Metody dokowania ligandów
|
|
|
- Maria Nawrocka
- 9 lat temu
- Przeglądów:
Transkrypt
1 Metody dokowania ligandów

2 Strategie projektowania leków Ligand-based drug design nieznana Budowanie modelu miejsc aktywnych liganda (farmakofor) Przeszukiwanie baz danych (screening) Struktura celu molekularnego znana 1D i 3D QSAR (pseudoreceptory i pola molekularne) Dopasowanie ligandów do miejsca aktywnego receptora (dokowanie) Budowa nowych ligandów ab-initio Receptor-based drug design Dynamika kompleksu receptor-ligand
 Budowa nowych ligandów ab-initio Receptor-based drug design Dynamika kompleksu")
 Energia lub tzw.")
3 Dokowanie znajdowanie struktury kompleksu o najniższej energii (najbardziej dopasowany) Energia lub tzw. scoring function (kształt i właściwości fizykochemiczne)



4 Energia a potencjał termodynamiczny mechanika molekularna Energia jest użyteczna przy porównywaniu różnych konformacji kompleksu

5 Trzy składniki programów do dokowania a) b) c) Atomic Surface Grid (najbardziej dokładna reprezentacja) Algorytmy szukania konformacji i wyznaczania energii są ściśle ze sobą związane


6 Dokowanie przez komplementarność powierzchni H hole, K knob; sposób używany w b. wielu programach dokujących

7 Reprezentacja białka przez grid (sieć) W każdym punkcie sieci mierzymy wielkość oddziaływań niewiążących: - Van der Waalsa - Elektrostatyczne Oczka sieci ok. 2 Å Zmierzone wielkości są zapamiętywane w tabeli Pierwsze użycie: Goodford 1985 r.

8 Użycie gridu do uproszczenia dokowania Dla każdego atomu liganda liczone jest oddziaływanie od otaczających punktów sieci (odcięcie cutoff ok. 10 Å) Dodatkowe przyspieszenie: Dla każdego typu atomu liganda w każdym węźle sieci energia jest obliczana i przechowywana w tabeli Potencjał dla danego atomu liganda jest obliczany przez interpolację z otaczających punktów gridu Metoda często używana w symulacjach typu virtul reality

9 Trudności w doborze scoring function Który enancjomer liganda jest aktywny a który nieaktywny?

10 Czynniki wpływające na wiązanie ligandów

11 Korzystne oddziaływania Oddziaływania kierunkowe i silne Oddziaływania van der Waalsa: bezkierunkowe i słabe lecz liczne

 Części hydrofobowe oddziałują ze sobą aby zmniejszyć oddziaływania z otaczającą")
12 Energie częściowej desolwatacji liganda i miejsca aktywnego Proces kierowany termodynamicznie (efekty entropowe) Części hydrofobowe oddziałują ze sobą aby zmniejszyć oddziaływania z otaczającą wodą

13 Konformacyjne efekty entropowe Konformacja bioaktywna (w miejscu aktywnym) może być inna niż na zewnątrz Dotyczy także konformacji białka: głównie konformacji łańcuchów bocznych aminokwasów z miejsca aktywnego Konieczne jest dokowanie wielu różnych konformacji liganda


14 Wiązanie liganda jako reakcja równowagowa Ln (K) Stała K jest mierzona doświadczalnie. K F stała tworzenia [M -1 ] K D stała dysocjacji [M]

 Pole siłowe skalibrowane dla kompleksów ze zbioru treningowego: znane są struktury 3D i aktywności biologiczne ligandów w tych")
15 Empiryczne funkcje oceniające Współczynniki wagowe k uzyskuje się w procesie parametryzacji (kalibracji) wybranego pola siłowego (k mogą być dodatnie lub ujemne) Pole siłowe skalibrowane dla kompleksów ze zbioru treningowego: znane są struktury 3D i aktywności biologiczne ligandów w tych kompleksach

 Obliczane są tylko niektóre składowe pola")
16 Przykład empirycznej funkcji oceniającej z programu AutoDock (obliczane z solvent excluded volumes) Obliczane są tylko niektóre składowe pola siłowego
 Obliczane są tylko niektóre")
 Solvent accessible surface area Promień kulki dla")
17 Powierzchnia dostępna dla rozpuszczalnika Connoly surface (wygładzona powierzchnia molekularna) Solvent accessible surface area Promień kulki dla wody = 1.4 Å


18 Funkcje oceniające oparte na wiedzy (knowledge based scoring functions) - wykorzystanie Bazują na odległościach w strukturach 3D kompleksów ligand-cel molekularny

 E ij ~ ln(g ij ) * kt E ij = ln (g ij / g ref ) Dla 126 typów atomów w białku i 34 typów atomów w ligandach obliczanych jest 4284")
19 Funkcje oceniające oparte na wiedzy otrzymywanie Dla każdej pary oddziałujących atomów (typów atomów) obliczany jest wykres PMF (Potential of Mean Force) K D rozkład Boltzmanna g ij ~ exp(-e ij /kt) E ij ~ ln(g ij ) * kt E ij = ln (g ij / g ref ) Dla 126 typów atomów w białku i 34 typów atomów w ligandach obliczanych jest 4284 wykresów PMF

20 Inne potencjały statystyczne Częstości dla kątów torsyjnych Wykresy 2D otrzymane dla częstości kątów dla aminokwasów

21 Przykład użycia funkcji oceniającej opartej na wiedzy w programie DrugScore 1400 kompleksów ligand-białko z bazy PDB (G. Klebe, Marburg University) G ref (R) wykres dla wszystkich ligandów (kolor czarny)

22 Funkcje oceniające są wrażliwe na wybór kompleksów ligand-białko do zbioru treningowego Większy zbiór kompleksów zapewnia większą dokładność funkcji oceniającej dla średniego białka

23 Inne funkcje oceniające wiązanie ligandów dokładność vs. szybkość
24 Komplementarność kształtu i własności w układzie ligand-cel molekularny Zwykle używane jako wstępne oszacowanie. Dodawane komplementarne własności: wiązania wodorowe, hydrofobowość, mostki solne itp.

25 Prosta metoda szacowania komplementarności powierzchni Metoda jest oparta na 3D grid
 2) Pozwala tylko porównywać ligandy 3) Daje dobre wyniki tylko przy małych zmianach liganda w miejscu")
26 FEP - Free Energy Perturbation najbardziej dokładna z funkcji oceniających 1) Wymaga długiego czasu obliczeń (wymaga dynamiki molekularnej) 2) Pozwala tylko porównywać ligandy 3) Daje dobre wyniki tylko przy małych zmianach liganda w miejscu aktywnym
27 Free Energy Perturbation Cykl termodynamiczny dla obliczania energii wymiany liganda w białku (FEP Free Energy Perturbation) G X + G C = G F + G Y G wymiany liganda = G X - G Y = G F - G C

28 Zmiana położenia liganda w miejscu aktywnym Ligand i białko traktowane jako sztywne obiekty

29 Podział algorytmów dokujących Algorytm szuka komplementarnych własności Algorytm eksploruje całą dostępną przestrzeń

30 Feature-based matching - idea Pattern recognition (algorytmy z analizy obrazów, robotyki, )

31 Dopasowanie oparte na komplementarności i podobieństwie

32 Etapy feature-based matching Podobne do układania puzzli

33 Programy stosujące feature-based matching Pierwszy program DOCK z 1982 r.

34 Program DOCK (algorytm clique-search) - kieszeń receptora jest odwzorowana sferami; - min. 4 atomy liganda muszą się nałożyć na środki sfer.

35 Cel algorytmu dokującego programu DOCK 1. Metoda najprostsza i najdłuższa: sprawdzić nakładanie wszystkich par; 2. Użycie grafu dokującego i szukanie tzw. maximum clique.

36 Idea algorytmu clique-search (oparty na teorii grafów) W powyższym przykładzie: miejsca B-1 i C-2 mogą być dopasowane do siebie jednocześnie gruba linia na grafie

37 Znajdowanie maximum clique Miejsca D-1, C-2 i B-3 mogą być dopasowane do siebie jednocześnie. Linie reprezentują dopasowanie odległości.

38 Szybkie znajdowanie pasujących własności Przykład: poszukiwanie trójkątów ADD (Akceptor, Donor, Donor) z określonymi odległościami. Użycie hash tables znacznie przyspiesza poszukiwania (porównuje się tylko indeksy nie oblicza odległości)

39 Klastrowanie póz ligand-receptor Użycie geometrycznego haszowania (hashing) znacznie przyspiesza ten proces. Hash tables oblicza się tylko raz dla każdego liganda.
 (klastrowanie wg.")
40 Program PatchDock dokowanie ligand-białko i białko-białko (Connoly dot surface) (klastrowanie wg. RMSD) PatchDock dokowanie łatek
41 Przygotowanie białka do dokowania formy tautomeryczne i minimalizacja energii

 (H)N-N cis-trans Analiza")
42 Przygotowanie liganda do dokowania 2D 3D, usuwanie soli itp. Dla struktur alternatywnych NH 2 NH 3+, COOH COO - chiralne lub N-N(H) (H)N-N cis-trans Analiza konformacyjna w próżni lub w roztworze wodnym

43 Dokowanie molekularne pozwala wyjaśnić jak małe zmiany struktury liganda mogą powodować duże zmiany aktywności
44 Metody optymalizacyjne znajdowania najlepszego dopasowania liganda

45 Schemat metody Monte Carlo

46 Schemat metody Simulated Annealing Cykl ogrzewania i powolnego oziębiania układu jest powtarzany wiele razy aż do znalezienia wielu prawie optymalnych rozwiązań. Ogrzewanie powoduje przekraczanie barier energetycznych a oziębianie pozwala na znalezienie układów z najniższą energią. Zaimplementowana po raz pierwszy w AutoDocku.

