnętrzna); 2. dyfuzja adsorptywu i poprzez porowatą strukturę adsorbentu do wnętrza ziarna (dyfuzja wewnętrzna); 3-4. adsorpcja składnika i na powierzchni adsorbentu (proces powierzchniowy) - równowaga termodynamiczna procesu powierzchniowego równoległa dyfuzja powierzchniowa adsorbatu A 1 2,5 A B A 6 Płyn 1 2 A Konwekcja, dyspersja A Dyfuzja zewnętrzna przenoszenie masy z płynu do powierzchni ziarna 2. Dyfuzja wewnętrzna (w porach) 3. Adsorpcja Dyfuzja powierzchniowa 4. Desorpcja 5. Dyfuzja wewnętrzna odwrotna 6. Dyfuzja zewnętrzna odwrotna Powierzchnia ziarna (zewnętrzna) Powierzchnia porów (wewnętrzna) 89
90 N ' A F ' k a C C ( r R ) Kinetyka procesu adsorpcji To są przykłady zapisu równań szybkości poszczególnych procesów cząstkowych w zapisie z Modelu ogólnego GR (dla chromatografii) Szybkość dyfuzji zewnętrznej wnikania masy w płynie na zewnątrz ziarna: ext, i p i pi p Szybkość dyfuzji wewnętrznej dyfuzji w porach ziarna adsorbentu: N ' A D 1 C effi 2 pi r 2 p r r r Szybkość procesu powierzchniowego adsorpcja- desorpcja k A S 1 A( S) k Przypadek podany dla izotermy Langmuira! -1 q * k K c q q (1 ) N' A k1ic pi (qi qi ) k1i i 1i ri pi i i 90
91 Dynamika chromatografii - Modele uproszczone Model Równowagowo-Dyspersyjny (ED) Różniczkowy bilans masy dla i-tego składnika w fazie ruchomej: gdzie: C q C C t t x x F Założenia upraszczające: * 2 i i i i F w Da 2 ; 1 t t N w L 2D ; a (*) wszystkie opory procesów cząstkowych ruchu masy ukryte w D a, (*) układ osiąga równowagę termodynamiczną procesu powierzchniowego praktycznie natychmiast. a u w t 1) D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.3. 91

; Kri k1i k1i 1i jest stałą równowagi adsorpcyjnej: k (1 K c ) ri pi q * i q i")
92 Model Kinetyczno-Dyspersyjny (TD) Różniczkowy bilans masy dla i-tego składnika w fazie ruchomej: C q C D C t t x x 2 i i i e L i F w 2 t Kinetyka zewnętrznego i wewnętrznego transportu masy a także kinetyka procesu powierzchniowego adsorpcja-desorpcja są przedstawione łącznie za pomocą równania szybkości procesu: qi t * ' * mi i i mi i i k ( q q )= k ( C C ) gdzie: k mi jest zastępczym współczynnikiem przenikania masy gdzie: q t i q i k Model Reakcyjno-Dyspersyjny (RD) 1 icpi ( qi qi ) k1i q i jest pojemnością adsorbentu (maksymalnym pokryciem złoża miejscami aktywnymi); Kri k1i k1i 1i jest stałą równowagi adsorpcyjnej: k (1 K c ) ri pi q * i q i 92
93 Model Idealny (ID) Bilans masowy tego modelu jest następujący: * Ci qi Ci F w t t x 0 MODELE DYSKRETNE Model Craiga Równanie bilansu masowego w modelu Craiga dla i-tego składnika w j-tym stopniu teoretycznym oraz w k-tym etapie: C C F q q k1 k k1 k i, j i, j1 i, j i, j 0 Związek pomiędzy ilością stopni teoretycznych N c a sprawnością kolumny N a (model ED): Nc Na 1 k k k F H - to współczynnik retencji składnika chromatografowanego powiązany z równowagą adsorpcyjną (tu dla izotermy liniowej) 93
94 Parametry kinetyczne modeli transportu masy W omówionych modelach występują parametry kinetyczne charakteryzujące szybkość transportu (przenikania) masy, a zatem decydujące o ogólnej (efektywnej) szybkości procesu adsorpcji. Na ogólną szybkość tego procesu mają wpływ szybkości następujących etapów cząstkowych (powtórzenie!): 1.wnikania masy adsorptywu od płynu do zewnętrznej powierzchni adsorbentu (dyfuzja zewnętrzna) określonego wartością zewnętrznego współczynnika wnikania masy, k ext, 2.dyfuzji adsorptywu poprzez porowatą strukturę adsorbentu do wnętrza ziarna (dyfuzja wewnętrzna) określonej efektywnym współczynnikiem dyfuzji D eff, 3.kinetyki procesu adsorpcji-desorpcji składnika na powierzchni adsorbentu wyrażonej równaniem (proces powierzchniowy). Ponadto kształt pików chromatograficznych jest zdeterminowany wartością współczynnika dyspersji D L. 94
95 Współczynnik zewnętrznego wnikania masy (dyfuzji zewnętrznej) Wartość zewnętrznego współczynnika wnikania masy k ext można obliczyć bezpośrednio z równań kryterialnych, podanych w literaturze. Najczęściej używanym równaniem jest korelacja Wilsona-Geankoplisa (korelacja przykładowa): Sh = k ext d p D m = 1.09 ε e Re Sc Kryteria użycia: 1. Równanie modułowe obowiązuje dla wartości liczby Reynoldsa z zakresu < Re < Poszczególne moduły bezwymiarowe: moduł Sherwooda (Sh), Schmidta (Sc) i Reynoldsa (Re) są zdefiniowane najczęściej następująco: Sh = k ext d p D m ; Re = u d p ρ m ηm ; Sc = ηm ρ m ; D m przy czym: d p to przeciętna średnica ziarna adsorbentu, D m - współczynnik dyfuzji molekularnej adsorptywu w płynie (fazie ruchomej), u to prędkość fazy ruchomej liczona na pusty przekrój aparatu, m to dynamiczny współczynnik lepkości fazy ruchomej, m to gęstość fazy ruchomej, e to porowatość zewnętrzna złoża. 95
96 Współczynnik dyfuzji molekularnej Obliczenie współczynnika wnikania masy wymaga znajomości wartości współczynnika dyfuzji molekularnej D m dla pary adsorptyw-rozpuszczalnik. Wartość D m dla układów dyfuzyjnych, dla których brak jest danych eksperymentalnych, najczęściej oblicza się z równania Wilke-Changa, zaadoptowanego dla mieszanin rozpuszczalników przez Perkinsa i Geankoplisa. Korelacja ta jest polecana do obliczeń wartości współczynnika dyfuzji molekularnej dla małocząsteczkowych adsorptywów w podstawowych rozpuszczalnikach, a błąd obliczeń nie przekracza 10%: T D m = Φm M m η m v i 0.6 gdzie: T to absolutna temperatura, v i to objętość molowa adsorptywu w jego temperaturze wrzenia [cm3/mol], rozumiana jako wielkość addytywna, m to współczynnik dynamiczny lepkości fazy ruchomej. Iloczyn ( m M m ) jest obliczany jako funkcja ułamka molowego danej dyfundującej pary oraz odpowiednich współczynników asocjacji i, gdy roztwór (mieszanina) odbiega w swym zachowaniu od ideału: Φ m M m = x A φ A M A + x B φ B M 96
97 Współczynnik dyfuzji molekularnej dla związków wielkocząsteczkowych, w tym białek Dyfuzja dużych sferycznych molekuł może być obliczona z równania Stokesa-Einsteina: D m = k B T 6 π η m R s gdzie: k B jest stałą Boltzmana, R s jest promieniem Stokesa. Zależność współczynnika dyfuzji od temperatury wynika bezpośrednio z zależności współczynnika lepkości od temperatury. Jeżeli makromolekuła ma kształt kulisty, wówczas promień Stokesa R s może być przyrównany do promienia molekuły, R 0, który to dla oblicza się z równania: R 0 = 3 M 4 π ρ N A Τ 1 3 gdzie: M jest masą molekularną białka, gęstością molekuły a N A liczbą Avogadro. 97
98 Współczynnik dyfuzji molekularnej dla związków wielkocząsteczkowych, w tym białek Young i współautorzy na bazie równania Stokesa-Einsteina opracowali następującą korelację do obliczania współczynnika dyfuzji dla białek: D m = T η m M gdzie: D m jest wyrażony w [cm 2 /s]]. Dla makrocząsteczek, które nie mają kształtu sferycznego współczynnik dyfuzji proporcjonalny jest do ich masy molekularnej w potędze zbliżonej do 0.5: D m = M exp T Obliczenie wartości współczynnika dyfuzji w fazie ciekłej oraz określenie wpływu zmiany jego wartości z temperaturą można przeprowadzić przez tzw. grupę dyfuzyjną F A (metoda Wilkego), równą: F A = T D m η m = f v i = const. 98
99 Efektywny współczynnik dyfuzji Szybkość transportu masy do wnętrza ziarna adsorbentu (Dyfuzja wewnętrzna (w porach) to szybkość dyfuzji masy i jest ona charakteryzowana wartością efektywnego współczynnika dyfuzji D eff w nieruchomym płynie w porach adsorbentu. Wartość D eff obejmuje skomplikowany proces, w którego skład wchodzi: dyfuzja w makroporach, mikroporach oraz tzw. dyfuzja powierzchniowa. W przypadku przewagi makroporów w ziarnie, to jest takich porów, w których średnia droga swobodna dyfundującej cząstki jest dużo mniejsza od średnicy poru, mechanizm dyfuzji jest identyczny z mechanizmem dyfuzji molekularnej w płynie na zewnątrz ziarna, charakteryzowanej wartością współczynnika dyfuzji D m. W sytuacji odwrotnej, gdy przeważają w ziarnie mikropory o efektywnej szybkości dyfuzji decydować będzie ich kształt i stosunek średnicy poru do średnicy dyfundującej molekuły. W tym przypadku należy się też spodziewać istotnej roli dyfuzji powierzchniowej. Zjawisko dyfuzji powierzchniowej wystąpi wówczas, gdy bariera energetyczna występująca między sąsiadującymi miejscami aktywnymi powierzchni jest mniejsza od ciepła adsorpcji danej substancji. Molekuły adsorbatu mogą wtedy przemieszczać się skokowo po powierzchni adsorbentu między centrami aktywnymi. 99
100 Efektywny współczynnik dyfuzji Jeżeli dyfuzja powierzchniowa może być zaniedbana wówczas efektywny współczynnik dyfuzji D eff oblicza się z równania: D eff = ε p D p gdzie: D p jest współczynnikiem dyfuzji w porach, a porowatość ziarna uwzględnia fakt, że molekuła dyfunduje nie poprzez cały przekrój poprzeczny ziarna, a jedynie przez jego część, proporcjonalną do p. Gdyby pory w ziarnie adsorbentu były prostymi szerokimi kanałami, wówczas współczynnik D p równałby się współczynnikowi dyfuzji molekularnej D m w płynie. W rzeczywistości pory tworzą skomplikowane kształty, przez co droga molekuły do środka ziarna może znacznie przewyższać geometryczną odległość między powierzchnią ziarna i jej wnętrzem. Fakt ten uwzględnia się wprowadzając do równania współczynnik krętości porów, : D eff = ε p D m χ Do obliczania wartości współczynnika krętości porów zaproponowano szereg korelacji, z których otrzymuje się dość rozbieżne wartości (PRZYKŁAD!): χ = 2 ε p ε p 2 100
101 Efektywny współczynnik dyfuzji W przypadkach, gdy zjawisko dyfuzji powierzchniowej nie może być zaniedbane, wówczas efektywny współczynnik dyfuzji D eff oblicza się z równania: D eff = ε p D m χ + 1 ε p D s Miyabe opublikował następującą zależność na D s (PRZYKŁAD!): D s0 = D 0 exp α Q st,0 R T exp α β q R T gdzie: D 0 jest współczynnikiem empirycznym, Q st,0 jest izosterycznym ciepłem adsorpcji przy zerowym pokryciu powierzchni, jest współczynnikiem proporcjonalności; jest parametrem empirycznym; R to stała gazowa. 101
 i spada N t.")