47 Zasada działania Algorytmów Genetycznych

48 Krzyżowanie i mutacje w GA przy dokowaniu Dokowanie sztywne (tylko rotacje i translacje)

49 Algorytmy Genetyczne Lamarcka (LGA) z lokalną optymalizacją Procedura stosowana obecnie w AutoDocku

50 Tabu search (TS) Pozwala na wyjście z lokalnych minimów kosztem przejściowego zaburzenia układu (stopniowe zasypywanie lokalnych minimów energii)

51 Metoda hybrydowa dokowania (TS+GA)
52 Dokowanie giętkich ligandów Rozwój metod giętkiego dokowania zapoczątkował nową dziedzinę projektowania leków: structure-based drug design

53 Metody do giętkiego dokowania ligandów

54 Dokowanie zespołowe/całościowe (ensemble docking) Efektywne unikanie błędnych dokowań Łatwa implementacja do dokowania podobnych związków z dużych baz danych
 - Inrcremental approach (fragmenty dokowane zależne od siebie)")
55 Dokowanie fragmentowe (fragmentation docking) Metody dokowania fragmentowego: - Place-and-join (fragmenty dokowane niezależne) - Inrcremental approach (fragmenty dokowane zależne od siebie)

56 Metoda ułóż i połącz (place-and-join) Wybór nieoptymalnych dokowań cząstkowych aby połączyć fragmenty (wskazówki do modyfikacji struktury liganda)
57 Metoda stopniowego dokowania fragmentów (incremental approach) Najpierw dokuje się rdzeń liganda a następnie po kolei pozostałe fragmenty minimalizując energię powstającego liganda

58 Wybór położenia pierwszego fragmentu ma wpływ na wynik dokowania Wybór najlepszego dopasowania na podstawie funkcji oceniającej poszczególne pozy
59 Przykłady zastosowań dokowania giętkiego w programach GOLD algorytmy genetyczne (GA) MOE-Dock Monte Carlo (MC), Tabu search PRO_LEADS algorytmy genetyczne AutoDock algorytmy genetyczne, Tabu search Kąty torsyjne giętkiego liganda wprowadza się do chromosomów w GA lub do parametrów do optymalizacji w MC

60 GOLD skład chromosomów w GA część chromosomu dotycząca giętkiego dokowania Dodatkowe śledzenie każdego potencjalnego wiązania wodorowego pomiędzy ligandem i białkiem w celu maksymalizacji ich liczby

61 Ważność giętkiego dokowania W 50% przypadków sztywne kros-dokowanie prowadziło do błędnych wyników
62 Inhibitory trombiny (proteaza serynowa tnie fibrynogen krzepliwość krwi) pochodna tetrazolowa porównanie struktur trombiny + CXCR4 62

63 Uwzględnianie giętkości białka


64 Miękkie dokowanie ligandów Pozwala na niewielkie nakładanie się struktur liganda i receptora Modyfikacja potencjału Lennarda-Jonesa (LJ) aby siły van der Waalsa były mniej odpychające (plastyczność receptora bez zmiany jego konformacji)


65 Miękkie dokowanie ligandów Zmniejszanie promieni van der Waalsa (rozmiarów atomów) dla liganda i receptora Modyfikacja potencjału elektrostatycznego
 Soft scoring jest stosowana")

66 Miękkie funkcje oceniające (soft scoring) Soft scoring jest stosowana jako wstępna funkcja oceniająca do szybkiego odsiania niepasujących ligandów

 eksplozja")
67 Uwzględnianie ruchów łańcuchów bocznych w białku Rozwiązanie: biblioteka konformerów aminokwasów (1987 Ponder & Richards) eksplozja konformacyjna

68 Złożoność obliczeniowa przy giętkim dokowaniu Oszacowanie na jeden CPU
69 Metoda Multiple protein structure (MPS) Białko CDK2 (Cyclin-Dependent Kinase 2 )
70 Źródła struktur do metody MPS Metoda Zalety Wady Metody doświadczalne (X-ray, NMR) Biblioteki rotamerów Dynamika molekularna Analiza drgań normalnych Rzeczywiste rotamery Dostępność Dostępność, konformery o niskiej energii Dostępne duże zmiany strukturalne Mała dostępność Tylko standardowe łańcuchy boczne Czas obliczeniowy, brak weryfikacji dośw. Brak weryfikacji dośw.

71 Metoda łączonych białek united protein approach Program FlexE. Nałożone struktury białka pozwalają na zbudowaniu wielu wirtualnych konformacji Sposób dokowania ligandów


72 Metoda uśrednionego gridu Energie oddziaływania liczone dla każdej struktury białka oddzielnie Uśredniony grid


73 Tolerancja dokowania ligandów
74 Uwzględnianie dużych ruchów białka wiązanie substratu przez kalmodulinę

75 Badania zmian w białku metodami symulacyjnymi możliwości i ograniczenia MD kompleksu biotyna-streptawidyna

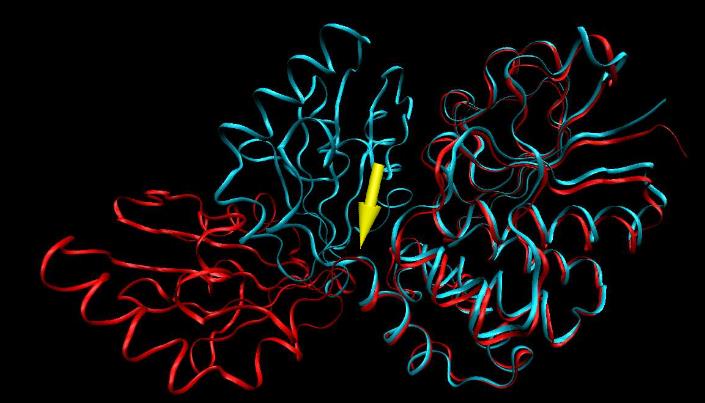
76 Ruchy zawiasowe białek Identyfikacja zawiasów

77 Dokowanie ze zginaniem domen Bardziej efektywny algorytm
78 Zastosowania dokowania hamowanie oddziaływania dwu białek Analiza oddziaływań
79 Budowanie zapytania do baz danych ligandów


80 Sprawdzanie hipotez wiązania ligandów Inhibitor kinazy CDK2 Dokowanie pozwala sprawdzić hipotezy


81 Identyfikacja błędnej hipotezy Pochodne adeniny Wiązanie się części adeninowej w innej orientacji
82 Wyznaczanie potencjału związku do jego późniejszej modyfikacji
83 Modyfikacje liganda (inhibitory trombiny join.sce) Modyfikacje ligandów aby zwiększyć oddziaływania z receptorem Manualne modyfikacje liganda Automatyczne budowanie de-novo z fragmentów

84 Przewidywanie konformacji bioaktywnej i tworzenie farmakoforów

85 Programy do dokowania
86 Program GOLD Własności: - Algorytmy genetyczne do dokowania ligandu - Giętki ligand i częściowo giętkie białko - Funkcje oceniające oparte na strukturach realnych ligandów - Możliwość wyboru funkcji oceniającej Dokowanie inhibitora do metaloproteazy (porównanie ze strukturą krystaliczną)
87 Program GLIDE (Schrödinger Inc.) Stosowana szybka metoda systematycznego przeszukiwania (powrót do tej metody za sprawą komputerów wieloprocesorowych i obliczeń masywnie równoległych)

88 Procentowe użycie poszczególnych programów do dokowania

89 Przykłady leków uzyskanych przy użyciu dokowania
90 Dynamika molekularna schemat obliczeń F(t) i = m i * a(t) i II zasada ruchu Newtona a(t) i = F(t) i / m i a(t) i v(t) i r(t+ t) i t = 1 fs = s r(t+ t) i F(t+ t) i
91 Metody symulacyjne - Dynamika Molekularna Zgodnie z prawami dynamiki ruchu Newtona. Trajektoria ruchu jest otrzymywana przez rozwiązanie równań różniczkowych: (F = ma) d x dt Algorytm całkowania numerycznego szybkość obliczeń, stałość energii całkowitej (zasada zachowania energii) 2 i 2 F x m r(t+ t) = r(t) + t v(t) + ½ ( t) 2 a(t) +... v(t+ t) = v(t) + t a(t) +... i i metoda skończonych różnic: algorytm Verleta (najbardziej popularny) [Verlet 1967] i modyfikacje: leap-frog [Hockney 1970], metoda Beemana [Beeman 1976]; metoda predykcyjno-korekcyjna [Gear 1971]

92 Symulacje MD energia układu E całkowita E kinetyczna i potencjalna różnice E całk zależą też od wyboru kroku czasowego obliczeń t.
.")
93 Wybór kroku czasowego obliczeń Jeśli energia kinetyczna przekroczy progową wielkość (tu: przyciąganie się atomów Ar) atomy rozbiegają się. Krok czasowy powinien być porównywalny z częstotliwością najszybszych ruchów w cząsteczce (jeśli nie są one celem badawczym można je "zamrozić" np. stałe długości wiązań i wydłużyć krok obliczeń). (Ar 2 ) układ atomy sztywne cząsteczki giętkie cząsteczki, sztywne wiązania giętkie cząsteczki, giętkie wiązania Typ ruchu translacje translacje, rotacje translacje, rotacje, oscylacje translacje, rotacje, oscylacje cząsteczki i oscylacje wiązań Krok czasowy 10fs 5fs 2fs 1fs lub 0.5fs (z H)
94 Przypisanie prędkości początkowych T = 0K i stopniowe ogrzewanie do 300K lub prędkości przypadkowe zgodnie z rozkładem prędkości Maxwella-Boltzmanna. Monitorowanie parametrów układu należy rozpocząć po fazie równowagowania (equilibration). Dotyczy szczególnie układów heterogenicznych np. białko w otoczeniu rozpuszczalnika: minimalizacja rozpuszczalnika z przeciwjonami (białko nieruchome) dynamika rozpuszczalnika (białko nieruchome); czas > czas relaksacji samego rozpuszczalnika (dla H 2 O 10ps). minimalizacja całego układu start MD
95 Obliczanie temperatury E kin N i 1 2 p i 2m i kbt 2 3N N c N c - liczba więzów 3N - N c - całkowita liczba stopni swobody W układach izolowanych - całkowity pęd i moment pędu są zachowane i równe 0 przez dobór początkowych prędkości (N c = 6). W układach z periodic box moment pędu nie jest zachowany (N c = 3) albo można co jakiś czas odpowiednio skalować prędkości. Dodatkowe więzy na dowolnych współrzędnych wewnętrznych: SHAKE [Tobias & Brooks 1988] (łatwiejszy w implementacji) RATTLE [Anderson 1983]
96 Wybór warunków prowadzenia procesu Zespół mikrokanoniczny kanoniczny izotermalnoizobaryczny Wielkości stałe NVE NVT NPT Stan równowagi max. entropii (S) tradycyjna MD min. wolnej energii (A) tradycyjna MC min. pot. Gibbsa (G)
97 Zastosowanie ciągłego rozpuszczalnika dynamika stochastyczna (równanie ruchu Langevina): m i 2 d xi ( t) 2 dt F { x i i ( t)} i dxi ( t) dt m i R i ( t) - uwzględnienie sił tarcia ( m i v i ) - oraz ruchów Browna (przypadkowych zderzeń z cząsteczkami rozpuszczalnika pozwala na dłuższy krok czasowy umożliwia dużo dłuższe obliczenia niż z rozpuszczalnikiem explicite (szczególnie do polimerów) nie uwzględnia specyficznych oddziaływań z rozpuszczalnikiem (np. wiązania wodorowe)
 - napięcie powierzchniowe, parowanie.")
98 Pudło periodyczne periodic box Liczba cząsteczek w pudle pozostaje stała. Periodic box pozwala na pozbycie się efektów brzegowych przy symulacjach w próżni (sfera rozpuszczalnika otacza badaną cząsteczkę) - napięcie powierzchniowe, parowanie. Aby zmniejszyć liczbę cząsteczek rozpuszczalnika lub dostosować do kształtu badanej cząsteczki (sfera, helisa) stosuje się różne kształty periodic box. Muszą wypełniać całą przestrzeń poprzez odpowiednie translacje komórki elementarnej. Periodic box dla symulacji ciał stałych: Powierzchnia kryształu jest granicą rzeczywistą. Kiedy molekuła wychodzi górą jest odbijana od górnej granicy.
 enzym 2500 atomów w periodic box (odległość powierzchni białka do granicy pudła 10 Å) -")
, rezerwuar (pomiędzy R 1 i R 2 ) dozwolony ruch atomów tylko wewnątrz tej sfery.")
99 Nieperiodyczne warunki brzegowe gdy bardzo duża cząsteczka lub gdy warunki nierównowagowe 1. symulacja w "kropli" rozpuszczalnika (10Å lub 5Å powłoka wokół badanej cząsteczki) enzym 2500 atomów w periodic box (odległość powierzchni białka do granicy pudła 10 Å) - razem 20,000 atomów kropla 10 Å - 14,700 kropla 5 Å - 8, Podział cząsteczki na miejsce aktywne (atomy mogą się poruszać), rezerwuar (pomiędzy R 1 i R 2 ) dozwolony ruch atomów tylko wewnątrz tej sfery. Reszta atomów nieruchoma lub ograniczona potencjałem harmonicznym do położeń początkowych. W obu tych metodach możliwe jest wystąpienie dużych efektów ubocznych w postaci sztucznych zjawisk nie obserwowanych w rzeczywistości
100 Promienie odcięcia (cutoff) dla oddziaływań van der Waalsa i elektrostatycznych Metoda PME (Particle Mesh Ewald) pozwala na obliczenia elektrostatyczne dalekozasięgowe (multipole zamiast ładunków cząstkowych na atomach) zamiast cutoff el-stat
 (H 2 O) 2 (H 2 O) 2 cutoff = 8Å")
101 Efekty działania potencjału - cutoff obliczenia oddziaływań niewiążących jest najbardziej czasochłonną częścią MD lub MC (ilość obliczeń N 2 ) dlatego wprowadza się do nich sferyczną granicę działania potencjału cutoff powinien być tak dobrany aby cząsteczka nie widziała swego własnego odbicia (mniejszy niż połowa długości najkrótszego boku komórki) (H 2 O) 2 (H 2 O) 2 cutoff = 8Å group-based cutoff potencjały przesuwane lub przełączane (np. na wielomian) Particle Mesh Ewald (PME)
102 Gładkie wyłączanie potencjału na granicy cutoff

103 Tworzenie listy par atomów dla oddziaływań niewiążących
104 Wybór długości symulacji
![metoda Monte Carlo Algorytm Metropolisa [Metropolis et al. 1953]: nowy stan układu jest akceptowany zawsze jeśli ma energię niższą od poprzedniego stanu.](/docs-images/30/13959166/images/105-0.png "jeśli ma wyższą energię - tylko wtedy jeśli czynnik Boltzmanna nie jest mniejszy od przypadkowej liczby z zakresu 0 1: Rozmiar każdego kroku musi być nie większy niż r max który jest tak określony,")
105 metoda Monte Carlo Algorytm Metropolisa [Metropolis et al. 1953]: nowy stan układu jest akceptowany zawsze jeśli ma energię niższą od poprzedniego stanu. jeśli ma wyższą energię - tylko wtedy jeśli czynnik Boltzmanna nie jest mniejszy od przypadkowej liczby z zakresu 0 1: Rozmiar każdego kroku musi być nie większy niż r max który jest tak określony, aby ok. 50% próbnych układów było akceptowanych. Jest on szacowany na początku i korygowany w trakcie obliczeń Podobnie jak w MD wymagana jest faza równowagowania układu przed fazą zbierania danych. W klasycznej netodzie MC T=const. i V=const. Można także używać zespołu NPT

106 Zmiana struktury w metodzie Monte Carlo we współrzędnych kartezjańskich: tylko małe zmiany są dozwolone aby nie zniekształcić cząsteczki - dużo więcej obliczeń. we współrzędnych wewnętrznych: można ustalić długości wiązań i kąty płaskie. Także małe zmiany kątów torsyjnych. MC jest stosowana głównie do modelowania uproszczonych modeli polimerów i biopolimerów rozpiętych na sieciach: sieć sześcienna sieć typu diamentu

107 Typy ruchów MC stosowanych w sieciach Przy szczególnie ciasno upakowanych polimerach jedynym możliwym ruchem może być ruch "wężowy": Modelowanie nieskończonych łańcuchów polimerów:

108 Próbkowanie przestrzeni konformacyjnej przy użyciu metod symulacyjnych Stosując odpowiednio wysokie temperatury (fizycznie niemożliwe) w MD i MC można przejść przez bardzo wysokie bariery energetyczne i zbadać całą przestrzeń fazową. Minimalizacja pozwala na uzyskanie lokalnych minimów. Simulated annealing [Kirkpatrick et al., 1983] Proces, w którym temperatura jest powoli obniżana a układ ma wystarczająco dużo czasu aby krystalizować bez defektów sieci. W wysokiej temperaturze układ jest równowagowany (MD lub MC) - może przechodzić przez wysokie bariery energetyczne. W miarę obniżania temperatury do 0 K osiąga się konformację o możliwie najniższej energii. Nie ma gwarancji osiągnięcia minimum globalnego ale stosując różne konformacje startowe można uzyskać prawie optymalne struktury.