102 Współczynnik dyspersji wzdłużnej Ostatnim parametrem niezbędnym do rozwiązania modeli kolumn chromatograficznych GR, POR lub TD jest współczynnik dyspersji wzdłużnej D L w kolumnie. Wiele zjawisk przyczynia się do dyspersji stref rozdzielanych substancji. Wzrost dyspersji wzdłużnej = spadek sprawności kolumny wzrasta H (HETP) i spada N t. Im niższa wartość wysokości równoważnej półce teoretycznej, tym wyższa wartość liczby półek teoretycznych tym wyższa sprawność rozdzielania - także - kolumny 102
103 Współczynnik dyspersji wzdłużnej Chung i Wen do obliczeń tego parametru zaproponowali korelację na wartość liczby kryterialnej Pecleta: Pe = u d p D L ε e = Re 0.48 Częściej stosowana jest korelacja Gunna, która wykazuje lepszą zgodność z doświadczeniem: przy czym: ε e D L d p u = B 1 + σ v σ v 2 + ε e χ Re Sc B = 2 Re Sc 4 α 2 1 p ε e Re Sc 2 16 α ε 2 p 1 p 3 exp 4α ε e e p 1 p Re Sc 1 p = exp( 24ΤReሻ gdzie: 1 jest pierwszym pierwiastkiem funkcji Bessela zerowego rzędu, jest współczynnikiem krętości σ v 2 Re = u d p ρ m ηm ; Sc = ηm ρ m ; D m dla Re >0.05 porów wypełnienia ( = 1.4), jest bezwymiarową wariancją rozkładu lokalnej prędkości w odniesieniu do średniej prędkości eluentu. W warunkach chromatografii kolumnowej wartość ta jest w przybliżeniu równa zeru. 103
104 Stałe szybkości adsorpcji i desorpcji W znakomitej większości przypadków powierzchniowy proces adsorpcji można traktować jako nieskończenie szybki. Gdy jednak szybkość procesu powierzchniowego adsorpcja-desorpcja jest porównywalna z szybkością transportu masy, wówczas modele dynamiki kolumny chromatograficznej muszą być rozwiązywane z odpowiednim modelem kinetyki procesu adsorpcji-desorpcji składnika na powierzchni adsorbentu. Stałe szybkości adsorpcji oraz desorpcji k +1i, k -1i można wyznaczyć na bazie doświadczalnych profili stężenia i dalej doborze modelu izotermy adsorpcji i znalezionej wartości stałej równowagi adsorpcyjnej K r, np. metodą dopasowania do piku. 104
105 F = KRYTERIA KOMPATYBILNOŚCI MODELI GR ORAZ ED I TD Porównania dokładności obliczeń otrzymanych za pomocą różnych modeli można dokonać bezpośrednio porównując wyniki rozwiązań. Jest to sposób pracochłonny ze względu na analizę wyników symulacji komputerowych dla szerokiego zakresu zmian parametrów. Analityczne rozwiązanie modelu ogólnego, GR, w dziedzinie zmiennych rzeczywistych nie jest jak dotąd możliwe. O ile jednak izoterma adsorpcji jest liniowa to możliwe jest rozwiązanie tego modelu w tak zwanym obrazie Laplace a, przy wykorzystaniu całkowego przekształcenia Laplace a. Znając rozwiązanie modelu kolumny w obrazie Laplace a, dla liniowej izotermy adsorpcji, można obliczyć n-ty moment, m n, piku chromatograficznego. Z kolei momenty piku chromatograficznego związane są z momentami normalizowanymi, takimi jak: pierwszy moment absolutny 1 i drugi moment centralny 2. Obydwa momenty umożliwiają wyznaczenie z danych doświadczalnych liczby półek teoretycznych, N t oraz wysokość półki teoretycznej, HEPT: HETP = L/N t = L σ2 μ 1 2 gdzie: μ 1 2 to drugi moment centralny; σ 2 to wariancja profilu stężenia 1) D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz
106 KRYTERIA KOMPATYBILNOŚCI MODELI GR ORAZ ED Podstawiając wyrażenia na pierwszy i drugi moment do równania na HETP otrzymuje się wzór wiążący wysokość równoważną półce teoretycznej z parametrami kinetycznymi modelu kolumny. Równanie poniżej jest to już równanie porównujące rozwiązania modeli GR i ED za pomocą metody momentów: 2 L 2D 2 1 HETP GR HETP a DL e k udp d p 1 ED 2 N a w u 1 k ' 1 6 F e 10 Deff kext Dla chromatografii liniowej rozwiązania modelu ED są kompatybilne z rozwiązaniami modelu ogólnego GR, jeśli GR spełniona jest równość N t = N a = N t, czyli liczba Pecleta w modelu ED jest równa zastępczej liczbie Pecleta, ' k F 1 p 1 p F' 1 ; e e dq * dc F'' 1 p = N GR D a ε t Τ2 t Jak widać ze wzoru, w przypadku chromatografii liniowej wszystkie efekty kinetyczne powodujące rozmycie frontu sorpcji są addytywne i można je uwzględnić w równaniu na wysokość półki teoretycznej. Wysokość HETP jest sumą niezależnych udziałów: dyspersji wzdłużnej, zewnętrznych oraz wewnętrznych oporów wnikania masy. 106 Pe a = ul p
107 Równanie Van Deemtera Dyspersja wzdłużna - zjawisko niekorzystne, jednak, nieuniknione Zjawiska powodujące dyspersję: Dyfuzja wirowa (A); Dyfuzja molekularna (B); Opory przenoszenia masy (C) 1. w fazie ruchomej (C m ), 2. w fazie stacjonarnej (C S ) Równanie Van Deemter a, HETP = A + B/u + C u 1Τk ext Jeśli opór dyfuzji zewnętrznej jest mały w porównaniu z innymi oporami transportu masy to można go wyeliminować z równania. Podstawiając do liczby Pecleta Pe, równanie opisujące współczynnik dyspersji zapisane w postaci sumy członu dyfuzyjnego oraz członu hydrodynamicznego: D L = γ 1 D m + γ 2 d p u 107
108 Równanie Van Deemter a L N t b = HETP a d p c u gdzie: 1 oraz 2 to stałe równania, d p to średnica ziarna adsorbentu; a = 2 2 e ; b = 2 1 e D m ; k 1 c2 1 k ' 60F ed eff u d Pierwsze dwa człony w równaniu reprezentują udział dyspersji wzdłużnej, ostatni człon udział oporów wymiany masy. 2 p H E P T [ c m ] * 1 e u [ c m / s ] Rys. Porównanie doświadczalnego przebiegu krzywej Van Deemter a oraz wyników symulacji modelem GR. Otrzymano metodą impulsu przy plateau c = 0.1 [g/dm 3 ] dla benzoesanu butylu, faza ruchoma 65% vol. MeOH : H 2 O Piątkowski, W. Gritti, F., Kaczmarski, K., Guiochon, G., Influence of the particle porosity on chromatographic band profiles, J. Chromatogr. A, 989 (2003)
109 KRYTERIA KOMPATYBILNOŚCI MODELI GR ORAZ TD Alternatywna postać równania na wysokość półki teoretycznej może być otrzymana z wykorzystaniem modelu TD. Lapidus i Amundson wykazali, że wpływ oporów przenoszenia masy na rozmycie profilu stężenia może być uwzględniony w sposób pośredni poprzez zastępczy współczynnik przenikania masy k m. Równanie wysokości półki teoretycznej odpowiadające modelowi TD można zapisać: 2 L e k e 2D u HETP GR HETP TD 2 u 1 k k k Porównując równania na HETP dla obu modeli otrzymuje się: 2 1 d t p A ' k 1 m e F 60 D eff q t i t k m mi( qi * qi ) gdzie: A k1 1 k k 2 k 1 k * i dq k F F H dc i 109
![PRZYKŁAD (1) ZASTĄPIENIA MODELU GR MODELEM TD 1.2 1.0 3 2 C 0.8 c [mol/cm 3 ] 10 5 0.6 0.4 1 0.2 Piątkowski, W., Antos D., Kaczmarski, K.](/docs-images/62/47809867/images/110-1.jpg ", Modeling of preparative chromatography processes with slow intraparticle 0.0 mass transport kinetics, 0 J. 100 200 300 400 500 600 700 800 900 1000 1100 Chromatogr. A, 988 (2003) 219 231. t [s] Rys.")
110 PRZYKŁAD (1) ZASTĄPIENIA MODELU GR MODELEM TD C 0.8 c [mol/cm 3 ] Piątkowski, W., Antos D., Kaczmarski, K., Modeling of preparative chromatography processes with slow intraparticle 0.0 mass transport kinetics, 0 J Chromatogr. A, 988 (2003) t [s] Rys. Porównanie doświadczalnego profilu stężenia (punkty) cholanu metylu oraz wyników symulacji dla następujących stężeń: składnika chromatografowanego: c f = 1.89*10-5 [mol/cm 3 ], oraz następujących stężeń modyfikatora eluentu: 0 C = 2.83*10-3 (1); 2.25*10-3 (2);1.17 *10-3 [mol/cm 3 ] mod (3); punkty - doświadczenie; linia ciągła symulacje modelami TD oraz GR z estymowaną wartością współczynnika dyfuzji powierzchniowej D s. 110
 73 89. Rys.")
 C 0 = 14.496 [g/cm 3 ].")
111 PRZYKŁAD (2) ZASTĄPIENIA MODELU GR MODELEM TD Piątkowski, W., Antos D., Gritti, F., Guiochon, G., Study of the competitive isotherm model and the mass transfer kinetics for a BET binary system, J. Chromatogr. A, 1003, (2003) Rys. Porównanie pomiędzy danymi doświadczalnymi i symulowanymi profilami stężenia dla fenetolu oraz benzoesanu propylu. Faza ruchoma: 65% vol. roztwór metanolu w wodzie. D m = [cm 2 /s]; (a) C 0 = [g/cm 3 ]. Profile stężenia symulowane za pomocą modeli: GR oraz TD z wprowadzoną izotermą konkurencyjną według teorii IAS opartą na pojedynczych izotermach BET 111
112 PRZYKŁAD (3) ZASTOSOWANIA TEORII IAS c [ g / d m 3 ] mau t [ s ] 200 Piątkowski, W., Antos D., Gritti, F., Guiochon, G., Study of the competitive isotherm model and the mass transfer kinetics for a BET binary system, J. Chromatogr. A, 1003, (2003) Rys. Porównanie danych doświadczalnych z symulowanymi profilami stężenia dla mieszaniny fenetolu oraz benzoesanu propylu. Profile symulowane za pomocą modelu TD z wyprowadzoną na podstawie izoterm pojedynczych BET izotermą konkurencyjną według teorii IAS. C 01 = C 02 = [g/cm3]; t [s] 112
113 Zakłada się następujące warunki początkowe (WP): t = 0, Analityczne rozwiązywanie Modelu Idealnego (ID) Metoda charakterystyk oparta na tzw. Teorii Równowagi Izoterma liniowa * C q i F i w C i 0 C C i i 0 t t x t x gdzie: w 1 k const. D. Antos, K. Kaczmarski, W. Piątkowski Chromatografia preparatywna jako proces rozdzielania mieszanin rozdz.4. * i dq k F F H dc Zakłada się następujące warunki początkowe (WP): t = 0; ; C i = 0, które odpowiadają kolumnie wolnej od składnika chromatografowanego w chwili rozpoczęcia procesu. Oraz warunki brzegowe (WB): x= 0; 0 t timp ; C i = C F, które opisują prostokątny impuls próbki o stężeniu C F podawany na wlot kolumny przez czas impulsu t imp. i 0 xl Graficzną reprezentacją tego zjawiska są równoległe linie proste (charakterystyki) o nachyleniu: w dt 1 const. 1 k dx 113
114 Metoda charakterystyk oparta na tzw. Teorii Równowagi t x = L t k c m = 0 (WB) c F t s c m t imp m = cf (WB) z= x/l 0 0 z 1 c m = 0 (WP) Przedstawiono w układzie współrzędnych (t,z). Ponieważ stężenie wlotowe jest podawane w postaci impulsu prostokątnego, warunki brzegowe zawierają dwie nieciągłości w punktach x = 0 i t = 0 oraz dla x = 0 i t = t imp. Nieciągłości te odpowiadają dwom liniom granicznym zaznaczonym na rys. Linie ciągłe pomiędzy liniami granicznymi obrazują przemieszczanie się impulsu wzdłuż kolumny chromatograficznej (fala prosta). W okresie czasowym t < t s na wylocie kolumny stężenie składnika wynosi C i = 0, po czasie t s pojawia się na wylocie kolumny (x = L) pik impulsu i stężenie wzrasta skokowo do C i = C F, po czasie t k = t s + t imp stężenie na wylocie spada znowu do zera. Na podstawie wykresu charakterystyk można skonstruować profil stężenia na wylocie z kolumny t t s t k 114
115 c m Metoda charakterystyk oparta na tzw. Teorii Równowagi Izoterma nieliniowa - przykład Langmuira szok fala w ci 1 FH / 1 K C r F 1.2 c F c m 0.6 szok fala t s t fp t fk t t s t fk t c F - przykład anty-langmuira 0.7 c m fala szok t t fp t s 115
116 Metoda charakterystyk oparta na tzw. Teorii Równowagi Chromatografia gradientowa z = x L CV tv V Wpisz tutaj równanie. Gradient schodkowy Gradienty liniowe trafiony nietrafiony 116
117 Wpływ składu fazy ruchomej w adsorpcji konkurencyjnej Przegląd metod stosowanych do opisu procesu Chromatografia analityczna 1. Teoria LSST (ang. Linear Solvent Strength Theory) lnk lnkw gdzie: ψ to parametr opisujący siłę elucji rozpuszczalnika, φ to udział objętościowy aktywnego rozpuszczalnika (modyfikatora), k w to współczynnik retencji substancji rozpuszczonej w wodzie. Chromatografia preparatywna (nieliniowa) 2a. Podejście pół- lub całkowicie empiryczne q * i 0 i mod i H C C 1 K C C 0 i mod i 2b. Podejście poprzez izotermy wieloskładnikowe PATRZ PRZYKŁAD NA NASTĘPNYM PRZEŹROCZU 117
 65 75.")
118 PRZYKŁAD (4) ZASTOSOWANIA TEORII AST * q K a q A 1 i exp A ln 2 h 1 K ai exp h h x A mole ratio *1e C5 - exp. Unilan isotherm 1 Piątkowski W., Petrushka I., Antos D., Adsorbed solution model for prediction of normal-phase chromatography process with varying composition of the mobile phase, J. Chromatogr. A, 1092 (2005) t [s] Rys. Porównanie doświadczalnych profili stężenia cyklopentanonu (C5) rejestrowane przy różnym stężeniu modyfikatora eluentu: octanu etylu (EA) w fazie ruchomej odpowiednio: x EA = 0 (1); (2); (3); 0.25 (4); (5); 1 (6); eluent EA n-heksan - z wynikami symulacji za pomocą modelu ED ze wstawioną izotermą konkurencyjną opartą na teorii AST otrzymanej z nadmiarowych izoterm pojedynczych UNILAN dla każdego z chromatografowanych składników. Wlotowe stężenie cyclopentanonu x C5,F = = const dla wszystkich przebiegów. 118
119 Wpływ temperatury na wartości parametrów izotermy Chromatografia analityczna i preparatywna Równanie van t Hoffa Związek między stałą równowagi i temperaturą ln( K) H / R T S / R lnk H RT 0 S R 0 lnf K exp( H / R T S / R) q * T A, [mol/cm 3 ]* o C 15 o C 20 o C 35 o C 50 o C 0.0 C x A Izotermy nadmiarowe metanolu w wodzie; układ RP-18e Poplewska I., Kramarz R., Piatkowski W., Seidel-Morgenstern A., Antos D., "Influence of preferential adsorption of mobile phase on retention behavior of amino acids on the teicoplanin chiral selector", J. Chromatogr. A, 1173 (2007)
120 Wpływ temperatury na wartości parametrów izoterm c.d. Różniczkując równanie van t Hoff a po (1/T), przy założeniu stałej koncentracji adsorbatu, otrzymuje się: ( Qst ) d(ln C) R d(1/ T) q const. gdzie izosteryczne ciepło adsorpcji (-Q st ) równe jest zmianie entalpii. Można go obliczyć z nachylenia linii, obliczyć ciepło adsorpcji 4 3 q * 1 ln(c) q * 2 q * N 0, , , , , /T 120
121 Wpływ ciśnienia na wartości parametrów izoterm ' ln k V 1 ln F p RT F p gdzie: k = F H jest współczynnikiem retencji, F to stosunek faz, V jest zmianą objętości molowej cząsteczki związanej z jej przejściem od fazy cieczowej do stałej. Zmiana objętości molowej może być istotna dla dużych cząsteczek, takich jak BIAŁKA. Wpływ ciśnienia na czas retencji zaobserwowano między innymi dla insuliny. 121
122 ORGANIZACJA PROCEDURY PRZENOSZENIA SKALI ORAZ OPTYMALIZACJI CHROMATOGRAFII 1. Przeprowadzenie procedury budowania modelu dynamiki chromatografii zaczynając od cyklu badań doświadczalnych na kolumnie analitycznej: Wyznaczenie parametrów modelu izotermy pojedynczej dla każdego ze składników, uwzględniającej czy adsorpcja jest idealna czy rzeczywista w obu fazach układu. Jeśli zastosuje się chromatografię z gradientem wyznaczenie tych parametrów przy różnym stężeniu modyfikatora eluentu czy też w różnej temperaturze, Wyznaczenie modelu izotermy wieloskładnikowej, uwzględniającego fakt idealności lub rzeczywistości adsorpcji w obu fazach układu. Wybranie adekwatnego typu modelu dynamiki procesu. Dyskusja możliwości doboru modelu prostszego. Uzyskanie na bazie eksperymentalnej, także na bazie obliczeniowej, wszystkich innych, potrzebnych danych fizykochemicznych dla optymalizowanego, wieloskładnikowego układu chromatograficznego. Wyznaczenie parametrów modelu dynamiki procesu. Im bardziej skomplikowany model dynamiki tym większa ilość parametrów modelu koniecznych do wyznaczenia. 122
123 ORGANIZACJA PROCEDURY PRZENOSZENIA SKALI ORAZ OPTYMALIZACJI CHROMATOGRAFII c.d. 2. Podjęcie decyzji dotyczącej parametrów optymalizacyjnych. W tym celu należy: Wybrać funkcję celu oraz składnik kluczowy procesu. Wybrać ograniczenia i zmienne decyzyjne optymalizacji. 3. Wybór metody optymalizacji. 123
124 Definicje najczęściej używanych w chromatografii funkcji celu: Pr i t m F i col t c Pfi Pri Yi mi Yi 100% V C inj i, F EC V Pr elu Zmienne decyzyjne ciągłe oraz zmienne decyzyjne dyskretne optymalizacji: C mod L f, i V C inj i, F t 1 t Vcol q wash parametr d p2 /L i Ograniczenia procesu optymalizacji: produkt kluczowy Pui Pu i min Pu i C C i C 1 2 p p max u k L m 2 0 d p 124
125 PRZENOSZENIE SKALI CHROMATOGRAFII 0.5 c m [g/dm 3 ] t [s] Rys. Przenoszenie skali chromatografii: z kolumny 4 mm; d p = 5 m na kolumnę 10 mm; d p = 12 m. L = 10cm. Mieszanina izomerów cis- (C) oraz trans- (T) analogów terpenów. Symulacja modelem ED; izoterma konkurencyjna Langmuira Piątkowski W., Problemy przenoszenia skali w procesie chromatografii preparatywnej, Inż. Chem. i Proc. 26, (2005),
126 ADSORPCJA I CHROMATOGRAFIA Dynamika procesu ciągłego 126
127 Dynamika adsorpcji ciągłej Bilans wylotu: m A V masowy dla kolumny od wlotu do C C m s q q A0 Ak Ak Ap Adsorber współprądowy Szybkość transportu masy: N' m A A k k z z A C s A C C* A A C* A Wynik dla izotermy liniowej oraz dla k C C C C Ak A0 Ak A0 1 1 ' gdzie: ' m s k ' ' exp[ ( ) ] ' 1 H V dla k rzeczywistego R. Petrus; G. Aksielrud; J. Gumnicki; W. Piątkowski Wymiana masy w układzie ciało stałe ciecz, rozdz
128 Dynamika adsorpcji ciągłej c.d. Bilans wylotu: masowy dla kolumny od wlotu do Adsorber przeciwprądowy m A V C C m s q q A0 Ak Szybkość transportu masy: N' m A A k k z z A C s A C C* A A C* A Wynik dla izotermy liniowej oraz dla k Ap Ak C C C C Ak A0 Ak A0 0 gdzie: ' m s V H ' ' ( 1) exp[ 3( 1) k ] ' ' exp[ 3( 1) ] dla k rzeczywistego k 128
129 Przeciwprądowa Chromatografia ciągła (TMB) - IDEA A + B Zone IV A A+B Zone III B A Zone II B Zone I Zone II Zone III Desorbent 130
130 Chromatografia ciągła - TMB Rozwiązanie dla izotermy liniowej q* = HC Jeśli współczynnik rozdzielenia chromatograficznego: i, j H H 2 1 comp. 1 comp. 2 V R V F V E comp. 1 feed comp. 2 Zone IV Zone III Zone II V V IV sh1c1 V III C1 II C 1 VS H1C1 V C m H2 2 VsH2C2 m IV H1 m III II H 1 Zone I H 2C I S 2 V C2 m I H 2 V V S. k V m V k. s V K V D m k to bezwymiarowy stosunek przepływów faz w danej strefie: K = I - IV 131
131 Sekcja m k I m I > H 1 ; m I > H 2 II H 1 < m II < H 2 III H 1 < m III < H 2 IV m IV < H 1 ; m IV < H 2 Rys. pokazuje graficznie ograniczenia zred. przepływu m k w strefach II i III a także rozwiązanie analityczne modelu idealnego dynamiki chromatografii w postaci tzw. trójkąta operacyjnego dla chromatografii ciągłej TMB (SMB). m III H 2 H 1. k V m V k. s Trójkąt operacyjny (region pełnego rozdzielenia) jest zaznaczony w funkcji m k w sekcjach II oraz III H 1 H 2 m II M. Mazzotti, G. Storti, M. Morbidelli, AIChE J, 40 (1994),
 V F C mod, F Zone III. V R Fluid flow Zone II Solid flow Zone IV V. E Extract (2) Zone I Cmod, D Desorbent.")
132 Idea symulowanego ruchu złoża - SMB Desorbent I II III IV A B Solid stream Extract (A) Feed (A,B) Raffinate (B) Feed (1+2) Raffinate (1) V F C mod, F Zone III. V R Fluid flow Zone II Solid flow Zone IV V. E Extract (2) Zone I Cmod, D Desorbent. V D 133
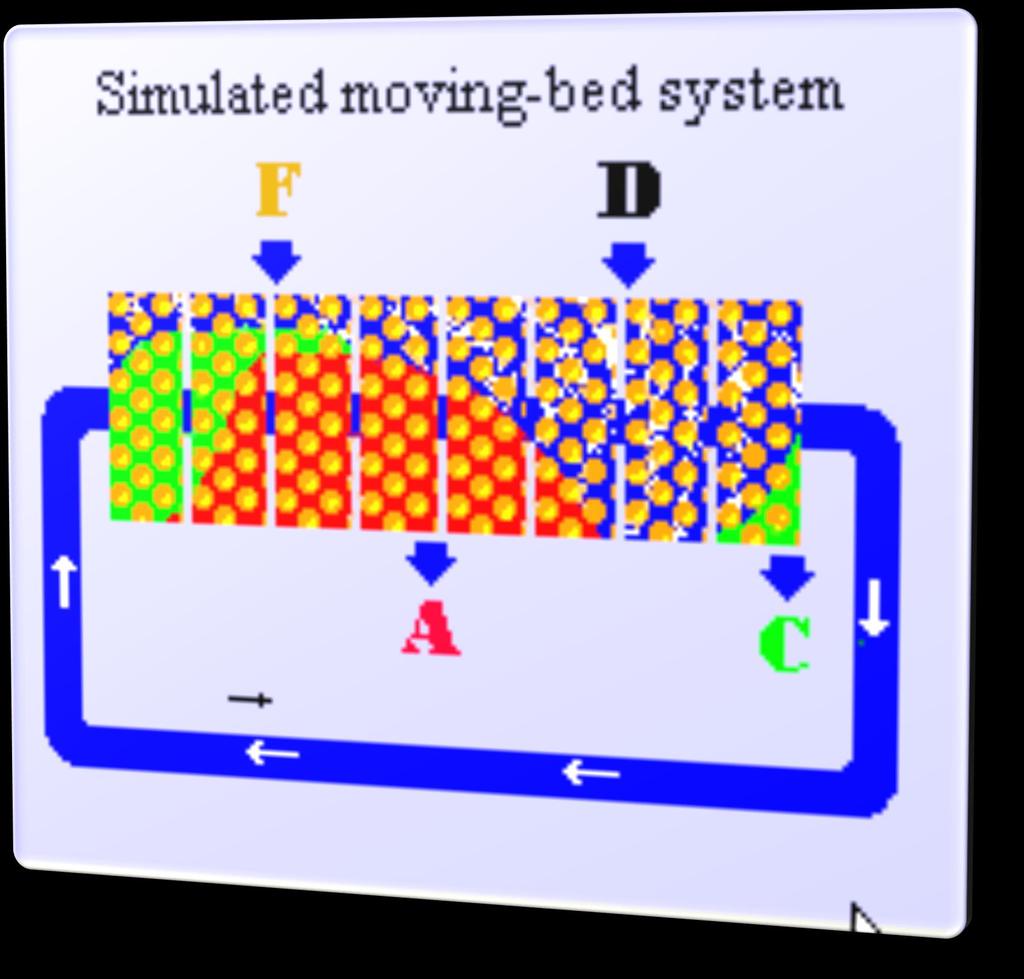
133 134
134 Przykłady przemysłowe 135


135 Kolumny HPLC Ciśnienie do 1000 bar Kolumny HPLC Ciśnienie do 200 bar 136

136 Kolumna preparatywna Hipersep, Prochrom DAC, NOVASEP. Za zgodą NOVASEP, 137

137 80 cm SMB system Produktywność 1 10 kg czystej subst./dzień. 138

138 2 m średnicy 0.5 m wysokości złoża 1,570 L objetości 12,000 kg wagi (Amersham Biosciences) 139
139 Podsumowanie Chromatografia jest praktycznie jedyną przemysłową metodą rozdzielania tzw. mieszanin trudnych, które nie dają się rozdzielić alternatywnymi metodami rozdzielania. Chromatografia może być z punktu widzenia ekonomiki procesu najważniejszą drogą do produkcji substancji czynnych farmacja; Chromatografia jest często najszybszą oraz pewną drogą do otrzymywania pożądanych enancjomerów farmacja, biotechnologia; Chromatografia jest obecnie najważniejszym procesem do oczyszczania oraz otrzymywania pożądanych białek, w tym np. przeciwciał. Przeciwciała stosowane są w diagnostyce oraz w nowych metodach terapeutycznych, zwłaszcza dotyczących leczenia nowotworów, chorób serca, chorób układu immunologicznego oraz alergii. farmacja, biotechnologia. 140
140 Podsumowanie c.d. Sprawność pojedynczej kolumny chromatograficznej okresowej jest bardzo wysoka przy jednoczesnej wysokiej czystości produktu kluczowego. Jednocześnie produktywność procesu chromatografii okresowej jest bardzo niska w porównaniu do altermatywnych metod rozdzielania. Zastosowanie chromatografii ciągłej (SMB) pozwala znacząco zredukować tę różnicę produktywności. 141
141 Wybrany dorobek zespołu w zakresie chromatografii w tym rozdzielania białek 1. Petrus R., Aksielrud G., Gumnicki J., Piątkowski W., Wymiana masy w układzie ciało stałe-ciecz, monografia, Oficyna Wyd. PRz., Rzeszów Antos D., Kaczmarski K., Piątkowski W., Chromatografia preparatywna jako proces rozdzielania mieszanin, monografia, wyd.2 zmienione Oficyna WNT, Warszawa Muca R., Piątkowski W., Antos D., Effects of thermal heterogeneity in hydrophobic interaction chromatography, J. Chromatogr (2009) Muca R., Piątkowski W., Antos D., Altering efficiency of hydrophobic interaction chromatography by combined salt and temperature effects, J. Chromatogr (2009) Muca R., Marek W., Piątkowski W., Antos D., Influence of the sample-solvent on protein retention, mass transfer and unfolding kinetics in hydrophobic interaction chromatography, J. Chromatogr (2010) Marek W., Piątkowski W., Antos D., Multiple-injection technique for isolating a target protein from multicomponent mixtures, J. Chromatogr (2011) Marek W., Muca R., Woś S., Piątkowski W., Antos D., Isolation of monoclonal antibody from a CHO supernatant.i. Assessment of different separation concepts, J. Chromatogr. A 1305 (2013) Marek W., Muca R., Woś S., Piątkowski W., Antos D., Isolation of monoclonal antibody from a CHO supernatant.ii. Dynamics of the integrated separation on IEC and HIC column, J. Chromatogr. A 1305 (2013) Poplewska I., Muca R., Strachota A., Piatkowski W., Antos D., Adsorption behavior of proteins on temperature-responsive resins, J. Chromatogr. A, 1324 (2014) Poplewska I., Piątkowski W., Antos D., Overcoming solubility limits in overloaded gradient hydrophobic interaction chromatography, J. Chromatogr. A, 1386 (2015) Ryś S., Piątkowski W., Antos D., Predictions of matrix-assisted refolding of α-lactalbumin: Process efficiency versus batch dilution method, Eng. Life Sci., 15 (2015) Ryś S., Muca R., Kołodziej M., Piątkowski W., Dürauer A., Jungbauer A., Antos D., Design and optimization of protein refolding with crossflow ultrafiltration, Chem. Eng. Sc., 130 (2015), Antos D., Piątkowski W., Band deformation in non-isocratic liquid Chromatography, Trends Anal. Chem., 81 (2016), 69 78, 14. Gorczyca R., Marek W., Bochenek R., Piątkowski W., Antos D., Protein separation in carousel multicolumn setup. Performance analysis and experimental validation, J. Chromatogr. A, 1460 (2016), 40 50, 15. Antos D., Piątkowski W., Thermodynamics of Multicomponent Liquid Chromatography, Chem. Ing. Tech. 2016, 88, (11), 1 13, 16. Muca R., Piątkowski W., Antos D., A short-cut method for evaluation of yield reduction due to protein deposition onto the membrane surface in crossflow ultrafiltration, Eng. Life Sci., 0 (2016) 1 12.