109 Dynamika Molekularna z więzami Restrained Molecular Dynamics RMD i SA są wykorzystywane w X-ray i NMR do uzyskiwania końcowych struktur dużych cząsteczek. Dodatkowa funkcja kary jest dodawana do potencjału - podwyższa potencjał jeśli konformacja wykracza poza więzy. miara dopasowania - czynnik R struktury: R F - amplitudowe czynniki struktury F obs F obs F calc Metoda RMD zastąpiła metodę najmniejszych kwadratów prowadząc do lepszych struktur końcowych. Klejone potencjały stosowane w więzach: Dopasowywanie struktury cząsteczki do map gęstości elektronowej z X-ray
110 Porównanie algorytmów znajdowania konformacji Metoda systematyczne przeszukiwanie random search (cartesian) random search (internal) distance geometry molekularna dynamika Liczba konformacji [Saunders et al., 1990] C 17 H 34 Całkowita liczba konformacji w zakresie 3 kcal/mol od minimum globalnego wyniosła 262 żadna z metod nie znalazła wszystkich konformacji zgodnie z rozkładem Boltzmanna konformacje minimum globalnego stanowią tylko 8% wszystkich konformacji (zakładając równość entropii tych konformacji).
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład IV Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład IV Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Strategie projektowania leków
 Strategie projektowania leków Ligand-based drug design nieznana Budowanie modelu miejsc aktywnych liganda (farmakofor) Przeszukiwanie baz danych (screening) Struktura celu molekularnego znana 1D i 3D QSAR
Strategie projektowania leków Ligand-based drug design nieznana Budowanie modelu miejsc aktywnych liganda (farmakofor) Przeszukiwanie baz danych (screening) Struktura celu molekularnego znana 1D i 3D QSAR
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład V Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład V Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład VI Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład VI Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowania leków
 Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Dokowanie molekularne. Karol Kamel Uniwersytet Warszawski
 molekularne Wstęp Dokowanie metoda modelowania molekularnego, pozwalająca na znalezienie położenia (i konformacji) liganda w miejscu wiążącym receptora. Informacja ta pozwala na ocenę energii swobodnej
molekularne Wstęp Dokowanie metoda modelowania molekularnego, pozwalająca na znalezienie położenia (i konformacji) liganda w miejscu wiążącym receptora. Informacja ta pozwala na ocenę energii swobodnej
QSAR i związki z innymi metodami. Karol Kamel Uniwersytet Warszawski
 QSAR i związki z innymi metodami Wstęp QSAR Quantitative Structure-Activity Relationship. Jest to metoda polegająca na znalezieniu (i analizie) zależności pomiędzy strukturą chemiczną (geometria cząsteczki,
QSAR i związki z innymi metodami Wstęp QSAR Quantitative Structure-Activity Relationship. Jest to metoda polegająca na znalezieniu (i analizie) zależności pomiędzy strukturą chemiczną (geometria cząsteczki,
cz. VII Metody projektowania leków i modelowanie molekularne
 Oddziaływanie leków z celami molekularnymi cz. VII Metody projektowania leków i modelowanie molekularne Prof. dr hab. Sławomir Filipek Uniwersytet Warszawski, Wydział Chemii 1 Strategie projektowania leków
Oddziaływanie leków z celami molekularnymi cz. VII Metody projektowania leków i modelowanie molekularne Prof. dr hab. Sławomir Filipek Uniwersytet Warszawski, Wydział Chemii 1 Strategie projektowania leków
Optymalizacja optymalizacji
 7 maja 2008 Wstęp Optymalizacja lokalna Optymalizacja globalna Algorytmy genetyczne Badane czasteczki Wykorzystane oprogramowanie (Algorytm genetyczny) 2 Sieć neuronowa Pochodne met-enkefaliny Optymalizacja
7 maja 2008 Wstęp Optymalizacja lokalna Optymalizacja globalna Algorytmy genetyczne Badane czasteczki Wykorzystane oprogramowanie (Algorytm genetyczny) 2 Sieć neuronowa Pochodne met-enkefaliny Optymalizacja
17.1 Podstawy metod symulacji komputerowych dla klasycznych układów wielu cząstek
 Janusz Adamowski METODY OBLICZENIOWE FIZYKI 1 Rozdział 17 KLASYCZNA DYNAMIKA MOLEKULARNA 17.1 Podstawy metod symulacji komputerowych dla klasycznych układów wielu cząstek Rozważamy układ N punktowych cząstek
Janusz Adamowski METODY OBLICZENIOWE FIZYKI 1 Rozdział 17 KLASYCZNA DYNAMIKA MOLEKULARNA 17.1 Podstawy metod symulacji komputerowych dla klasycznych układów wielu cząstek Rozważamy układ N punktowych cząstek
Podstawy projektowania leków wykład 12
 Podstawy projektowania leków wykład 12 Łukasz Berlicki Projektowanie wspomagane komputerowo Ligand-based design QSAR i 3D-QSAR Structure-based design projektowanie oparte na strukturze celu molekularnego
Podstawy projektowania leków wykład 12 Łukasz Berlicki Projektowanie wspomagane komputerowo Ligand-based design QSAR i 3D-QSAR Structure-based design projektowanie oparte na strukturze celu molekularnego
Badanie długości czynników sieciujących metodami symulacji komputerowych
 Badanie długości czynników sieciujących metodami symulacji komputerowych Agnieszka Obarska-Kosińska Prof. dr hab. Bogdan Lesyng Promotorzy: Dr hab. Janusz Bujnicki Zakład Biofizyki, Instytut Fizyki Doświadczalnej,
Badanie długości czynników sieciujących metodami symulacji komputerowych Agnieszka Obarska-Kosińska Prof. dr hab. Bogdan Lesyng Promotorzy: Dr hab. Janusz Bujnicki Zakład Biofizyki, Instytut Fizyki Doświadczalnej,
Bioinformatyka wykład 3.I.2008
 Bioinformatyka wykład 3.I.2008 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2008-01-03 1 Plan wykładu analiza i porównywanie struktur białek. doświadczalne metody badania struktur
Bioinformatyka wykład 3.I.2008 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2008-01-03 1 Plan wykładu analiza i porównywanie struktur białek. doświadczalne metody badania struktur
Projektowanie Nowych Chemoterapeutyków
 Jan Mazerski Katedra Technologii Leków i Biochemii Wydział Chemiczny Projektowanie Nowych Chemoterapeutyków XV. QSAR 3D QSAR w przestrzeni Rozwój metod ustalania struktury 3D dla białek i ich kompleksów.
Jan Mazerski Katedra Technologii Leków i Biochemii Wydział Chemiczny Projektowanie Nowych Chemoterapeutyków XV. QSAR 3D QSAR w przestrzeni Rozwój metod ustalania struktury 3D dla białek i ich kompleksów.
Ćwiczenie 4: Modelowanie reakcji chemicznych. Stan przejściowy.
 Ćwiczenie 4: Modelowanie reakcji chemicznych. Stan przejściowy. Celem ćwiczenia jest wymodelowanie przebiegu reakcji chemicznej podstawienia nukleofilowego zachodzącego zgodnie z mechanizmem SN2. Wprowadzenie:
Ćwiczenie 4: Modelowanie reakcji chemicznych. Stan przejściowy. Celem ćwiczenia jest wymodelowanie przebiegu reakcji chemicznej podstawienia nukleofilowego zachodzącego zgodnie z mechanizmem SN2. Wprowadzenie:
Modelowanie molekularne
 Ck08 Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 10 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody
Ck08 Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 10 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody
Program MC. Obliczyć radialną funkcję korelacji. Zrobić jej wykres. Odczytać z wykresu wartość radialnej funkcji korelacji w punkcie r=
 Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
Ogólny schemat postępowania
 Ogólny schemat postępowania 1. Należy zdecydować, który rozkład prawdopodobieństwa chcemy badać. Rozkład oznaczamy przez P; zależy od zespołu statystycznego. 2. Narzucamy warunek równowagi szczegółowej,
Ogólny schemat postępowania 1. Należy zdecydować, który rozkład prawdopodobieństwa chcemy badać. Rozkład oznaczamy przez P; zależy od zespołu statystycznego. 2. Narzucamy warunek równowagi szczegółowej,
Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d.
 Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 11.01.11 1 Dopasowanie strukturalne (alignment) odległość: d ij = (x i -x J ) 2 + (y i -y J ) 2
Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 11.01.11 1 Dopasowanie strukturalne (alignment) odległość: d ij = (x i -x J ) 2 + (y i -y J ) 2
Structure and Charge Density Studies of Pharmaceutical Substances in the Solid State
 Maura Malińska Wydział Chemii, Uniwersytet Warszawski Promotorzy: prof. dr hab. Krzysztof Woźniak, prof. dr hab. Andrzej Kutner Structure and Charge Density Studies of Pharmaceutical Substances in the
Maura Malińska Wydział Chemii, Uniwersytet Warszawski Promotorzy: prof. dr hab. Krzysztof Woźniak, prof. dr hab. Andrzej Kutner Structure and Charge Density Studies of Pharmaceutical Substances in the
Model wiązania kowalencyjnego cząsteczka H 2
 Model wiązania kowalencyjnego cząsteczka H 2 + Współrzędne elektronu i protonów Orbitale wiążący i antywiążący otrzymane jako kombinacje orbitali atomowych Orbital wiążący duża gęstość ładunku między jądrami
Model wiązania kowalencyjnego cząsteczka H 2 + Współrzędne elektronu i protonów Orbitale wiążący i antywiążący otrzymane jako kombinacje orbitali atomowych Orbital wiążący duża gęstość ładunku między jądrami
Wykład 5 Widmo rotacyjne dwuatomowego rotatora sztywnego
 Wykład 5 Widmo rotacyjne dwuatomowego rotatora sztywnego W5. Energia molekuł Przemieszczanie się całych molekuł w przestrzeni - Ruch translacyjny - Odbywa się w fazie gazowej i ciekłej, w fazie stałej
Wykład 5 Widmo rotacyjne dwuatomowego rotatora sztywnego W5. Energia molekuł Przemieszczanie się całych molekuł w przestrzeni - Ruch translacyjny - Odbywa się w fazie gazowej i ciekłej, w fazie stałej
Podstawy projektowania leków wykład 13
 odstawy projektowania leków wykład 13 Łukasz Berlicki rojektowanie wspomagane komputerowo rojektowanie leków oparte na strukturze często wykorzystuje metody komputerowe, aby: rzeanalizować duŝą liczbę
odstawy projektowania leków wykład 13 Łukasz Berlicki rojektowanie wspomagane komputerowo rojektowanie leków oparte na strukturze często wykorzystuje metody komputerowe, aby: rzeanalizować duŝą liczbę
Podstawowe prawa opisujące właściwości gazów zostały wyprowadzone dla gazu modelowego, nazywanego gazem doskonałym (idealnym).
.") Spis treści 1 Stan gazowy 2 Gaz doskonały 21 Definicja mikroskopowa 22 Definicja makroskopowa (termodynamiczna) 3 Prawa gazowe 31 Prawo Boyle a-mariotte a 32 Prawo Gay-Lussaca 33 Prawo Charlesa 34 Prawo
Spis treści 1 Stan gazowy 2 Gaz doskonały 21 Definicja mikroskopowa 22 Definicja makroskopowa (termodynamiczna) 3 Prawa gazowe 31 Prawo Boyle a-mariotte a 32 Prawo Gay-Lussaca 33 Prawo Charlesa 34 Prawo
Czym się różni ciecz od ciała stałego?
 Szkła Czym się różni ciecz od ciała stałego? gęstość Czy szkło to ciecz czy ciało stałe? Szkło powstaje w procesie chłodzenia cieczy. Czy szkło to ciecz przechłodzona? kryształ szkło ciecz przechłodzona
Szkła Czym się różni ciecz od ciała stałego? gęstość Czy szkło to ciecz czy ciało stałe? Szkło powstaje w procesie chłodzenia cieczy. Czy szkło to ciecz przechłodzona? kryształ szkło ciecz przechłodzona
WIĄZANIA. Co sprawia, że ciała stałe istnieją i są stabilne? PRZYCIĄGANIE ODPYCHANIE
 WIĄZANIA Co sprawia, że ciała stałe istnieją i są stabilne? PRZYCIĄGANIE ODPYCHANIE Przyciąganie Wynika z elektrostatycznego oddziaływania między elektronami a dodatnimi jądrami atomowymi. Może to być
WIĄZANIA Co sprawia, że ciała stałe istnieją i są stabilne? PRZYCIĄGANIE ODPYCHANIE Przyciąganie Wynika z elektrostatycznego oddziaływania między elektronami a dodatnimi jądrami atomowymi. Może to być
Kacper Kulczycki. Dynamika molekularna atomów oddziałujących siłami van der Waalsa
 Kacper Kulczycki Dynamika molekularna atomów oddziałujących siłami van der Waalsa Warszawa 2007 Spis treści: Spis treści 1 Wstęp 2 Teoria 2 Algorytm 3 Symulacje 4 Wyniki 24 Wnioski 47 1 Wstęp Ćwiczenie
Kacper Kulczycki Dynamika molekularna atomów oddziałujących siłami van der Waalsa Warszawa 2007 Spis treści: Spis treści 1 Wstęp 2 Teoria 2 Algorytm 3 Symulacje 4 Wyniki 24 Wnioski 47 1 Wstęp Ćwiczenie
Modelowanie molekularne
 Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 4 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody chemii
Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 4 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody chemii
- parametry geometryczne badanego związku: współrzędne i typy atomów, ich masy, ładunki, prędkości początkowe itp. (w NAMD plik.
 Avogadro Tworzenie i manipulacja modelami związków chemicznych. W symulacjach dynamiki molekularnej kluczowych elementem jest przygotowanie układu do symulacji tzn. stworzyć pliki wejściowe zawierające
Avogadro Tworzenie i manipulacja modelami związków chemicznych. W symulacjach dynamiki molekularnej kluczowych elementem jest przygotowanie układu do symulacji tzn. stworzyć pliki wejściowe zawierające
Żwirki i Wigury 93, Warszawa TEL.: , FAX: , E- MAIL: Dr hab. Joanna T
 Żwirki i Wigury 93, 02-089 Warszawa TEL.: + 48 22 55 40 800, FAX: +48 22 55 40 801, E- MAIL: sekretariat@uw.edu.pl www.cent.uw.edu.pl Dr hab. Joanna Trylska, prof. UW tel. (22) 5540843 e- mail: joanna@cent.uw.edu.pl
Żwirki i Wigury 93, 02-089 Warszawa TEL.: + 48 22 55 40 800, FAX: +48 22 55 40 801, E- MAIL: sekretariat@uw.edu.pl www.cent.uw.edu.pl Dr hab. Joanna Trylska, prof. UW tel. (22) 5540843 e- mail: joanna@cent.uw.edu.pl
Problemy i rozwiązania
 Problemy i rozwiązania Znakomita większość układów, które badamy liczy sobie co najmniej mol cząsteczek >> 10 23 Typowy krok czasowy symulacji to 10-15 s natomiast zjawiska, które zachodzą wokół nas trwają
Problemy i rozwiązania Znakomita większość układów, które badamy liczy sobie co najmniej mol cząsteczek >> 10 23 Typowy krok czasowy symulacji to 10-15 s natomiast zjawiska, które zachodzą wokół nas trwają
Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }
![Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }](/thumbs/64/51033976.jpg "Zespół kanoniczny N,V, T. acc o n =min {1, exp [ U n U o ] }") Zespół kanoniczny Zespół kanoniczny N,V, T acc o n =min {1, exp [ U n U o ] } Zespół izobaryczno-izotermiczny Zespół izobaryczno-izotermiczny N P T acc o n =min {1, exp [ U n U o ] } acc o n =min {1, exp[
Zespół kanoniczny Zespół kanoniczny N,V, T acc o n =min {1, exp [ U n U o ] } Zespół izobaryczno-izotermiczny Zespół izobaryczno-izotermiczny N P T acc o n =min {1, exp [ U n U o ] } acc o n =min {1, exp[
Atomy wieloelektronowe
 Wiązania atomowe Atomy wieloelektronowe, obsadzanie stanów elektronowych, układ poziomów energii. Przykładowe konfiguracje elektronów, gazy szlachetne, litowce, chlorowce, układ okresowy pierwiastków,
Wiązania atomowe Atomy wieloelektronowe, obsadzanie stanów elektronowych, układ poziomów energii. Przykładowe konfiguracje elektronów, gazy szlachetne, litowce, chlorowce, układ okresowy pierwiastków,
Bioinformatyka wykład 8, 27.XI.2012
 Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Modelowanie molekularne
 Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 4 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody chemii
Modelowanie molekularne metodami chemii kwantowej Dr hab. Artur Michalak Zakład Chemii Teoretycznej Wydział Chemii UJ Wykład 4 http://www.chemia.uj.edu.pl/~michalak/mmod2007/ Podstawowe idee i metody chemii
Elementy teorii powierzchni metali
 prof. dr hab. Adam Kiejna Elementy teorii powierzchni metali Wykład 4 v.16 Wiązanie metaliczne Wiązanie metaliczne Zajmujemy się tylko metalami dlatego w zasadzie interesuje nas tylko wiązanie metaliczne.
prof. dr hab. Adam Kiejna Elementy teorii powierzchni metali Wykład 4 v.16 Wiązanie metaliczne Wiązanie metaliczne Zajmujemy się tylko metalami dlatego w zasadzie interesuje nas tylko wiązanie metaliczne.
Optymalizacja ciągła
 Optymalizacja ciągła 5. Metoda stochastycznego spadku wzdłuż gradientu Wojciech Kotłowski Instytut Informatyki PP http://www.cs.put.poznan.pl/wkotlowski/ 04.04.2019 1 / 20 Wprowadzenie Minimalizacja różniczkowalnej
Optymalizacja ciągła 5. Metoda stochastycznego spadku wzdłuż gradientu Wojciech Kotłowski Instytut Informatyki PP http://www.cs.put.poznan.pl/wkotlowski/ 04.04.2019 1 / 20 Wprowadzenie Minimalizacja różniczkowalnej
Analiza zderzeń dwóch ciał sprężystych
 Ćwiczenie M5 Analiza zderzeń dwóch ciał sprężystych M5.1. Cel ćwiczenia Celem ćwiczenia jest pomiar czasu zderzenia kul stalowych o różnych masach i prędkościach z nieruchomą, ciężką stalową przeszkodą.
Ćwiczenie M5 Analiza zderzeń dwóch ciał sprężystych M5.1. Cel ćwiczenia Celem ćwiczenia jest pomiar czasu zderzenia kul stalowych o różnych masach i prędkościach z nieruchomą, ciężką stalową przeszkodą.
Wiązania chemiczne. Związek klasyfikacji ciał krystalicznych z charakterem wiązań atomowych. 5 typów wiązań
 Wiązania chemiczne Związek klasyfikacji ciał krystalicznych z charakterem wiązań atomowych 5 typów wiązań wodorowe A - H - A, jonowe ( np. KCl ) molekularne (pomiędzy atomami gazów szlachetnych i małymi
Wiązania chemiczne Związek klasyfikacji ciał krystalicznych z charakterem wiązań atomowych 5 typów wiązań wodorowe A - H - A, jonowe ( np. KCl ) molekularne (pomiędzy atomami gazów szlachetnych i małymi
Systemy wbudowane. Uproszczone metody kosyntezy. Wykład 11: Metody kosyntezy systemów wbudowanych
 Systemy wbudowane Wykład 11: Metody kosyntezy systemów wbudowanych Uproszczone metody kosyntezy Założenia: Jeden procesor o znanych parametrach Znane parametry akceleratora sprzętowego Vulcan Początkowo