142 DZIĘKUJĘ ZA UWAGĘ
4. WYZNACZENIE IZOTERMY ADSORPCJI METODĄ ECP
 4. WYZNACZENIE IZOTERMY ADSORPCJI METODĄ ECP Opracował: Krzysztof Kaczmarski I. WPROWADZENIE W chromatografii adsorpcyjnej rozdzielanie mieszanin jest uwarunkowane różnym powinowactwem adsorpcyjnym składników
4. WYZNACZENIE IZOTERMY ADSORPCJI METODĄ ECP Opracował: Krzysztof Kaczmarski I. WPROWADZENIE W chromatografii adsorpcyjnej rozdzielanie mieszanin jest uwarunkowane różnym powinowactwem adsorpcyjnym składników
Zjawiska powierzchniowe
 Zjawiska powierzchniowe Adsorpcja Model Langmuira Model BET 1 Zjawiska powierzchniowe Adsorpcja Proces gromadzenia się substancji z wnętrza fazy na granicy międzyfazowej; Wynika z tego, że w obszarze powierzchniowym
Zjawiska powierzchniowe Adsorpcja Model Langmuira Model BET 1 Zjawiska powierzchniowe Adsorpcja Proces gromadzenia się substancji z wnętrza fazy na granicy międzyfazowej; Wynika z tego, że w obszarze powierzchniowym
1. MODELOWANIE I SYMULACJA PRACY PREPARATYWNEJ KOLUMNY CHROMATOGRAFICZNEJ I KOLUMNY ADSORPCYJNEJ PROGRAMEM Kolumna Chromatograficzna
 1. MODELOWANIE I SYMULACJA PRACY PREPARATYWNEJ KOLUMNY CHROMATOGRAFICZNEJ I KOLUMNY ADSORPCYJNEJ PROGRAMEM Kolumna Chromatograficzna opracował: Krzysztof Kaczmarski I. WPROWADZENIE Najprostszym modelem
1. MODELOWANIE I SYMULACJA PRACY PREPARATYWNEJ KOLUMNY CHROMATOGRAFICZNEJ I KOLUMNY ADSORPCYJNEJ PROGRAMEM Kolumna Chromatograficzna opracował: Krzysztof Kaczmarski I. WPROWADZENIE Najprostszym modelem
5. WYZNACZENIE KRZYWEJ VAN DEEMTER a I WSPÓŁCZYNNIKA ROZDZIELENIA DLA KOLUMNY CHROMATOGRAFICZNEJ
 5. WYZNACZENIE KRZYWEJ VAN DEEMTER a I WSPÓŁCZYNNIKA ROZDZIELENIA DLA KOLUMNY CHROMATOGRAFICZNEJ Opracował: Krzysztof Kaczmarski I. WPROWADZENIE Sprawność kolumn chromatograficznych określa się liczbą
5. WYZNACZENIE KRZYWEJ VAN DEEMTER a I WSPÓŁCZYNNIKA ROZDZIELENIA DLA KOLUMNY CHROMATOGRAFICZNEJ Opracował: Krzysztof Kaczmarski I. WPROWADZENIE Sprawność kolumn chromatograficznych określa się liczbą
Wykład 1. Anna Ptaszek. 5 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Chemia fizyczna - wykład 1. Anna Ptaszek 1 / 36
 Wykład 1 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 5 października 2015 1 / 36 Podstawowe pojęcia Układ termodynamiczny To zbiór niezależnych elementów, które oddziałują ze sobą tworząc integralną
Wykład 1 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 5 października 2015 1 / 36 Podstawowe pojęcia Układ termodynamiczny To zbiór niezależnych elementów, które oddziałują ze sobą tworząc integralną
Termodynamika fazy powierzchniowej Zjawisko sorpcji Adsorpcja fizyczna: izoterma Langmuira oraz BET Zjawiska przylegania
 ermodynamika zjawisk powierzchniowych 3.6.1. ermodynamika fazy powierzchniowej 3.6.2. Zjawisko sorpcji 3.6.3. Adsorpcja fizyczna: izoterma Langmuira oraz BE 3.6.4. Zjawiska przylegania ZJAWISKA PWIERZCHNIWE
ermodynamika zjawisk powierzchniowych 3.6.1. ermodynamika fazy powierzchniowej 3.6.2. Zjawisko sorpcji 3.6.3. Adsorpcja fizyczna: izoterma Langmuira oraz BE 3.6.4. Zjawiska przylegania ZJAWISKA PWIERZCHNIWE
Pytania z Wysokosprawnej chromatografii cieczowej
 Pytania z Wysokosprawnej chromatografii cieczowej 1. Jak wpłynie 50% dodatek MeOH do wody na retencję kwasu propionowego w układzie faz odwróconych? 2. Jaka jest kolejność retencji kwasów mrówkowego, octowego
Pytania z Wysokosprawnej chromatografii cieczowej 1. Jak wpłynie 50% dodatek MeOH do wody na retencję kwasu propionowego w układzie faz odwróconych? 2. Jaka jest kolejność retencji kwasów mrówkowego, octowego
Roztwory rzeczywiste (1)
") Roztwory rzeczywiste (1) Również w temp. 298,15K, ale dla CCl 4 () i CH 3 OH (). 2 15 1 5-5 -1-15 Τ S H,2,4,6,8 1 G -2 Chem. Fiz. TCH II/12 1 rzyczyny dodatnich i ujemnych odchyleń od prawa Raoulta konsekwencja
Roztwory rzeczywiste (1) Również w temp. 298,15K, ale dla CCl 4 () i CH 3 OH (). 2 15 1 5-5 -1-15 Τ S H,2,4,6,8 1 G -2 Chem. Fiz. TCH II/12 1 rzyczyny dodatnich i ujemnych odchyleń od prawa Raoulta konsekwencja
Równowagi fazowe. Zakład Chemii Medycznej Pomorski Uniwersytet Medyczny
 Równowagi fazowe Zakład Chemii Medycznej Pomorski Uniwersytet Medyczny Równowaga termodynamiczna Przemianom fazowym towarzyszą procesy, podczas których nie zmienia się skład chemiczny układu, polegają
Równowagi fazowe Zakład Chemii Medycznej Pomorski Uniwersytet Medyczny Równowaga termodynamiczna Przemianom fazowym towarzyszą procesy, podczas których nie zmienia się skład chemiczny układu, polegają
2. POMIAR IZOTERMY ADSORPCJI W UKŁADZIE CIECZ CIAŁO STAŁE
 2. POMIAR IZOTERMY ADSORPCJI W UKŁADZIE CIECZ CIAŁO STAŁE Opracował: Wojciech Zapała I. WPROWADZENIE I.1. Izotermy adsorpcji. Według Brunaur a istnieje pięć zasadniczych typów izoterm adsorpcji [1]. JednakŜe
2. POMIAR IZOTERMY ADSORPCJI W UKŁADZIE CIECZ CIAŁO STAŁE Opracował: Wojciech Zapała I. WPROWADZENIE I.1. Izotermy adsorpcji. Według Brunaur a istnieje pięć zasadniczych typów izoterm adsorpcji [1]. JednakŜe
Podstawowe prawa opisujące właściwości gazów zostały wyprowadzone dla gazu modelowego, nazywanego gazem doskonałym (idealnym).
.") Spis treści 1 Stan gazowy 2 Gaz doskonały 21 Definicja mikroskopowa 22 Definicja makroskopowa (termodynamiczna) 3 Prawa gazowe 31 Prawo Boyle a-mariotte a 32 Prawo Gay-Lussaca 33 Prawo Charlesa 34 Prawo
Spis treści 1 Stan gazowy 2 Gaz doskonały 21 Definicja mikroskopowa 22 Definicja makroskopowa (termodynamiczna) 3 Prawa gazowe 31 Prawo Boyle a-mariotte a 32 Prawo Gay-Lussaca 33 Prawo Charlesa 34 Prawo
Odwracalność przemiany chemicznej
 Odwracalność przemiany chemicznej Na ogół wszystkie reakcje chemiczne są odwracalne, tzn. z danych substratów tworzą się produkty, a jednocześnie produkty reakcji ulegają rozkładowi na substraty. Fakt
Odwracalność przemiany chemicznej Na ogół wszystkie reakcje chemiczne są odwracalne, tzn. z danych substratów tworzą się produkty, a jednocześnie produkty reakcji ulegają rozkładowi na substraty. Fakt
Przedmiot: Chemia budowlana Zakład Materiałoznawstwa i Technologii Betonu
 Przedmiot: Chemia budowlana Zakład Materiałoznawstwa i Technologii Betonu Ćw. 4 Kinetyka reakcji chemicznych Zagadnienia do przygotowania: Szybkość reakcji chemicznej, zależność szybkości reakcji chemicznej
Przedmiot: Chemia budowlana Zakład Materiałoznawstwa i Technologii Betonu Ćw. 4 Kinetyka reakcji chemicznych Zagadnienia do przygotowania: Szybkość reakcji chemicznej, zależność szybkości reakcji chemicznej
Wykład 3. Fizykochemia biopolimerów- wykład 3. Anna Ptaszek. 30 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego
 Wykład 3 - wykład 3 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 30 października 2013 1/56 Warunek równowagi fazowej Jakich układów dotyczy równowaga fazowa? Równowaga fazowa dotyczy układów: jednoskładnikowych
Wykład 3 - wykład 3 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 30 października 2013 1/56 Warunek równowagi fazowej Jakich układów dotyczy równowaga fazowa? Równowaga fazowa dotyczy układów: jednoskładnikowych
Wykład 5. Anna Ptaszek. 9 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Chemia fizyczna - wykład 5. Anna Ptaszek 1 / 20
 Wykład 5 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 9 października 2015 1 / 20 Zjawiska powierzchniowe Adsorpcja na powierzchni ciała stałego (adsorbentu): adsorpcja fizyczna: substancja adsorbująca
Wykład 5 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 9 października 2015 1 / 20 Zjawiska powierzchniowe Adsorpcja na powierzchni ciała stałego (adsorbentu): adsorpcja fizyczna: substancja adsorbująca
Technologie wytwarzania metali. Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe
 Technologie wytwarzania metali Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe KRYSTALIZACJA METALI I STOPÓW Krzepnięcie - przemiana fazy
Technologie wytwarzania metali Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe KRYSTALIZACJA METALI I STOPÓW Krzepnięcie - przemiana fazy
Technologie wytwarzania metali. Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe
 Technologie wytwarzania metali Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe KRYSTALIZACJA METALI I STOPÓW Krzepnięcie - przemiana fazy
Technologie wytwarzania metali Odlewanie Metalurgia proszków Otrzymywanie monokryształów Otrzymywanie materiałów superczystych Techniki próżniowe KRYSTALIZACJA METALI I STOPÓW Krzepnięcie - przemiana fazy
Wykład 13. Anna Ptaszek. 4 stycznia Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Fizykochemia biopolimerów - wykład 13.