Systemy wbudowane Wykład 11: Metody kosyntezy systemów wbudowanych Uproszczone metody kosyntezy Założenia: Jeden procesor o znanych parametrach Znane parametry akceleratora sprzętowego Vulcan Początkowo
Wykładnicze grafy przypadkowe: teoria i przykłady zastosowań do analizy rzeczywistych sieci złożonych
 Gdańsk, Warsztaty pt. Układy Złożone (8 10 maja 2014) Agata Fronczak Zakład Fizyki Układów Złożonych Wydział Fizyki Politechniki Warszawskiej Wykładnicze grafy przypadkowe: teoria i przykłady zastosowań
Gdańsk, Warsztaty pt. Układy Złożone (8 10 maja 2014) Agata Fronczak Zakład Fizyki Układów Złożonych Wydział Fizyki Politechniki Warszawskiej Wykładnicze grafy przypadkowe: teoria i przykłady zastosowań
Temat Ocena dopuszczająca Ocena dostateczna Ocena dobra Ocena bardzo dobra Ocena celująca. Uczeń:
 Chemia - klasa I (część 2) Wymagania edukacyjne Temat Ocena dopuszczająca Ocena dostateczna Ocena dobra Ocena bardzo dobra Ocena celująca Dział 1. Chemia nieorganiczna Lekcja organizacyjna. Zapoznanie
Chemia - klasa I (część 2) Wymagania edukacyjne Temat Ocena dopuszczająca Ocena dostateczna Ocena dobra Ocena bardzo dobra Ocena celująca Dział 1. Chemia nieorganiczna Lekcja organizacyjna. Zapoznanie
Krystalografia. Analiza wyników rentgenowskiej analizy strukturalnej i sposób ich prezentacji
 Krystalografia Analiza wyników rentgenowskiej analizy strukturalnej i sposób ich prezentacji Opis geometrii Symetria: kryształu: grupa przestrzenna cząsteczki: grupa punktowa Parametry geometryczne współrzędne
Krystalografia Analiza wyników rentgenowskiej analizy strukturalnej i sposób ich prezentacji Opis geometrii Symetria: kryształu: grupa przestrzenna cząsteczki: grupa punktowa Parametry geometryczne współrzędne
Analiza zderzeń dwóch ciał sprężystych
 Ćwiczenie M5 Analiza zderzeń dwóch ciał sprężystych M5.1. Cel ćwiczenia Celem ćwiczenia jest pomiar czasu zderzenia kul stalowych o różnych masach i prędkościach z nieruchomą, ciężką stalową przeszkodą.
Ćwiczenie M5 Analiza zderzeń dwóch ciał sprężystych M5.1. Cel ćwiczenia Celem ćwiczenia jest pomiar czasu zderzenia kul stalowych o różnych masach i prędkościach z nieruchomą, ciężką stalową przeszkodą.
Metody rozwiązania równania Schrödingera
 Metody rozwiązania równania Schrödingera Równanie Schrödingera jako algebraiczne zagadnienie własne Rozwiązanie analityczne dla skończonej i nieskończonej studni potencjału Problem rozwiązania równania
Metody rozwiązania równania Schrödingera Równanie Schrödingera jako algebraiczne zagadnienie własne Rozwiązanie analityczne dla skończonej i nieskończonej studni potencjału Problem rozwiązania równania
Spis treści. Przedmowa... XI. Rozdział 1. Pomiar: jednostki miar... 1. Rozdział 2. Pomiar: liczby i obliczenia liczbowe... 16
 Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
WYMAGANIA EDUKACYJNE Z FIZYKI
 WYMAGANIA EDUKACYJNE Z FIZYKI KLASA I Budowa materii Wymagania na stopień dopuszczający obejmują treści niezbędne dla dalszego kształcenia oraz użyteczne w pozaszkolnej działalności ucznia. Uczeń: rozróżnia
WYMAGANIA EDUKACYJNE Z FIZYKI KLASA I Budowa materii Wymagania na stopień dopuszczający obejmują treści niezbędne dla dalszego kształcenia oraz użyteczne w pozaszkolnej działalności ucznia. Uczeń: rozróżnia
Bioinformatyka wykład 9
 Bioinformatyka wykład 9 14.XII.21 białkowa bioinformatyka strukturalna krzysztof_pawlowski@sggw.pl 211-1-17 1 Plan wykładu struktury białek dlaczego? struktury białek geometria i fizyka modyfikacje kowalencyjne
Bioinformatyka wykład 9 14.XII.21 białkowa bioinformatyka strukturalna krzysztof_pawlowski@sggw.pl 211-1-17 1 Plan wykładu struktury białek dlaczego? struktury białek geometria i fizyka modyfikacje kowalencyjne
Oddziaływanie leków z celami molekularnymi i projektowanie leków
 Oddziaływanie leków z celami molekularnymi i projektowanie leków Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania (biomodellab.eu) Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych
Oddziaływanie leków z celami molekularnymi i projektowanie leków Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania (biomodellab.eu) Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych
Oddziaływanie leków z celami molekularnymi i projektowanie leków
 Oddziaływanie leków z celami molekularnymi i projektowanie leków Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania (biomodellab.eu) Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych
Oddziaływanie leków z celami molekularnymi i projektowanie leków Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania (biomodellab.eu) Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych
Bioinformatyka wykład 10
 Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład II Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład II Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Równoległe symulacje Monte Carlo na współdzielonej sieci
 Równoległe symulacje Monte Carlo na współdzielonej sieci Szymon Murawski, Grzegorz Musiał, Grzegorz Pawłowski Wydział Fizyki, Uniwersytet im. Adama Mickiewicza 12 maja 2015 S. Murawski, G. Musiał, G. Pawłowski
Równoległe symulacje Monte Carlo na współdzielonej sieci Szymon Murawski, Grzegorz Musiał, Grzegorz Pawłowski Wydział Fizyki, Uniwersytet im. Adama Mickiewicza 12 maja 2015 S. Murawski, G. Musiał, G. Pawłowski
Metody numeryczne. materiały do wykładu dla studentów. 7. Całkowanie numeryczne
 Metody numeryczne materiały do wykładu dla studentów 7. Całkowanie numeryczne 7.1. Całkowanie numeryczne 7.2. Metoda trapezów 7.3. Metoda Simpsona 7.4. Metoda 3/8 Newtona 7.5. Ogólna postać wzorów kwadratur
Metody numeryczne materiały do wykładu dla studentów 7. Całkowanie numeryczne 7.1. Całkowanie numeryczne 7.2. Metoda trapezów 7.3. Metoda Simpsona 7.4. Metoda 3/8 Newtona 7.5. Ogólna postać wzorów kwadratur
Dokowanie molekularne. Andrzej Bąk Instytut Chemii UŚ chemoinformatyka wykład 1
 Dokowanie molekularne Andrzej Bąk Instytut Chemii UŚ chemoinformatyka wykład 1 Zarys Oddziaływanie ligand-receptor Modelowanie struktury receptora Reprezentacja makromolekuł Opis energii oddziaływań ligand-receptor
Dokowanie molekularne Andrzej Bąk Instytut Chemii UŚ chemoinformatyka wykład 1 Zarys Oddziaływanie ligand-receptor Modelowanie struktury receptora Reprezentacja makromolekuł Opis energii oddziaływań ligand-receptor
Modelowanie jako sposób opisu rzeczywistości. Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka
 Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
FIZYKA klasa 1 Liceum Ogólnokształcącego (4 letniego)
") 2019-09-01 FIZYKA klasa 1 Liceum Ogólnokształcącego (4 letniego) Treści z podstawy programowej przedmiotu POZIOM ROZSZERZONY (PR) SZKOŁY BENEDYKTA Podstawa programowa FIZYKA KLASA 1 LO (4-letnie po szkole
2019-09-01 FIZYKA klasa 1 Liceum Ogólnokształcącego (4 letniego) Treści z podstawy programowej przedmiotu POZIOM ROZSZERZONY (PR) SZKOŁY BENEDYKTA Podstawa programowa FIZYKA KLASA 1 LO (4-letnie po szkole
KI + Pb(NO 3 ) 2 PbI 2 + KNO 3. fermentacja alkoholowa
 2 PbI 2 + KNO 3. fermentacja alkoholowa") Kinetyka chemiczna KI + Pb(NO 3 ) 2 PbI 2 + KNO 3 fermentacja alkoholowa czynniki wpływaj ywające na szybkość reakcji chemicznych stęż ężenie reagentów w (lub ciśnienie gazów w jeżeli eli reakcja przebiega
Kinetyka chemiczna KI + Pb(NO 3 ) 2 PbI 2 + KNO 3 fermentacja alkoholowa czynniki wpływaj ywające na szybkość reakcji chemicznych stęż ężenie reagentów w (lub ciśnienie gazów w jeżeli eli reakcja przebiega
e E Z = P = 1 Z e E Kanoniczna suma stanów Prawdopodobieństwo wystąpienia mikrostanu U E = =Z 1 Wartość średnia energii
 Metoda Metropolisa Z = e E P = 1 Z e E Kanoniczna suma stanów Prawdopodobieństwo wystąpienia mikrostanu U E = P E =Z 1 E e E Wartość średnia energii Średnia wartość A = d r N A r N exp[ U r N ] d r N exp[
Metoda Metropolisa Z = e E P = 1 Z e E Kanoniczna suma stanów Prawdopodobieństwo wystąpienia mikrostanu U E = P E =Z 1 E e E Wartość średnia energii Średnia wartość A = d r N A r N exp[ U r N ] d r N exp[
Komputerowe wspomaganie projektowania leków
 Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Wstęp. Krystalografia geometryczna
 Wstęp Przedmiot badań krystalografii. Wprowadzenie do opisu struktury kryształów. Definicja sieci Bravais go i bazy atomowej, komórki prymitywnej i elementarnej. Podstawowe typy komórek elementarnych.
Wstęp Przedmiot badań krystalografii. Wprowadzenie do opisu struktury kryształów. Definicja sieci Bravais go i bazy atomowej, komórki prymitywnej i elementarnej. Podstawowe typy komórek elementarnych.
Stany skupienia materii
 Stany skupienia materii Ciała stałe - ustalony kształt i objętość - uporządkowanie dalekiego zasięgu - oddziaływania harmoniczne Ciecze -słabo ściśliwe - uporządkowanie bliskiego zasięgu -tworzą powierzchnię