 Wykład 13 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 4 stycznia 2018 1 / 29 Układy wielofazowe FAZA rozpraszająca rozpraszana gaz ciecz ciało stałe gaz - piana piana stała ciecz mgła/aerozol
Wykład 13 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 4 stycznia 2018 1 / 29 Układy wielofazowe FAZA rozpraszająca rozpraszana gaz ciecz ciało stałe gaz - piana piana stała ciecz mgła/aerozol
SPECJALNE TECHNIKI ROZDZIELANIA W BIOTECHNOLOGII. Laboratorium nr1 CHROMATOGRAFIA ODDZIAŁYWAŃ HYDROFOBOWYCH
 SPECJALNE TECHNIKI ROZDZIELANIA W BIOTECHNOLOGII Laboratorium nr1 CHROMATOGRAFIA ODDZIAŁYWAŃ HYDROFOBOWYCH Opracowała: dr inż. Renata Muca I. WPROWADZENIE TEORETYCZNE Chromatografia oddziaływań hydrofobowych
SPECJALNE TECHNIKI ROZDZIELANIA W BIOTECHNOLOGII Laboratorium nr1 CHROMATOGRAFIA ODDZIAŁYWAŃ HYDROFOBOWYCH Opracowała: dr inż. Renata Muca I. WPROWADZENIE TEORETYCZNE Chromatografia oddziaływań hydrofobowych
ADSORPCJA PARACETAMOLU NA WĘGLU AKTYWNYM
 ADSORPCJA PARACETAMOLU NA WĘGLU AKTYWNYM CEL ĆWICZENIA Celem ćwiczenia jest analiza procesu adsorpcji paracetamolu na węglu aktywnym. Zadanie praktyczne polega na spektrofotometrycznym oznaczeniu stężenia
ADSORPCJA PARACETAMOLU NA WĘGLU AKTYWNYM CEL ĆWICZENIA Celem ćwiczenia jest analiza procesu adsorpcji paracetamolu na węglu aktywnym. Zadanie praktyczne polega na spektrofotometrycznym oznaczeniu stężenia
Podstawy termodynamiki
 Podstawy termodynamiki Organizm żywy z punktu widzenia termodynamiki Parametry stanu Funkcje stanu: U, H, F, G, S I zasada termodynamiki i prawo Hessa II zasada termodynamiki Kierunek przemian w warunkach
Podstawy termodynamiki Organizm żywy z punktu widzenia termodynamiki Parametry stanu Funkcje stanu: U, H, F, G, S I zasada termodynamiki i prawo Hessa II zasada termodynamiki Kierunek przemian w warunkach
Kolumnowa Chromatografia Cieczowa I. 1. Czym różni się (z punktu widzenia użytkownika) chromatografia gazowa od chromatografii cieczowej?
 chromatografia gazowa od chromatografii cieczowej?") Kolumnowa Chromatografia Cieczowa I 1. Czym różni się (z punktu widzenia użytkownika) chromatografia gazowa od chromatografii cieczowej? 2. Co jest miarą polarności rozpuszczalników w chromatografii cieczowej?
Kolumnowa Chromatografia Cieczowa I 1. Czym różni się (z punktu widzenia użytkownika) chromatografia gazowa od chromatografii cieczowej? 2. Co jest miarą polarności rozpuszczalników w chromatografii cieczowej?
Wykład 5. Anna Ptaszek. 30 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Fizykochemiczne podstawy procesów przemysłu
 Wykład 5 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 30 października 2018 1 / 22 Zjawiska powierzchniowe Adsorpcja na powierzchni ciała stałego (adsorbentu): adsorpcja fizyczna: substancja adsorbująca
Wykład 5 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 30 października 2018 1 / 22 Zjawiska powierzchniowe Adsorpcja na powierzchni ciała stałego (adsorbentu): adsorpcja fizyczna: substancja adsorbująca
CHROMATOGRAFIA W UKŁADACH FAZ ODWRÓCONYCH RP-HPLC
 CHROMATOGRAFIA W UKŁADACH FAZ ODWRÓCONYCH RP-HPLC MK-EG-AS Wydział Chemiczny Politechniki Gdańskiej Gdańsk 2009 Chromatograficzne układy faz odwróconych (RP) Potocznie: Układy chromatograficzne, w których
CHROMATOGRAFIA W UKŁADACH FAZ ODWRÓCONYCH RP-HPLC MK-EG-AS Wydział Chemiczny Politechniki Gdańskiej Gdańsk 2009 Chromatograficzne układy faz odwróconych (RP) Potocznie: Układy chromatograficzne, w których
Wykład 6. Klasyfikacja przemian fazowych
 Wykład 6 Klasyfikacja przemian fazowych JS Klasyfikacja Ehrenfesta Ehrenfest klasyfikuje przemiany fazowe w oparciu o potencjał chemiczny. nieciągłość Przemiany fazowe pierwszego rodzaju pochodne potencjału
Wykład 6 Klasyfikacja przemian fazowych JS Klasyfikacja Ehrenfesta Ehrenfest klasyfikuje przemiany fazowe w oparciu o potencjał chemiczny. nieciągłość Przemiany fazowe pierwszego rodzaju pochodne potencjału
Symulacja Monte Carlo izotermy adsorpcji w układzie. ciało stałe-gaz
 Ćwiczenie nr 2 Symulacja Monte Carlo izotermy adsorpcji w układzie ciało stałe-gaz I. Cel ćwiczenia Celem ćwiczenia jest określenie wpływu parametrów takich jak temperatura, energia oddziaływania cząsteczka-powierzchnia
Ćwiczenie nr 2 Symulacja Monte Carlo izotermy adsorpcji w układzie ciało stałe-gaz I. Cel ćwiczenia Celem ćwiczenia jest określenie wpływu parametrów takich jak temperatura, energia oddziaływania cząsteczka-powierzchnia
Techniki Rozdzielania Mieszanin
 Techniki Rozdzielania Mieszanin Techniki Sorpcji i Chromatografii cz. I prof. dr hab. inż. Marian Kamiński Gdańsk 2010 Chromatografia cieczowa jako technika analityki, przygotowania próbek, wsadów do rozdzielania,
Techniki Rozdzielania Mieszanin Techniki Sorpcji i Chromatografii cz. I prof. dr hab. inż. Marian Kamiński Gdańsk 2010 Chromatografia cieczowa jako technika analityki, przygotowania próbek, wsadów do rozdzielania,
Seria 2, ćwiczenia do wykładu Od eksperymentu do poznania materii
 Seria 2, ćwiczenia do wykładu Od eksperymentu do poznania materii 8.1.21 Zad. 1. Obliczyć ciśnienie potrzebne do przemiany grafitu w diament w temperaturze 25 o C. Objętość właściwa (odwrotność gęstości)
Seria 2, ćwiczenia do wykładu Od eksperymentu do poznania materii 8.1.21 Zad. 1. Obliczyć ciśnienie potrzebne do przemiany grafitu w diament w temperaturze 25 o C. Objętość właściwa (odwrotność gęstości)
TERMODYNAMIKA TECHNICZNA I CHEMICZNA
 TERMODYNAMIKA TECHNICZNA I CHEMICZNA WYKŁAD IX RÓWNOWAGA FAZOWA W UKŁADZIE CIAŁO STAŁE-CIECZ (krystalizacja) ADSORPCJA KRYSTALIZACJA, ADSORPCJA 1 RÓWNOWAGA FAZOWA W UKŁADZIE CIAŁO STAŁE-CIECZ (krystalizacja)
TERMODYNAMIKA TECHNICZNA I CHEMICZNA WYKŁAD IX RÓWNOWAGA FAZOWA W UKŁADZIE CIAŁO STAŁE-CIECZ (krystalizacja) ADSORPCJA KRYSTALIZACJA, ADSORPCJA 1 RÓWNOWAGA FAZOWA W UKŁADZIE CIAŁO STAŁE-CIECZ (krystalizacja)
MATERIAŁOZNAWSTWO Wydział Mechaniczny, Mechatronika, sem. I. dr inż. Hanna Smoleńska
 MATERIAŁOZNAWSTWO Wydział Mechaniczny, Mechatronika, sem. I dr inż. Hanna Smoleńska UKŁADY RÓWNOWAGI FAZOWEJ Równowaga termodynamiczna pojęcie stosowane w termodynamice. Oznacza stan, w którym makroskopowe
MATERIAŁOZNAWSTWO Wydział Mechaniczny, Mechatronika, sem. I dr inż. Hanna Smoleńska UKŁADY RÓWNOWAGI FAZOWEJ Równowaga termodynamiczna pojęcie stosowane w termodynamice. Oznacza stan, w którym makroskopowe
Ćwiczenie IX KATALITYCZNY ROZKŁAD WODY UTLENIONEJ
 Wprowadzenie Ćwiczenie IX KATALITYCZNY ROZKŁAD WODY UTLENIONEJ opracowanie: Barbara Stypuła Celem ćwiczenia jest poznanie roli katalizatora w procesach chemicznych oraz prostego sposobu wyznaczenia wpływu
Wprowadzenie Ćwiczenie IX KATALITYCZNY ROZKŁAD WODY UTLENIONEJ opracowanie: Barbara Stypuła Celem ćwiczenia jest poznanie roli katalizatora w procesach chemicznych oraz prostego sposobu wyznaczenia wpływu
Kontrola produktu leczniczego. Piotr Podsadni
 Kontrola produktu leczniczego Piotr Podsadni Kontrola Kontrola - sprawdzanie czegoś, zestawianie stanu faktycznego ze stanem wymaganym. Zakres czynności sprawdzający zapewnienie jakości. Jakość to stopień,
Kontrola produktu leczniczego Piotr Podsadni Kontrola Kontrola - sprawdzanie czegoś, zestawianie stanu faktycznego ze stanem wymaganym. Zakres czynności sprawdzający zapewnienie jakości. Jakość to stopień,
POLITECHNIKA GDAŃSKA
 POLITECHNIKA GDAŃSKA WYDZIAŁ CHEMICZNY KATEDRA TECHNOLOGII CHEMICZNEJ Ćwiczenia laboratoryjne CHEMIA I TECHNOLOGIA MATERIAŁÓW BARWNYCH USUWANIE BARWNIKÓW ZE ŚCIEKÓW PRZEMYSŁU TEKSTYLNEGO Z WYKORZYSTANIEM
POLITECHNIKA GDAŃSKA WYDZIAŁ CHEMICZNY KATEDRA TECHNOLOGII CHEMICZNEJ Ćwiczenia laboratoryjne CHEMIA I TECHNOLOGIA MATERIAŁÓW BARWNYCH USUWANIE BARWNIKÓW ZE ŚCIEKÓW PRZEMYSŁU TEKSTYLNEGO Z WYKORZYSTANIEM
PORÓWNANIE FAZ STACJONARNYCH STOSOWANYCH W HPLC
 PORÓWNANIE FAZ STACJONARNYCH STOSOWANYCH W HPLC Instrukcja do ćwiczeń opracowana w Katedrze Chemii Środowiska Uniwersytetu Łódzkiego 1. Wstęp Chromatografia jest techniką umożliwiającą rozdzielanie składników
PORÓWNANIE FAZ STACJONARNYCH STOSOWANYCH W HPLC Instrukcja do ćwiczeń opracowana w Katedrze Chemii Środowiska Uniwersytetu Łódzkiego 1. Wstęp Chromatografia jest techniką umożliwiającą rozdzielanie składników
relacje ilościowe ( masowe,objętościowe i molowe ) dotyczące połączeń 1. pierwiastków w związkach chemicznych 2. związków chemicznych w reakcjach
 dotyczące połączeń 1. pierwiastków w związkach chemicznych 2. związków chemicznych w reakcjach") 1 STECHIOMETRIA INTERPRETACJA ILOŚCIOWA ZJAWISK CHEMICZNYCH relacje ilościowe ( masowe,objętościowe i molowe ) dotyczące połączeń 1. pierwiastków w związkach chemicznych 2. związków chemicznych w reakcjach
1 STECHIOMETRIA INTERPRETACJA ILOŚCIOWA ZJAWISK CHEMICZNYCH relacje ilościowe ( masowe,objętościowe i molowe ) dotyczące połączeń 1. pierwiastków w związkach chemicznych 2. związków chemicznych w reakcjach
Wykład 10 Równowaga chemiczna
 Wykład 10 Równowaga chemiczna REAKCJA CHEMICZNA JEST W RÓWNOWADZE, GDY NIE STWIERDZAMY TENDENCJI DO ZMIAN ILOŚCI (STĘŻEŃ) SUBSTRATÓW ANI PRODUKTÓW RÓWNOWAGA CHEMICZNA JEST RÓWNOWAGĄ DYNAMICZNĄ W rzeczywistości
Wykład 10 Równowaga chemiczna REAKCJA CHEMICZNA JEST W RÓWNOWADZE, GDY NIE STWIERDZAMY TENDENCJI DO ZMIAN ILOŚCI (STĘŻEŃ) SUBSTRATÓW ANI PRODUKTÓW RÓWNOWAGA CHEMICZNA JEST RÓWNOWAGĄ DYNAMICZNĄ W rzeczywistości
Wykład 4. Przypomnienie z poprzedniego wykładu
 Wykład 4 Przejścia fazowe materii Diagram fazowy Ciepło Procesy termodynamiczne Proces kwazistatyczny Procesy odwracalne i nieodwracalne Pokazy doświadczalne W. Dominik Wydział Fizyki UW Termodynamika
Wykład 4 Przejścia fazowe materii Diagram fazowy Ciepło Procesy termodynamiczne Proces kwazistatyczny Procesy odwracalne i nieodwracalne Pokazy doświadczalne W. Dominik Wydział Fizyki UW Termodynamika
Warunki izochoryczno-izotermiczne
 WYKŁAD 5 Pojęcie potencjału chemicznego. Układy jednoskładnikowe W zależności od warunków termodynamicznych potencjał chemiczny substancji czystej definiujemy następująco: Warunki izobaryczno-izotermiczne