Stany skupienia materii Ciała stałe - ustalony kształt i objętość - uporządkowanie dalekiego zasięgu - oddziaływania harmoniczne Ciecze -słabo ściśliwe - uporządkowanie bliskiego zasięgu -tworzą powierzchnię
Ćwiczenie 14. Maria Bełtowska-Brzezinska KINETYKA REAKCJI ENZYMATYCZNYCH
 Ćwiczenie 14 aria Bełtowska-Brzezinska KINETYKA REAKCJI ENZYATYCZNYCH Zagadnienia: Podstawowe pojęcia kinetyki chemicznej (szybkość reakcji, reakcje elementarne, rząd reakcji). Równania kinetyczne prostych
Ćwiczenie 14 aria Bełtowska-Brzezinska KINETYKA REAKCJI ENZYATYCZNYCH Zagadnienia: Podstawowe pojęcia kinetyki chemicznej (szybkość reakcji, reakcje elementarne, rząd reakcji). Równania kinetyczne prostych
Rozszerzony konspekt preskryptu do przedmiotu Podstawy Robotyki
 Projekt współfinansowany przez Unię Europejską w ramach Europejskiego Funduszu Społecznego Rozszerzony konspekt preskryptu do przedmiotu Podstawy Robotyki dr inż. Marek Wojtyra Instytut Techniki Lotniczej
Projekt współfinansowany przez Unię Europejską w ramach Europejskiego Funduszu Społecznego Rozszerzony konspekt preskryptu do przedmiotu Podstawy Robotyki dr inż. Marek Wojtyra Instytut Techniki Lotniczej
DYNAMIKA dr Mikolaj Szopa
 dr Mikolaj Szopa 17.10.2015 Do 1600 r. uważano, że naturalną cechą materii jest pozostawanie w stanie spoczynku. Dopiero Galileusz zauważył, że to stan ruchu nie zmienia się, dopóki nie ingerujemy I prawo
dr Mikolaj Szopa 17.10.2015 Do 1600 r. uważano, że naturalną cechą materii jest pozostawanie w stanie spoczynku. Dopiero Galileusz zauważył, że to stan ruchu nie zmienia się, dopóki nie ingerujemy I prawo
Fizyka statystyczna Fenomenologia przejść fazowych. P. F. Góra
 Fizyka statystyczna Fenomenologia przejść fazowych P. F. Góra http://th-www.if.uj.edu.pl/zfs/gora/ 2015 Przejście fazowe transformacja układu termodynamicznego z jednej fazy (stanu materii) do innej, dokonywane
Fizyka statystyczna Fenomenologia przejść fazowych P. F. Góra http://th-www.if.uj.edu.pl/zfs/gora/ 2015 Przejście fazowe transformacja układu termodynamicznego z jednej fazy (stanu materii) do innej, dokonywane
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA. Marta Szachniuk
 Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Obliczenia polowe silnika przełączalnego reluktancyjnego (SRM) w celu jego optymalizacji
 w celu jego optymalizacji") Akademia Górniczo Hutnicza im. Stanisława Staszica w Krakowie Wydział Elektrotechniki, Automatyki, Informatyki i Elektroniki Studenckie Koło Naukowe Maszyn Elektrycznych Magnesik Obliczenia polowe silnika
Akademia Górniczo Hutnicza im. Stanisława Staszica w Krakowie Wydział Elektrotechniki, Automatyki, Informatyki i Elektroniki Studenckie Koło Naukowe Maszyn Elektrycznych Magnesik Obliczenia polowe silnika
Równa Równ n a i n e i ru r ch u u ch u po tor t ze (równanie drogi) Prędkoś ędkoś w ru r ch u u ch pros pr t os ol t i ol n i io i wym
 Prędkoś ędkoś w ru r ch u u ch pros pr t os ol t i ol n i io i wym") Mechanika ogólna Wykład nr 14 Elementy kinematyki i dynamiki 1 Kinematyka Dział mechaniki zajmujący się matematycznym opisem układów mechanicznych oraz badaniem geometrycznych właściwości ich ruchu, bez
Mechanika ogólna Wykład nr 14 Elementy kinematyki i dynamiki 1 Kinematyka Dział mechaniki zajmujący się matematycznym opisem układów mechanicznych oraz badaniem geometrycznych właściwości ich ruchu, bez
Modelowanie biomechaniczne. Dr inż. Sylwia Sobieszczyk Politechnika Gdańska Wydział Mechaniczny KMiWM 2005/2006
 Modelowanie biomechaniczne Dr inż. Sylwia Sobieszczyk Politechnika Gdańska Wydział Mechaniczny KMiWM 2005/2006 Zakres: Definicja modelowania Modele kinematyczne ruch postępowy, obrotowy, przemieszczenie,
Modelowanie biomechaniczne Dr inż. Sylwia Sobieszczyk Politechnika Gdańska Wydział Mechaniczny KMiWM 2005/2006 Zakres: Definicja modelowania Modele kinematyczne ruch postępowy, obrotowy, przemieszczenie,
Metody numeryczne I Równania nieliniowe
 Metody numeryczne I Równania nieliniowe Janusz Szwabiński szwabin@ift.uni.wroc.pl Metody numeryczne I (C) 2004 Janusz Szwabiński p.1/66 Równania nieliniowe 1. Równania nieliniowe z pojedynczym pierwiastkiem
Metody numeryczne I Równania nieliniowe Janusz Szwabiński szwabin@ift.uni.wroc.pl Metody numeryczne I (C) 2004 Janusz Szwabiński p.1/66 Równania nieliniowe 1. Równania nieliniowe z pojedynczym pierwiastkiem
ZAKŁAD CHEMII TEORETYCZNEJ
 ZAKŁAD CHEMII TEORETYCZNEJ Prof. Krzysztof Nieszporek Kierownik Zakładu Prof. Krzysztof Woliński Prof. Paweł Szabelski Dr Mariusz Barczak Dr Damian Nieckarz Dr Przemysław Podkościelny prof. Krzysztof Woliński
ZAKŁAD CHEMII TEORETYCZNEJ Prof. Krzysztof Nieszporek Kierownik Zakładu Prof. Krzysztof Woliński Prof. Paweł Szabelski Dr Mariusz Barczak Dr Damian Nieckarz Dr Przemysław Podkościelny prof. Krzysztof Woliński
Wymiana ciepła. Ładunek jest skwantowany. q=n. e gdzie n = ±1, ±2, ±3 [1C = 6, e] e=1, C
![Wymiana ciepła. Ładunek jest skwantowany. q=n. e gdzie n = ±1, ±2, ±3 [1C = 6, e] e=1, C](/thumbs/63/50333873.jpg "Wymiana ciepła. Ładunek jest skwantowany. q=n. e gdzie n = ±1, ±2, ±3 [1C = 6, e] e=1, C") Wymiana ciepła Ładunek jest skwantowany ładunek elementarny ładunek pojedynczego elektronu (e). Każdy ładunek q (dodatni lub ujemny) jest całkowitą wielokrotnością jego bezwzględnej wartości. q=n. e gdzie
Wymiana ciepła Ładunek jest skwantowany ładunek elementarny ładunek pojedynczego elektronu (e). Każdy ładunek q (dodatni lub ujemny) jest całkowitą wielokrotnością jego bezwzględnej wartości. q=n. e gdzie
Mgr inż. Wojciech Chajec Pracownia Kompozytów, CNT Mgr inż. Adam Dziubiński Pracownia Aerodynamiki Numerycznej i Mechaniki Lotu, CNT SMIL
 Mgr inż. Wojciech Chajec Pracownia Kompozytów, CNT Mgr inż. Adam Dziubiński Pracownia Aerodynamiki Numerycznej i Mechaniki Lotu, CNT SMIL We wstępnej analizie przyjęto następujące założenia: Dwuwymiarowość
Mgr inż. Wojciech Chajec Pracownia Kompozytów, CNT Mgr inż. Adam Dziubiński Pracownia Aerodynamiki Numerycznej i Mechaniki Lotu, CNT SMIL We wstępnej analizie przyjęto następujące założenia: Dwuwymiarowość
Metody Optymalizacji: Przeszukiwanie z listą tabu
 Metody Optymalizacji: Przeszukiwanie z listą tabu Wojciech Kotłowski Instytut Informatyki Politechniki Poznańskiej email: imię.nazwisko@cs.put.poznan.pl pok. 2 (CW) tel. (61)665-2936 konsultacje: wtorek
Metody Optymalizacji: Przeszukiwanie z listą tabu Wojciech Kotłowski Instytut Informatyki Politechniki Poznańskiej email: imię.nazwisko@cs.put.poznan.pl pok. 2 (CW) tel. (61)665-2936 konsultacje: wtorek
Korzystanie z podstawowych rozkładów prawdopodobieństwa (tablice i arkusze kalkulacyjne)
") Korzystanie z podstawowych rozkładów prawdopodobieństwa (tablice i arkusze kalkulacyjne) Przygotował: Dr inż. Wojciech Artichowicz Katedra Hydrotechniki PG Zima 2014/15 1 TABLICE ROZKŁADÓW... 3 ROZKŁAD
Korzystanie z podstawowych rozkładów prawdopodobieństwa (tablice i arkusze kalkulacyjne) Przygotował: Dr inż. Wojciech Artichowicz Katedra Hydrotechniki PG Zima 2014/15 1 TABLICE ROZKŁADÓW... 3 ROZKŁAD
Diagramy fazowe graficzna reprezentacja warunków równowagi
 Diagramy fazowe graficzna reprezentacja warunków równowagi Faza jednorodna część układu, oddzielona od innych części granicami faz, na których zachodzi skokowa zmiana pewnych własności fizycznych. B 0
Diagramy fazowe graficzna reprezentacja warunków równowagi Faza jednorodna część układu, oddzielona od innych części granicami faz, na których zachodzi skokowa zmiana pewnych własności fizycznych. B 0
KI + Pb(NO 3 ) 2 PbI 2 + KNO 3. fermentacja alkoholowa
 2 PbI 2 + KNO 3. fermentacja alkoholowa") Kinetyka chemiczna KI + Pb(NO 3 ) 2 PbI 2 + KNO 3 fermentacja alkoholowa czynniki wpływaj ywające na szybkość reakcji chemicznych stęż ężenie reagentów w (lub ciśnienie gazów w jeżeli eli reakcja przebiega
Kinetyka chemiczna KI + Pb(NO 3 ) 2 PbI 2 + KNO 3 fermentacja alkoholowa czynniki wpływaj ywające na szybkość reakcji chemicznych stęż ężenie reagentów w (lub ciśnienie gazów w jeżeli eli reakcja przebiega
Spis treści PRZEDMOWA DO WYDANIA PIERWSZEGO...
 Spis treści PRZEDMOWA DO WYDANIA PIERWSZEGO....................... XI 1. WPROWADZENIE DO GEODEZJI WYŻSZEJ..................... 1 Z historii geodezji........................................ 1 1.1. Kształt
Spis treści PRZEDMOWA DO WYDANIA PIERWSZEGO....................... XI 1. WPROWADZENIE DO GEODEZJI WYŻSZEJ..................... 1 Z historii geodezji........................................ 1 1.1. Kształt
Ciśnienie i temperatura model mikroskopowy
 Ciśnienie i temperatura model mikroskopowy Mikroskopowy model ciśnienia gazu wzór na ciśnienie gazu Mikroskopowa interpretacja temperatury Średnia energia cząsteczki gazu zasada ekwipartycji energii Czy