WYKŁAD 5 Pojęcie potencjału chemicznego. Układy jednoskładnikowe W zależności od warunków termodynamicznych potencjał chemiczny substancji czystej definiujemy następująco: Warunki izobaryczno-izotermiczne
chemia wykład 3 Przemiany fazowe
 Przemiany fazowe Przemiany fazowe substancji czystych Wrzenie, krzepnięcie, przemiana grafitu w diament stanowią przykłady przemian fazowych, które zachodzą bez zmiany składu chemicznego. Diagramy fazowe
Przemiany fazowe Przemiany fazowe substancji czystych Wrzenie, krzepnięcie, przemiana grafitu w diament stanowią przykłady przemian fazowych, które zachodzą bez zmiany składu chemicznego. Diagramy fazowe
Adsorpcyjne oczyszczanie gazów z zanieczyszczeń związkami organicznymi
 Pracownia: Utylizacja odpadów i ścieków dla MSOŚ Instrukcja ćwiczenia nr 17 Adsorpcyjne oczyszczanie gazów z zanieczyszczeń związkami organicznymi Uniwersytet Warszawski Wydział Chemii Zakład Dydaktyczny
Pracownia: Utylizacja odpadów i ścieków dla MSOŚ Instrukcja ćwiczenia nr 17 Adsorpcyjne oczyszczanie gazów z zanieczyszczeń związkami organicznymi Uniwersytet Warszawski Wydział Chemii Zakład Dydaktyczny
GraŜyna Chwatko Zakład Chemii Środowiska
 Chromatografia podstawa metod analizy laboratoryjnej GraŜyna Chwatko Zakład Chemii Środowiska Chromatografia gr. chromatos = barwa grapho = pisze Michaił Siemionowicz Cwiet 2 Chromatografia jest metodą
Chromatografia podstawa metod analizy laboratoryjnej GraŜyna Chwatko Zakład Chemii Środowiska Chromatografia gr. chromatos = barwa grapho = pisze Michaił Siemionowicz Cwiet 2 Chromatografia jest metodą
Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }
![Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }](/thumbs/64/51033976.jpg "Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }") Zespół kanoniczny Zespół kanoniczny N,V, T acc o n =min {1, exp [ U n U o ] } Zespół izobaryczno-izotermiczny Zespół izobaryczno-izotermiczny N P T acc o n =min {1, exp [ U n U o ] } acc o n =min {1, exp[
Zespół kanoniczny Zespół kanoniczny N,V, T acc o n =min {1, exp [ U n U o ] } Zespół izobaryczno-izotermiczny Zespół izobaryczno-izotermiczny N P T acc o n =min {1, exp [ U n U o ] } acc o n =min {1, exp[
Zastosowanie programu DICTRA do symulacji numerycznej przemian fazowych w stopach technicznych kontrolowanych procesem dyfuzji" Roman Kuziak
 Zastosowanie programu DICTRA do symulacji numerycznej przemian fazowych w stopach technicznych kontrolowanych procesem dyfuzji" Roman Kuziak Instytut Metalurgii Żelaza DICTRA jest pakietem komputerowym
Zastosowanie programu DICTRA do symulacji numerycznej przemian fazowych w stopach technicznych kontrolowanych procesem dyfuzji" Roman Kuziak Instytut Metalurgii Żelaza DICTRA jest pakietem komputerowym
OPTYMALIZACJA EFEKTÓW ROZDZIELANIA W KOLUMNACH KAPILARNYCH DOBÓR PRĘDKOŚCI PRZEPŁYWU GAZU
 OPTYMALIZACJA EFEKTÓW ROZDZIELANIA W KOLUMNACH KAPILARNYCH DOBÓR PRĘDKOŚCI PRZEPŁYWU GAZU 1. WPROWADZENIE W czasie swej wędrówki wzdłuż kolumny pasmo chromatograficzne ulega poszerzeniu, co jest zjawiskiem
OPTYMALIZACJA EFEKTÓW ROZDZIELANIA W KOLUMNACH KAPILARNYCH DOBÓR PRĘDKOŚCI PRZEPŁYWU GAZU 1. WPROWADZENIE W czasie swej wędrówki wzdłuż kolumny pasmo chromatograficzne ulega poszerzeniu, co jest zjawiskiem
Wykład 2. Anna Ptaszek. 7 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Chemia fizyczna - wykład 2. Anna Ptaszek 1 / 1
 Wykład 2 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 7 października 2015 1 / 1 Zjawiska koligatywne Rozpuszczenie w wodzie substancji nielotnej powoduje obniżenie prężności pary nasyconej P woda
Wykład 2 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 7 października 2015 1 / 1 Zjawiska koligatywne Rozpuszczenie w wodzie substancji nielotnej powoduje obniżenie prężności pary nasyconej P woda
RP WPROWADZENIE. M. Kamiński PG WCh Gdańsk Układy faz odwróconych RP-HPLC, RP-TLC gdy:
 RP WPRWADZENIE M. Kamiński PG WCh Gdańsk 2013 Układy faz odwróconych RP-HPLC, RP-TLC gdy: Nisko polarna (hydrofobowa) faza stacjonarna, względnie polarny eluent, składający się z wody i dodatku organicznego;
RP WPRWADZENIE M. Kamiński PG WCh Gdańsk 2013 Układy faz odwróconych RP-HPLC, RP-TLC gdy: Nisko polarna (hydrofobowa) faza stacjonarna, względnie polarny eluent, składający się z wody i dodatku organicznego;
Rzeszów, 16 kwietnia, 2018 r. RECENZJA
 Rzeszów, 16 kwietnia, 2018 r. RECENZJA rozprawy doktorskiej mgr inż. Agaty PRZEWŁOCKIEJ pt.: Biosorpcjne usuwanie mieszaniny jonów Ni(II), Pb(II) oraz Zn(II) z roztworu wodnego przy zastosowaniu złoża
Rzeszów, 16 kwietnia, 2018 r. RECENZJA rozprawy doktorskiej mgr inż. Agaty PRZEWŁOCKIEJ pt.: Biosorpcjne usuwanie mieszaniny jonów Ni(II), Pb(II) oraz Zn(II) z roztworu wodnego przy zastosowaniu złoża
Chemia fizyczna. Adsorpcja. Zakład Chemii Medycznej Pomorskiego Uniwersytetu Medycznego
 Chemia fizyczna Adsorpcja Zakład Chemii Medycznej Pomorskiego Uniwersytetu Medycznego Adsorpcja na granicy faz istnieją niewysycone siły oddziaływań międzycząsteczkowych cząsteczki znajdujące się na powierzchni
Chemia fizyczna Adsorpcja Zakład Chemii Medycznej Pomorskiego Uniwersytetu Medycznego Adsorpcja na granicy faz istnieją niewysycone siły oddziaływań międzycząsteczkowych cząsteczki znajdujące się na powierzchni
Chemia - laboratorium
 Chemia - laboratorium Wydział Geologii, Geofizyki i Ochrony Środowiska Studia stacjonarne, Rok I, Semestr zimowy 01/1 Dr hab. inż. Tomasz Brylewski e-mail: brylew@agh.edu.pl tel. 1-617-59 Katedra Fizykochemii
Chemia - laboratorium Wydział Geologii, Geofizyki i Ochrony Środowiska Studia stacjonarne, Rok I, Semestr zimowy 01/1 Dr hab. inż. Tomasz Brylewski e-mail: brylew@agh.edu.pl tel. 1-617-59 Katedra Fizykochemii
WYKAZ NAJWAŻNIEJSZYCH SYMBOLI
 SPIS TREŚCI WYKAZ NAJWAŻNIEJSZYCH SYMBOLI...7 PRZEDMOWA...8 1. WSTĘP...9 2. MATEMATYCZNE OPRACOWANIE WYNIKÓW POMIARÓW...10 3. LEPKOŚĆ CIECZY...15 3.1. Pomiar lepkości...16 3.2. Lepkość względna...18 3.3.
SPIS TREŚCI WYKAZ NAJWAŻNIEJSZYCH SYMBOLI...7 PRZEDMOWA...8 1. WSTĘP...9 2. MATEMATYCZNE OPRACOWANIE WYNIKÓW POMIARÓW...10 3. LEPKOŚĆ CIECZY...15 3.1. Pomiar lepkości...16 3.2. Lepkość względna...18 3.3.
III r. EiP (Technologia Chemiczna)
") AKADEMIA GÓRNICZO HUTNICZA WYDZIAŁ ENERGETYKI I PALIW III r. EiP (Technologia Chemiczna) INŻYNIERIA CHEMICZNA I PROCESOWA (przenoszenie pędu) Prof. dr hab. Leszek CZEPIRSKI Kontakt: A4, p. 424 Tel. 12
AKADEMIA GÓRNICZO HUTNICZA WYDZIAŁ ENERGETYKI I PALIW III r. EiP (Technologia Chemiczna) INŻYNIERIA CHEMICZNA I PROCESOWA (przenoszenie pędu) Prof. dr hab. Leszek CZEPIRSKI Kontakt: A4, p. 424 Tel. 12
= = Budowa materii. Stany skupienia materii. Ilość materii (substancji) n - ilość moli, N liczba molekuł (atomów, cząstek), N A
 n - ilość moli, N liczba molekuł (atomów, cząstek), N A") Budowa materii Stany skupienia materii Ciało stałe Ciecz Ciała lotne (gazy i pary) Ilość materii (substancji) n N = = N A m M N A = 6,023 10 mol 23 1 n - ilość moli, N liczba molekuł (atomów, cząstek),
Budowa materii Stany skupienia materii Ciało stałe Ciecz Ciała lotne (gazy i pary) Ilość materii (substancji) n N = = N A m M N A = 6,023 10 mol 23 1 n - ilość moli, N liczba molekuł (atomów, cząstek),
K02 Instrukcja wykonania ćwiczenia
 Katedra Chemii Fizycznej Uniwersytetu Łódzkiego K2 Instrukcja wykonania ćwiczenia Wyznaczanie krytycznego stężenia micelizacji (CMC) z pomiarów napięcia powierzchniowego Zakres zagadnień obowiązujących
Katedra Chemii Fizycznej Uniwersytetu Łódzkiego K2 Instrukcja wykonania ćwiczenia Wyznaczanie krytycznego stężenia micelizacji (CMC) z pomiarów napięcia powierzchniowego Zakres zagadnień obowiązujących
Sorpcyjne Systemy Energetyczne
 Sorpcyjne Systemy Energetyczne Procesy adsorpcji i desorpcji w systemach chłodniczych dr inż. Bartosz Zajączkowski bartosz.zajaczkowski@pwr.edu.pl, bud. D2, pok. 9b Wydział Mechaniczno-Energetyczny Katedra
Sorpcyjne Systemy Energetyczne Procesy adsorpcji i desorpcji w systemach chłodniczych dr inż. Bartosz Zajączkowski bartosz.zajaczkowski@pwr.edu.pl, bud. D2, pok. 9b Wydział Mechaniczno-Energetyczny Katedra
Wpływ ilości modyfikatora na współczynnik retencji w technice wysokosprawnej chromatografii cieczowej
 Wpływ ilości modyfikatora na współczynnik retencji w technice wysokosprawnej chromatografii cieczowej WPROWADZENIE Wysokosprawna chromatografia cieczowa (HPLC) jest uniwersalną techniką analityczną, stosowaną
Wpływ ilości modyfikatora na współczynnik retencji w technice wysokosprawnej chromatografii cieczowej WPROWADZENIE Wysokosprawna chromatografia cieczowa (HPLC) jest uniwersalną techniką analityczną, stosowaną
Chromatografia kolumnowa planarna
 Chromatografia kolumnowa planarna Znaczenie chromatografii w analizie i monitoringu środowiska lotne zanieczyszczenia organiczne (alifatyczne, aromatyczne) w powietrzu, glebie, wodzie Mikrozanieczyszczenia
Chromatografia kolumnowa planarna Znaczenie chromatografii w analizie i monitoringu środowiska lotne zanieczyszczenia organiczne (alifatyczne, aromatyczne) w powietrzu, glebie, wodzie Mikrozanieczyszczenia
Roztwory rzeczywiste (1)
") Roztwory rzeczywiste (1) Również w temp. 298,15K, ale dla CCl 4 () i CH 3 OH (). 2 15 1 5-5 -1-15 Τ S H,2,4,6,8 1 G -2 Chem. Fiz. TCH II/12 1 Roztwory rzeczywiste (2) Tym razem dla (CH 3 ) 2 CO () i CHCl
Roztwory rzeczywiste (1) Również w temp. 298,15K, ale dla CCl 4 () i CH 3 OH (). 2 15 1 5-5 -1-15 Τ S H,2,4,6,8 1 G -2 Chem. Fiz. TCH II/12 1 Roztwory rzeczywiste (2) Tym razem dla (CH 3 ) 2 CO () i CHCl
rodzajach chromatografii cieczowej w związku ze wszczętym na
 Katedra Chemii Analitycznej Wydział Chemiczny, Politechnika Gdańska, ul. G. Narutowicza 11/12, 80-233 Gdańsk tel. 058 347 10 10 Kierownik Katedry 058 347 19 10 Sekretariat 058 347 21 10 Laboratorium fax.
Katedra Chemii Analitycznej Wydział Chemiczny, Politechnika Gdańska, ul. G. Narutowicza 11/12, 80-233 Gdańsk tel. 058 347 10 10 Kierownik Katedry 058 347 19 10 Sekretariat 058 347 21 10 Laboratorium fax.
Instrukcja do ćwiczeń laboratoryjnych
 UNIWERSYTET GDAŃSKI Pracownia studencka Katedry Analizy Środowiska Instrukcja do ćwiczeń laboratoryjnych Ćwiczenie nr 2 Oznaczanie benzoesanu denatonium w skażonym alkoholu etylowym metodą wysokosprawnej
UNIWERSYTET GDAŃSKI Pracownia studencka Katedry Analizy Środowiska Instrukcja do ćwiczeń laboratoryjnych Ćwiczenie nr 2 Oznaczanie benzoesanu denatonium w skażonym alkoholu etylowym metodą wysokosprawnej
Wykład 1 i 2. Termodynamika klasyczna, gaz doskonały
 Wykład 1 i 2 Termodynamika klasyczna, gaz doskonały dr hab. Agata Fronczak, prof. PW Wydział Fizyki, Politechnika Warszawska 1 stycznia 2017 dr hab. A. Fronczak (Wydział Fizyki PW) Wykład: Elementy fizyki
Wykład 1 i 2 Termodynamika klasyczna, gaz doskonały dr hab. Agata Fronczak, prof. PW Wydział Fizyki, Politechnika Warszawska 1 stycznia 2017 dr hab. A. Fronczak (Wydział Fizyki PW) Wykład: Elementy fizyki
Materiały pomocnicze do laboratorium z przedmiotu Metody i Narzędzia Symulacji Komputerowej
 Materiały pomocnicze do laboratorium z przedmiotu Metody i Narzędzia Symulacji Komputerowej w Systemach Technicznych Symulacja prosta dyszy pomiarowej Bendemanna Opracował: dr inż. Andrzej J. Zmysłowski
Materiały pomocnicze do laboratorium z przedmiotu Metody i Narzędzia Symulacji Komputerowej w Systemach Technicznych Symulacja prosta dyszy pomiarowej Bendemanna Opracował: dr inż. Andrzej J. Zmysłowski
KRYSTALIZACJA METALI I STOPÓW. Publikacja współfinansowana ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego
 KRYSTALIZACJA METALI I STOPÓW Publikacja współfinansowana ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego Krzepnięcie przemiana fazy ciekłej w fazę stałą Krystalizacja przemiana
KRYSTALIZACJA METALI I STOPÓW Publikacja współfinansowana ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego Krzepnięcie przemiana fazy ciekłej w fazę stałą Krystalizacja przemiana
Wykład 3. Entropia i potencjały termodynamiczne
 Wykład 3 Entropia i potencjały termodynamiczne dr hab. Agata Fronczak, prof. PW Wydział Fizyki, Politechnika Warszawska 1 stycznia 2017 dr hab. A. Fronczak (Wydział Fizyki PW) Wykład: Elementy fizyki statystycznej
Wykład 3 Entropia i potencjały termodynamiczne dr hab. Agata Fronczak, prof. PW Wydział Fizyki, Politechnika Warszawska 1 stycznia 2017 dr hab. A. Fronczak (Wydział Fizyki PW) Wykład: Elementy fizyki statystycznej
Termodynamika Część 6 Związki i tożsamości termodynamiczne Potencjały termodynamiczne Warunki równowagi termodynamicznej Potencjał chemiczny
 Termodynamika Część 6 Związki i tożsamości termodynamiczne Potencjały termodynamiczne Warunki równowagi termodynamicznej Potencjał chemiczny Janusz Brzychczyk, Instytut Fizyki UJ Związek pomiędzy równaniem
Termodynamika Część 6 Związki i tożsamości termodynamiczne Potencjały termodynamiczne Warunki równowagi termodynamicznej Potencjał chemiczny Janusz Brzychczyk, Instytut Fizyki UJ Związek pomiędzy równaniem
Wykład 8. Równowaga fazowa Roztwory rzeczywiste
 Wykład 8 Równowaga fazowa Roztwory rzeczywiste Roztwory doskonałe Porównanie roztworów doskonałych i Roztwory Doskonałe rzeczywistych Roztwory Rzeczywiste Spełniają prawo Raoulta Mieszanie w warunkach
Wykład 8 Równowaga fazowa Roztwory rzeczywiste Roztwory doskonałe Porównanie roztworów doskonałych i Roztwory Doskonałe rzeczywistych Roztwory Rzeczywiste Spełniają prawo Raoulta Mieszanie w warunkach
Statyka Cieczy i Gazów. Temat : Podstawy teorii kinetyczno-molekularnej budowy ciał
 Statyka Cieczy i Gazów Temat : Podstawy teorii kinetyczno-molekularnej budowy ciał 1. Podstawowe założenia teorii kinetyczno-molekularnej budowy ciał: Ciała zbudowane są z cząsteczek. Pomiędzy cząsteczkami
Statyka Cieczy i Gazów Temat : Podstawy teorii kinetyczno-molekularnej budowy ciał 1. Podstawowe założenia teorii kinetyczno-molekularnej budowy ciał: Ciała zbudowane są z cząsteczek. Pomiędzy cząsteczkami
Stany równowagi i zjawiska transportu w układach termodynamicznych
 Stany równowagi i zjawiska transportu w układach termodynamicznych dr hab. Jerzy Nakielski Katedra Biofizyki i Biologii Komórki plan wykładu: 1. Funkcje stanu dla termodynamicznego układu otwartego 2.
Stany równowagi i zjawiska transportu w układach termodynamicznych dr hab. Jerzy Nakielski Katedra Biofizyki i Biologii Komórki plan wykładu: 1. Funkcje stanu dla termodynamicznego układu otwartego 2.
ZADANIE 1 W temperaturze 700 K gazowa mieszanina dwutlenku węgla i wodoru reaguje z wytworzeniem pary wodnej i tlenku węgla. Stała równowagi reakcji
 ZADANIE 1 W temperaturze 700 K gazowa mieszanina dwutlenku węgla i wodoru reaguje z wytworzeniem pary wodnej i tlenku węgla. Stała równowagi reakcji w tej temperaturze wynosi K p = 0,11. Reaktor został
ZADANIE 1 W temperaturze 700 K gazowa mieszanina dwutlenku węgla i wodoru reaguje z wytworzeniem pary wodnej i tlenku węgla. Stała równowagi reakcji w tej temperaturze wynosi K p = 0,11. Reaktor został
Termodynamika materiałów
 Termodynamika materiałów Plan wykładu 1. Funkcje termodynamiczne, pojemność cieplna. 2. Warunki równowagi termodynamicznej w układach jedno- i wieloskładnikowych, pojęcie potencjału chemicznego. 3. Modele
Termodynamika materiałów Plan wykładu 1. Funkcje termodynamiczne, pojemność cieplna. 2. Warunki równowagi termodynamicznej w układach jedno- i wieloskładnikowych, pojęcie potencjału chemicznego. 3. Modele
TRANSPORT NIEELEKTROLITÓW PRZEZ BŁONY WYZNACZANIE WSPÓŁCZYNNIKA PRZEPUSZCZALNOŚCI
 Ćwiczenie nr 7 TRANSPORT NIEELEKTROLITÓW PRZEZ BŁONY WYZNACZANIE WSPÓŁCZYNNIKA PRZEPUSZCZALNOŚCI Celem ćwiczenia jest zapoznanie się z podstawami teorii procesów transportu nieelektrolitów przez błony.
Ćwiczenie nr 7 TRANSPORT NIEELEKTROLITÓW PRZEZ BŁONY WYZNACZANIE WSPÓŁCZYNNIKA PRZEPUSZCZALNOŚCI Celem ćwiczenia jest zapoznanie się z podstawami teorii procesów transportu nieelektrolitów przez błony.
Podstawy teoretyczne technologii chemicznej / Józef Szarawara, Jerzy Piotrowski. Warszawa, Spis treści. Przedmowa 13
 Podstawy teoretyczne technologii chemicznej / Józef Szarawara, Jerzy Piotrowski. Warszawa, 2010 Spis treści Przedmowa 13 Wykaz waŝniejszych oznaczeń 16 1. Projektowanie i realizacja procesu technologicznego
Podstawy teoretyczne technologii chemicznej / Józef Szarawara, Jerzy Piotrowski. Warszawa, 2010 Spis treści Przedmowa 13 Wykaz waŝniejszych oznaczeń 16 1. Projektowanie i realizacja procesu technologicznego
Recenzja pracy doktorskiej mgr. inż. Wojciecha Marka zatytułowanej "Rozdział białek za pomocą łączonych technik chromatograficznych"
 Rzeszów, 02-06-2014 Krzysztof Kaczmarski Katedra Inżynierii Chemicznej i Procesowej Wydział Chemiczny, Politechnika Rzeszowska Recenzja pracy doktorskiej mgr. inż. Wojciecha Marka zatytułowanej "Rozdział
Rzeszów, 02-06-2014 Krzysztof Kaczmarski Katedra Inżynierii Chemicznej i Procesowej Wydział Chemiczny, Politechnika Rzeszowska Recenzja pracy doktorskiej mgr. inż. Wojciecha Marka zatytułowanej "Rozdział
Wykład Praca (1.1) c Całka liniowa definiuje pracę wykonaną w kierunku działania siły. Reinhard Kulessa 1
 c Całka liniowa definiuje pracę wykonaną w kierunku działania siły. Reinhard Kulessa 1") 1.6 Praca Wykład 2 Praca zdefiniowana jest jako ilość energii dostarczanej przez siłę działającą na pewnej drodze i matematycznie jest zapisana jako: W = c r F r ds (1.1) ds F θ c Całka liniowa definiuje
1.6 Praca Wykład 2 Praca zdefiniowana jest jako ilość energii dostarczanej przez siłę działającą na pewnej drodze i matematycznie jest zapisana jako: W = c r F r ds (1.1) ds F θ c Całka liniowa definiuje
Para pozostająca w równowadze z roztworem jest bogatsza w ten składnik, którego dodanie do roztworu zwiększa sumaryczną prężność pary nad nim.
 RÓWNOWAGA CIECZ-PARA DLA UKŁADÓW DWUSKŁADNIKOWYCH: 1) Zgodnie z regułą faz Gibbsa układ dwuskładnikowy osiąga największą liczbę stopni swobody (f max ), gdy znajduje się w nim najmniejsza możliwa liczba
RÓWNOWAGA CIECZ-PARA DLA UKŁADÓW DWUSKŁADNIKOWYCH: 1) Zgodnie z regułą faz Gibbsa układ dwuskładnikowy osiąga największą liczbę stopni swobody (f max ), gdy znajduje się w nim najmniejsza możliwa liczba
Materiały polimerowe laboratorium
 Materiały polimerowe laboratorium Wydział Chemiczny, Studia Stacjonarne II stopnia (magisterskie), rok 1, semestr 2 kierunek: INŻYNIERIA CHEMICZNA I PROCESOWA specjalność: Inżynieria procesów chemicznych
Materiały polimerowe laboratorium Wydział Chemiczny, Studia Stacjonarne II stopnia (magisterskie), rok 1, semestr 2 kierunek: INŻYNIERIA CHEMICZNA I PROCESOWA specjalność: Inżynieria procesów chemicznych
Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 6-1 w PWN. Warszawa, cop.
 Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 6-1 w PWN. Warszawa, cop. 2017 Spis treści Przedmowa 11 1. Wprowadzenie 13 1.1. Krótka historia
Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 6-1 w PWN. Warszawa, cop. 2017 Spis treści Przedmowa 11 1. Wprowadzenie 13 1.1. Krótka historia
Wykład z Chemii Ogólnej i Nieorganicznej
 Wykład z Chemii Ogólnej i Nieorganicznej Część 5 ELEMENTY STATYKI CHEMICZNEJ Katedra i Zakład Chemii Fizycznej Collegium Medicum w Bydgoszczy Uniwersytet Mikołaja Kopernika w Toruniu Prof. dr hab. n.chem.
Wykład z Chemii Ogólnej i Nieorganicznej Część 5 ELEMENTY STATYKI CHEMICZNEJ Katedra i Zakład Chemii Fizycznej Collegium Medicum w Bydgoszczy Uniwersytet Mikołaja Kopernika w Toruniu Prof. dr hab. n.chem.
Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 5, 4 dodr. Warszawa, 2015.
 Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 5, 4 dodr. Warszawa, 2015 Spis treści Przedmowa 11 1. Wprowadzenie 13 1.1. Krótka historia chromatografii
Podstawy chromatografii i technik elektromigracyjnych / Zygfryd Witkiewicz, Joanna Kałużna-Czaplińska. wyd. 5, 4 dodr. Warszawa, 2015 Spis treści Przedmowa 11 1. Wprowadzenie 13 1.1. Krótka historia chromatografii
dn dt C= d ( pv ) = d dt dt (nrt )= kt Przepływ gazu Pompowanie przez przewód o przewodności G zbiornik przewód pompa C A , p 1 , S , p 2 , S E C B
 = d dt dt (nrt )= kt Przepływ gazu Pompowanie przez przewód o przewodności G zbiornik przewód pompa C A , p 1 , S , p 2 , S E C B") Pompowanie przez przewód o przewodności G zbiornik przewód pompa C A, p 2, S E C B, p 1, S C [W] wydajność pompowania C= d ( pv ) = d dt dt (nrt )= kt dn dt dn / dt - ilość cząstek przepływających w ciągu
Pompowanie przez przewód o przewodności G zbiornik przewód pompa C A, p 2, S E C B, p 1, S C [W] wydajność pompowania C= d ( pv ) = d dt dt (nrt )= kt dn dt dn / dt - ilość cząstek przepływających w ciągu
Instrukcja do ćwiczeń laboratoryjnych
 UNIWERSYTET GDAŃSKI WYDZIAŁ CHEMII Pracownia studencka Katedra Analizy Środowiska Instrukcja do ćwiczeń laboratoryjnych Ćwiczenie nr 2 OPTYMALIZACJA ROZDZIELANIA MIESZANINY WYBRANYCH FARMACEUTYKÓW METODĄ
UNIWERSYTET GDAŃSKI WYDZIAŁ CHEMII Pracownia studencka Katedra Analizy Środowiska Instrukcja do ćwiczeń laboratoryjnych Ćwiczenie nr 2 OPTYMALIZACJA ROZDZIELANIA MIESZANINY WYBRANYCH FARMACEUTYKÓW METODĄ
TERMODYNAMIKA. przykłady zastosowań. I.Mańkowski I LO w Lęborku
 TERMODYNAMIKA przykłady zastosowań I.Mańkowski I LO w Lęborku 2016 UKŁAD TERMODYNAMICZNY Dla przykładu układ termodynamiczny stanowią zamknięty cylinder z ruchomym tłokiem, w którym znajduje się gaz tak
TERMODYNAMIKA przykłady zastosowań I.Mańkowski I LO w Lęborku 2016 UKŁAD TERMODYNAMICZNY Dla przykładu układ termodynamiczny stanowią zamknięty cylinder z ruchomym tłokiem, w którym znajduje się gaz tak
Kinetyka reakcji chemicznych. Dr Mariola Samsonowicz
 Kinetyka reakcji chemicznych Dr Mariola Samsonowicz 1 Czym zajmuje się kinetyka chemiczna? Badaniem szybkości reakcji chemicznych poprzez analizę eksperymentalną i teoretyczną. Zdefiniowanie równania kinetycznego
Kinetyka reakcji chemicznych Dr Mariola Samsonowicz 1 Czym zajmuje się kinetyka chemiczna? Badaniem szybkości reakcji chemicznych poprzez analizę eksperymentalną i teoretyczną. Zdefiniowanie równania kinetycznego
Chemia - laboratorium
 Chemia - laboratorium Wydział Geologii, Geofizyki i Ochrony Środowiska Studia stacjonarne, Rok I, Semestr zimowy 013/14 Dr hab. inż. Tomasz Brylewski e-mail: brylew@agh.edu.pl tel. 1-617-59 Katedra Fizykochemii
Chemia - laboratorium Wydział Geologii, Geofizyki i Ochrony Środowiska Studia stacjonarne, Rok I, Semestr zimowy 013/14 Dr hab. inż. Tomasz Brylewski e-mail: brylew@agh.edu.pl tel. 1-617-59 Katedra Fizykochemii
Badanie właściwości związków powierzchniowo czynnych
 POLITECHNIKA ŚLĄSKA WYDZIAŁ CHEMICZNY KATEDRA TECHNOLOGII CHEMICZNEJ ORGANICZNEJ I PETROCHEMII INSTRUKCJA DO ĆWICZEŃ LABORATORYJNYCH: Badanie właściwości związków powierzchniowo czynnych Laboratorium z
POLITECHNIKA ŚLĄSKA WYDZIAŁ CHEMICZNY KATEDRA TECHNOLOGII CHEMICZNEJ ORGANICZNEJ I PETROCHEMII INSTRUKCJA DO ĆWICZEŃ LABORATORYJNYCH: Badanie właściwości związków powierzchniowo czynnych Laboratorium z
Inżynieria Biomedyczna
 1.Obliczyć przy jakim stężeniu kwasu octowego stopień dysocjacji osiągnie wartość 3.%, jeżeli wiadomo, że stopień dysocjacji 15.%-wego roztworu (d=1.2 g/cm 3 ) w 2. Do 1 cm 3 2% (d=1.2 g/cm 3 ) roztworu
1.Obliczyć przy jakim stężeniu kwasu octowego stopień dysocjacji osiągnie wartość 3.%, jeżeli wiadomo, że stopień dysocjacji 15.%-wego roztworu (d=1.2 g/cm 3 ) w 2. Do 1 cm 3 2% (d=1.2 g/cm 3 ) roztworu
Fizyka statystyczna Fenomenologia przejść fazowych. P. F. Góra
 Fizyka statystyczna Fenomenologia przejść fazowych P. F. Góra http://th-www.if.uj.edu.pl/zfs/gora/ 2015 Przejście fazowe transformacja układu termodynamicznego z jednej fazy (stanu materii) do innej, dokonywane
Fizyka statystyczna Fenomenologia przejść fazowych P. F. Góra http://th-www.if.uj.edu.pl/zfs/gora/ 2015 Przejście fazowe transformacja układu termodynamicznego z jednej fazy (stanu materii) do innej, dokonywane
Kinetyka. Kinetyka. Stawia dwa pytania: 1)Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? energia swobodna, G. postęp reakcji.
Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? energia swobodna, G. postęp reakcji.") Kinetyka energia swobodna, G termodynamika stan 1 kinetyka termodynamika stan 2 postęp reakcji 1 Kinetyka Stawia dwa pytania: 1)Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? 2 Jak szybko
Kinetyka energia swobodna, G termodynamika stan 1 kinetyka termodynamika stan 2 postęp reakcji 1 Kinetyka Stawia dwa pytania: 1)Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? 2 Jak szybko
TERMODYNAMIKA PROCESOWA
 (pieczęć wydziału) KARTA PRZEDMIOTU 1. Nazwa przedmiotu: TERMODYNAMIKA PROCESOWA 2. Kod przedmiotu: 3. Karta przedmiotu ważna od roku akademickiego:2011/2012 4. Forma kształcenia: studia pierwszego stopnia
(pieczęć wydziału) KARTA PRZEDMIOTU 1. Nazwa przedmiotu: TERMODYNAMIKA PROCESOWA 2. Kod przedmiotu: 3. Karta przedmiotu ważna od roku akademickiego:2011/2012 4. Forma kształcenia: studia pierwszego stopnia
Chemia fizyczna/ termodynamika, 2015/16, zadania do kol. 2, zadanie nr 1 1
 Chemia fizyczna/ termodynamika, 2015/16, zadania do kol. 2, zadanie nr 1 1 [Imię, nazwisko, grupa] prowadzący Uwaga! Proszę stosować się do następującego sposobu wprowadzania tekstu w ramkach : pola szare
Chemia fizyczna/ termodynamika, 2015/16, zadania do kol. 2, zadanie nr 1 1 [Imię, nazwisko, grupa] prowadzący Uwaga! Proszę stosować się do następującego sposobu wprowadzania tekstu w ramkach : pola szare
Prężność pary nad roztworem
 Tomasz Lubera Układ: Prężność pary nad roztworem dwuskładnikowy (składniki I i II) dwufazowy (ciecz i gaz) w którym faza ciekła i gazowa to roztwory idealne W stanie równowagi prężności pary składników/układu
Tomasz Lubera Układ: Prężność pary nad roztworem dwuskładnikowy (składniki I i II) dwufazowy (ciecz i gaz) w którym faza ciekła i gazowa to roztwory idealne W stanie równowagi prężności pary składników/układu
DRUGA ZASADA TERMODYNAMIKI
 DRUGA ZASADA TERMODYNAMIKI Procesy odwracalne i nieodwracalne termodynamicznie, samorzutne i niesamorzutne Proces nazywamy termodynamicznie odwracalnym, jeśli bez spowodowania zmian w otoczeniu możliwy
DRUGA ZASADA TERMODYNAMIKI Procesy odwracalne i nieodwracalne termodynamicznie, samorzutne i niesamorzutne Proces nazywamy termodynamicznie odwracalnym, jeśli bez spowodowania zmian w otoczeniu możliwy
Technologia Wody. 1. Definicja. a) proces jednostkowy b) zjawisko. Wykład 6 - ADSORPCJA
 proces jednostkowy b) zjawisko. Wykład 6 - ADSORPCJA") Technologia Wody Wykład 6 - ADSORPCJA 1. Definicja a) proces jednostkowy b) zjawisko 2. Przyczyny a) energia wewntrzna układu b) ciepło adsorpcji c) hydrofobowe hydrofilowe (apolarne polarne) 3. Rodzaje
Technologia Wody Wykład 6 - ADSORPCJA 1. Definicja a) proces jednostkowy b) zjawisko 2. Przyczyny a) energia wewntrzna układu b) ciepło adsorpcji c) hydrofobowe hydrofilowe (apolarne polarne) 3. Rodzaje
- prędkość masy wynikająca z innych procesów, np. adwekcji, naprężeń itd.
 4. Równania dyfuzji 4.1. Prawo zachowania masy cd. Równanie dyfuzji jest prostą konsekwencją prawa zachowania masy, a właściwie to jest to prawo zachowania masy zapisane dla procesu dyfuzji i uwzględniające
4. Równania dyfuzji 4.1. Prawo zachowania masy cd. Równanie dyfuzji jest prostą konsekwencją prawa zachowania masy, a właściwie to jest to prawo zachowania masy zapisane dla procesu dyfuzji i uwzględniające
Plan zajęć. Sorpcyjne Systemy Energetyczne. Podstawowe definicje. Definicje. Podstawowe definicje. Procesy adsorpcji
 Plan zajęć Sorpcyjne Systemy Energetyczne Procesy adsorpcji i desorpcji w systemach chłodniczych dr inż. Bartosz Zajączkowski Wydział Mechaniczno-Energetyczny Katedra Termodynamiki, Teorii Maszyn i Urządzeń
Plan zajęć Sorpcyjne Systemy Energetyczne Procesy adsorpcji i desorpcji w systemach chłodniczych dr inż. Bartosz Zajączkowski Wydział Mechaniczno-Energetyczny Katedra Termodynamiki, Teorii Maszyn i Urządzeń
Projekt Inżynier mechanik zawód z przyszłością współfinansowany ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego
 Zajęcia wyrównawcze z fizyki -Zestaw 4 -eoria ermodynamika Równanie stanu gazu doskonałego Izoprzemiany gazowe Energia wewnętrzna gazu doskonałego Praca i ciepło w przemianach gazowych Silniki cieplne
Zajęcia wyrównawcze z fizyki -Zestaw 4 -eoria ermodynamika Równanie stanu gazu doskonałego Izoprzemiany gazowe Energia wewnętrzna gazu doskonałego Praca i ciepło w przemianach gazowych Silniki cieplne
Kinetyka. energia swobodna, G. postęp reakcji. stan 1 stan 2. kinetyka
 Kinetyka postęp reakcji energia swobodna, G termodynamika kinetyka termodynamika stan 1 stan 2 Kinetyka Stawia dwa pytania: 1) Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? 1) Jak szybko
Kinetyka postęp reakcji energia swobodna, G termodynamika kinetyka termodynamika stan 1 stan 2 Kinetyka Stawia dwa pytania: 1) Jak szybko biegną reakcje? 2) W jaki sposób przebiegają reakcje? 1) Jak szybko
Termodynamiczny opis przejść fazowych pierwszego rodzaju
 Wykład II Przejścia fazowe 1 Termodynamiczny opis przejść fazowych pierwszego rodzaju Woda występuje w trzech stanach skupienia jako ciecz, jako gaz, czyli para wodna, oraz jako ciało stałe, a więc lód.
Wykład II Przejścia fazowe 1 Termodynamiczny opis przejść fazowych pierwszego rodzaju Woda występuje w trzech stanach skupienia jako ciecz, jako gaz, czyli para wodna, oraz jako ciało stałe, a więc lód.
WPŁYW ILOŚCI MODYFIKATORA NA WSPÓŁCZYNNIK RETENCJI W TECHNICE WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ
 WPŁYW ILOŚCI MODYFIKATORA NA WSPÓŁCZYNNIK RETENCJI W TECHNICE WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ Wprowadzenie Wysokosprawna chromatografia cieczowa (HPLC) jest uniwersalną technika analityczną, stosowaną
WPŁYW ILOŚCI MODYFIKATORA NA WSPÓŁCZYNNIK RETENCJI W TECHNICE WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ Wprowadzenie Wysokosprawna chromatografia cieczowa (HPLC) jest uniwersalną technika analityczną, stosowaną
Równowaga. równowaga metastabilna (niepełna) równowaga niestabilna (nietrwała) równowaga stabilna (pełna) brak równowagi rozpraszanie energii
 równowaga niestabilna (nietrwała) równowaga stabilna (pełna) brak równowagi rozpraszanie energii") Równowaga równowaga stabilna (pełna) równowaga metastabilna (niepełna) równowaga niestabilna (nietrwała) brak równowagi rozpraszanie energii energia swobodna Co jest warunkiem równowagi? temperatura W
Równowaga równowaga stabilna (pełna) równowaga metastabilna (niepełna) równowaga niestabilna (nietrwała) brak równowagi rozpraszanie energii energia swobodna Co jest warunkiem równowagi? temperatura W
ROZDZIELENIE OD PODSTAW czyli wszystko (?) O KOLUMNIE CHROMATOGRAFICZNEJ
 O KOLUMNIE CHROMATOGRAFICZNEJ") ROZDZIELENIE OD PODSTAW czyli wszystko (?) O KOLUMNIE CHROMATOGRAFICZNEJ Prof. dr hab. inż. Agata Kot-Wasik Katedra Chemii Analitycznej Wydział Chemiczny, Politechnika Gdańska agawasik@pg.gda.pl ROZDZIELENIE
ROZDZIELENIE OD PODSTAW czyli wszystko (?) O KOLUMNIE CHROMATOGRAFICZNEJ Prof. dr hab. inż. Agata Kot-Wasik Katedra Chemii Analitycznej Wydział Chemiczny, Politechnika Gdańska agawasik@pg.gda.pl ROZDZIELENIE