Ciśnienie i temperatura model mikroskopowy Mikroskopowy model ciśnienia gazu wzór na ciśnienie gazu Mikroskopowa interpretacja temperatury Średnia energia cząsteczki gazu zasada ekwipartycji energii Czy
Symulacje komputerowe
 Fizyka w modelowaniu i symulacjach komputerowych Jacek Matulewski (e-mail: jacek@fizyka.umk.pl) http://www.fizyka.umk.pl/~jacek/dydaktyka/modsym/ Symulacje komputerowe Dynamika bryły sztywnej Wersja: 8
Fizyka w modelowaniu i symulacjach komputerowych Jacek Matulewski (e-mail: jacek@fizyka.umk.pl) http://www.fizyka.umk.pl/~jacek/dydaktyka/modsym/ Symulacje komputerowe Dynamika bryły sztywnej Wersja: 8
Katarzyna Jesionek Zastosowanie symulacji dynamiki cieczy oraz ośrodków sprężystych w symulatorach operacji chirurgicznych.
 Katarzyna Jesionek Zastosowanie symulacji dynamiki cieczy oraz ośrodków sprężystych w symulatorach operacji chirurgicznych. Jedną z metod symulacji dynamiki cieczy jest zastosowanie metody siatkowej Boltzmanna.
Katarzyna Jesionek Zastosowanie symulacji dynamiki cieczy oraz ośrodków sprężystych w symulatorach operacji chirurgicznych. Jedną z metod symulacji dynamiki cieczy jest zastosowanie metody siatkowej Boltzmanna.
Wyznaczanie modułu Younga metodą strzałki ugięcia
 Ćwiczenie M12 Wyznaczanie modułu Younga metodą strzałki ugięcia M12.1. Cel ćwiczenia Celem ćwiczenia jest wyznaczenie wartości modułu Younga różnych materiałów poprzez badanie strzałki ugięcia wykonanych
Ćwiczenie M12 Wyznaczanie modułu Younga metodą strzałki ugięcia M12.1. Cel ćwiczenia Celem ćwiczenia jest wyznaczenie wartości modułu Younga różnych materiałów poprzez badanie strzałki ugięcia wykonanych
Kryteria samorzutności procesów fizyko-chemicznych
 Kryteria samorzutności procesów fizyko-chemicznych 2.5.1. Samorzutność i równowaga 2.5.2. Sens i pojęcie entalpii swobodnej 2.5.3. Sens i pojęcie energii swobodnej 2.5.4. Obliczanie zmian entalpii oraz
Kryteria samorzutności procesów fizyko-chemicznych 2.5.1. Samorzutność i równowaga 2.5.2. Sens i pojęcie entalpii swobodnej 2.5.3. Sens i pojęcie energii swobodnej 2.5.4. Obliczanie zmian entalpii oraz
S. Baran - Podstawy fizyki materii skondensowanej Wiązania chemiczne w ciałach stałych. Wiązania chemiczne w ciałach stałych
 Wiązania chemiczne w ciałach stałych Wiązania chemiczne w ciałach stałych typ kowalencyjne jonowe metaliczne Van der Waalsa wodorowe siła* silne silne silne pochodzenie uwspólnienie e- (pary e-) przez
Wiązania chemiczne w ciałach stałych Wiązania chemiczne w ciałach stałych typ kowalencyjne jonowe metaliczne Van der Waalsa wodorowe siła* silne silne silne pochodzenie uwspólnienie e- (pary e-) przez
Teoria kinetyczna gazów
 Teoria kinetyczna gazów Mikroskopowy model ciśnienia gazu wzór na ciśnienie gazu Mikroskopowa interpretacja temperatury Średnia energia cząsteczki gazu zasada ekwipartycji energii Czy ciepło właściwe przy
Teoria kinetyczna gazów Mikroskopowy model ciśnienia gazu wzór na ciśnienie gazu Mikroskopowa interpretacja temperatury Średnia energia cząsteczki gazu zasada ekwipartycji energii Czy ciepło właściwe przy
Zasady oceniania karta pracy
 Zadanie 1.1. 5) stosuje zasadę zachowania energii oraz zasadę zachowania pędu do opisu zderzeń sprężystych i niesprężystych. Zderzenie, podczas którego wózki łączą się ze sobą, jest zderzeniem niesprężystym.
Zadanie 1.1. 5) stosuje zasadę zachowania energii oraz zasadę zachowania pędu do opisu zderzeń sprężystych i niesprężystych. Zderzenie, podczas którego wózki łączą się ze sobą, jest zderzeniem niesprężystym.
Wykład 1. Anna Ptaszek. 5 października Katedra Inżynierii i Aparatury Przemysłu Spożywczego. Chemia fizyczna - wykład 1. Anna Ptaszek 1 / 36
 Wykład 1 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 5 października 2015 1 / 36 Podstawowe pojęcia Układ termodynamiczny To zbiór niezależnych elementów, które oddziałują ze sobą tworząc integralną
Wykład 1 Katedra Inżynierii i Aparatury Przemysłu Spożywczego 5 października 2015 1 / 36 Podstawowe pojęcia Układ termodynamiczny To zbiór niezależnych elementów, które oddziałują ze sobą tworząc integralną
ALGORYTMY GENETYCZNE (wykład + ćwiczenia)
") ALGORYTMY GENETYCZNE (wykład + ćwiczenia) Prof. dr hab. Krzysztof Dems Treści programowe: 1. Metody rozwiązywania problemów matematycznych i informatycznych.. Elementarny algorytm genetyczny: definicja
ALGORYTMY GENETYCZNE (wykład + ćwiczenia) Prof. dr hab. Krzysztof Dems Treści programowe: 1. Metody rozwiązywania problemów matematycznych i informatycznych.. Elementarny algorytm genetyczny: definicja
Optymalizacja. Przeszukiwanie lokalne
 dr hab. inż. Instytut Informatyki Politechnika Poznańska www.cs.put.poznan.pl/mkomosinski, Maciej Hapke Idea sąsiedztwa Definicja sąsiedztwa x S zbiór N(x) S rozwiązań, które leżą blisko rozwiązania x
dr hab. inż. Instytut Informatyki Politechnika Poznańska www.cs.put.poznan.pl/mkomosinski, Maciej Hapke Idea sąsiedztwa Definicja sąsiedztwa x S zbiór N(x) S rozwiązań, które leżą blisko rozwiązania x
Modelowanie białek ab initio / de novo
 Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Elementy wspo łczesnej teorii inwersji
 Elementy wspo łczesnej teorii inwersji Metoda optymalizacyjna (2) W. Debski, 8.01.2015 Liniowy problem odwrotny m est (λ) = m apr + (G T G + λi) 1 G T ( dobs G m apr) +δ d est d o = + λ I ( G T G + λi
Elementy wspo łczesnej teorii inwersji Metoda optymalizacyjna (2) W. Debski, 8.01.2015 Liniowy problem odwrotny m est (λ) = m apr + (G T G + λi) 1 G T ( dobs G m apr) +δ d est d o = + λ I ( G T G + λi
Ćwiczenie 4: Ciepło właściwe monokryształu fcc argonu
 Ćwiczenie 4: Ciepło właściwe monokryształu fcc argonu Tym razem zajmiemy się już problemem bardziej złożonym. Celem ćwiczenia jest wyznaczenie dla monokryształu fcc argonu ciepła właściwego c, tj. ciepła
Ćwiczenie 4: Ciepło właściwe monokryształu fcc argonu Tym razem zajmiemy się już problemem bardziej złożonym. Celem ćwiczenia jest wyznaczenie dla monokryształu fcc argonu ciepła właściwego c, tj. ciepła
Różne typy wiązań mają ta sama przyczynę: energia powstającej stabilnej cząsteczki jest mniejsza niż sumaryczna energia tworzących ją, oddalonych
 Wiązania atomowe Atomy wieloelektronowe, obsadzanie stanów elektronowych, układ poziomów energii. Przykładowe konfiguracje elektronów, gazy szlachetne, litowce, chlorowce, układ okresowy pierwiastków,
Wiązania atomowe Atomy wieloelektronowe, obsadzanie stanów elektronowych, układ poziomów energii. Przykładowe konfiguracje elektronów, gazy szlachetne, litowce, chlorowce, układ okresowy pierwiastków,
Podstawy fizyki wykład 8
 Podstawy fizyki wykład 8 Dr Piotr Sitarek Instytut Fizyki, Politechnika Wrocławska Ładunek elektryczny Grecy ok. 600 r p.n.e. odkryli, że bursztyn potarty o wełnę przyciąga inne (drobne) przedmioty. słowo
Podstawy fizyki wykład 8 Dr Piotr Sitarek Instytut Fizyki, Politechnika Wrocławska Ładunek elektryczny Grecy ok. 600 r p.n.e. odkryli, że bursztyn potarty o wełnę przyciąga inne (drobne) przedmioty. słowo
Techniki optymalizacji
 Techniki optymalizacji Symulowane wyżarzanie Maciej Hapke maciej.hapke at put.poznan.pl Wyżarzanie wzrost temperatury gorącej kąpieli do takiej wartości, w której ciało stałe topnieje powolne zmniejszanie
Techniki optymalizacji Symulowane wyżarzanie Maciej Hapke maciej.hapke at put.poznan.pl Wyżarzanie wzrost temperatury gorącej kąpieli do takiej wartości, w której ciało stałe topnieje powolne zmniejszanie
Techniki Optymalizacji: Stochastyczny spadek wzdłuż gradientu I
 Techniki Optymalizacji: Stochastyczny spadek wzdłuż gradientu I Wojciech Kotłowski Instytut Informatyki Politechniki Poznańskiej email: imię.nazwisko@cs.put.poznan.pl pok. 2 (CW) tel. (61)665-2936 konsultacje:
Techniki Optymalizacji: Stochastyczny spadek wzdłuż gradientu I Wojciech Kotłowski Instytut Informatyki Politechniki Poznańskiej email: imię.nazwisko@cs.put.poznan.pl pok. 2 (CW) tel. (61)665-2936 konsultacje:
Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci
 Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu 1. Sieci jako modele interakcji
Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu 1. Sieci jako modele interakcji
8. PODSTAWY ANALIZY NIELINIOWEJ
 8. PODSTAWY ANALIZY NIELINIOWEJ 1 8. 8. PODSTAWY ANALIZY NIELINIOWEJ 8.1. Wprowadzenie Zadania nieliniowe mają swoje zastosowanie na przykład w rozwiązywaniu cięgien. Przyczyny nieliniowości: 1) geometryczne:
8. PODSTAWY ANALIZY NIELINIOWEJ 1 8. 8. PODSTAWY ANALIZY NIELINIOWEJ 8.1. Wprowadzenie Zadania nieliniowe mają swoje zastosowanie na przykład w rozwiązywaniu cięgien. Przyczyny nieliniowości: 1) geometryczne: