Żele agarozowe i poliakylamidowe: jak je przechować na później?
|
|
|
- Radosław Nowakowski
- 7 lat temu
- Przeglądów:
Transkrypt
1 Żele agarozowe i poliakylamidowe: jak je przechować na później? Wielu z nas korzysta w rutynowej pracy z klasycznych żeli PA i agarozowych. Jeżeli często puszczamy elektroforezę, warto się zastanowić, czy każdorazowe przygotowywanie żelu przed elektroforezą to nie strata czasu. Żele można bowiem do tygodnia, a nawet dłużej, składować w lodówce bez straty ich parametrów! Oto bardzo łatwy sposób na przechowanie żelu: 1. Przygotuj woreczki strunowe i szczelny pojemnik plastikowy na żywność. 2. Upewnij się, że masz nadmiar buforu do elektroforezy na podorędziu. 3. Potrzebna będzie także folia aluminiowa lub stretch spożywczy. Żel nie może wyschnąć, a w przypadku dodatku bromku etydyny nie może on też być eksponowany na światło. Warto także pamiętać, że bromkofobom nie spodoba się obecność żelu w ogólnodostępnej lodówce. Dlatego pojemniki, w których przechowywać będziemy nasączone bromkiem agarozy powinny być szczelne i dobrze myte. Układamy żel na folii aluminiowej, w kuwecie lub w plastikowym pojemniku. Następnie skrapiamy go buforem na którym został zrobiony i zawijamy w folię. Do kuwety/pojemnika wlewamy cieniutką warstewkę tegoż buforu a następnie zamykamy w takim pojemniku żel. Nie moczmy żelu w wodzie, powinien on pozostać wilgotny i przebywać w mokrej komorze, ale zbyt duża ilość płynu spowoduje wypłukanie z niego bromku.

2 Komorę z żelem umieszczamy w lodówce, podpisujemy datą produkcji i nie zapominamy zużyć go w ciągu najbliższego tygodnia. Przed wykorzystaniem żelu sprawdzamy, czy jego brzegi nie są zmarszczone i przesuszone, bo taki żel nie nada się do doświadczeń. Uwaga: żeli poliakrylamidowych nienatywnych, z domieszką formamidu, formaldehydu, SDS, czy też enzymów (DNAza) i sond, a także żeli gradientowych nie należy przechowywać, gdyż zmogą zmienić one swoje parametry! Ta metoda ma zastosowanie wyłącznie do żeli natywnych! Projektowanie reakcji multiplexpcr Dzięki nowoczesnym, wydajnym zestawom do PCR, amplifikacja DNA nie jest już takim wyzwaniem, jak w latach 80-tych i 90-tych. Wraz z poprawą metodyki, pojawiły się ciekawe pomysły na unowocześnienie reakcji PCR i zwiększenie jej wydajności i rzetelności. Przykładem modyfikacji PCR, która wprowadza cały wachlarz nowych możliwości, jest multiplex-pcr. Czym jest multiplex? Przez multiplex-pcr rozumiemy każdą modyfikację PCR, która polega na zmieszaniu co najmniej trzech primerów tak, by amplifikacji naraz ulegały dwie różne (lub więcej) matryce. Multipleksowanie można przetłumaczyć na polski jako scalanie kilku reakcji. Poniżej przedstawiliśmy poglądową rycinę z dwoma podstawowymi przykładami multipleksów.
3 Zalety i ograniczenia metod typu multiplex-pcr Wiele różnych dziedzin biologii molekularnej doceniło multiplex: prowadzenie równocześnie kilku reakcji skraca czas i zmniejsza koszt wykonania oznaczeń (zamiast kilku reakcji PCR wykonujemy jedną), pozwala też na oszczędność matrycy, co może być szczególnie ważne, gdy dysponujemy minimalnymi ilościami unikatowego materiału (tkanka nowotworowa, mikroślady); kolejną zaletą reakcji w multipleksie jest możliwość zastosowania wewnętrznej kontroli amplifikacji w reakcjach, w których stwierdzamy obecność lub brak produktu (dodatni/ujemny). Wówczas poza właściwą reakcją wykonujemy jednocześnie PCR dla jakiegoś fragmentu kontrolnego DNA, który niezależnie od obecności lub braku badanego amplikonu, zawsze będzie dawał produkt. Taka kontrola pozwoli nam odróżnić wynik fałszywie ujemny (nie dodana matryca, nieudana reakcja) od prawdziwie ujemnego; podejście multiplex-pcr jest pomocne podczas wykrywania kilku wariantów tego samego genu jednocześnie; dzięki scalaniu kilku reakcji możliwe stały się rzetelne pomiary metodą RealTime PCR, w których z zastosowaniem sond molekularnych możemy monitorować ilościowo naraz kilka transkryptów lub wariantów allelicznych. Ograniczenia podejścia multiplex są nieliczne, należą do nich: trudność wstępnej optymalizacji reakcji; problemy w doborze odpowiednich proporcji starterów reakcji w niektórych przypadkach w ogóle nie ma możliwości dobrania kilku par starterów tak, by nie tworzyły między sobą par typu primer-dimer; w przypadku amplifikacji z cdna genów o bardzo różnym poziomie ekspresji może dojść do faworyzowania amplifikacji tylko genu o wyższym poziomie ekspresji.
4 Optymalizacja reakcji w multipleksie Są 2 podejścia do tworzenia tego typu reakcji: pierwsza polega na wstępnym doborze warunków reakcji dla odrębnych amplikonów osobno, a następnie interpolowaniu stężeń odczynników i protokołu amplifikacji tak, by pogodzić ze sobą obie pary starterów. Drugie podejście polega na pójściu na żywioł - nie przejmujemy się wydajnością reakcji rozumianych osobno, od razu mieszamy startery i patrzymy, co z tego wyniknie. Podejście pierwsze jest bardziej racjonalne, jednak więcej kosztuje i jest bardziej czasochłonne. Nie zawsze też opracowanie dobrze działającej reakcji singleplex (pojedynczej) ma przełożenie na kinetykę reakcji multiplex. Drugie podejście powinno być stosowane przez osoby, które mają już doświadczenie w optymalizacji PCR i znają doskonale chemię, na której pracują. Takie osoby mogą sobie pozwolić na ryzykowne próbowanie na chybił trafił warunków reakcji zgodnie z poprzednimi doświadczeniami własnymi. Podstawowe zasady multipleksowania reakcji
5 Spróbujmy zatem wypracować jakiś schemat optymalizacji zgodnie ze strategią pierwszą. 1. Wybór amplikonów -jeżeli mamy luksus wyboru wśród kilku par starterów, wybierzmy dwie dla każdego amplikonu tak, by teoretycznie wyliczona temperatura topnienia była jak najbardziej zbliżona. Idealna różnica wielkości obu amplikonów, powinna wynosić nt, chociaż nie trzeba się trzymać bardzo rygorystycznie tego kryterium. Taka różnica wielkości amplikonów pozwala na łatwy rozdział na żelu, nie doprowadzając do faworyzowania krótszego produktu reakcji i jego preferencyjnej amplifikacji 2. Po otrzymaniu starterów przeprowadzamy optymalizację osobno dla każdej z reakcji PCR, zgodnie z naszym poradnikiem. Trudność polega na dobraniu wspólnych lub zbliżonych warunków termicznych i poziomu magnezu dla obu naszych reakcji. Częściowo te parametry są od siebie zależne i zwiększona ilość magnezu pozwala na podniesienie niewielkie temperatury bez straty wydajności (lecz nie jest to regułą.) 3. Scalamy obie reakcje i po rozdziale żelowym analizujemy, czy otrzymaliśmy wyraźne i specyficzne produkty, czy ich stężenie jest zbliżone, czy występują produkty niespecyficzne. 4. Zwykle za pierwszym razem nie udaje się dobrać idealnych warunków. Jeżeli widzimy tylko jeden z produktów, lub jest on w dużej przewadze nad drugim, zmniejszamy o połowę stężenie starterów dla przeważającego produktu. Alternatywnie, zwiększamy o 50% stężenie starterów dla słabszego. 5. Jeżeli otrzymujemy oba produkty, lecz poza nimi występuje produkt niespecyficzny, wykonujemy zwykły gradient termiczny i/lub magnezu dla multipleksu (zgodnie z Poradnikiem) 6. Jeżeli nie otrzymujemy żadnego produktu lub bardzo słabe, pierwszym krokiem jest obniżenie temperatury amplifikacji reakcji (gradient termiczny). Gdy nie daje to efektu, wracamy do temperatury wyjściowej i dodajemy magnezu (o 25-50% w pojedynczej reakcji lub stopniowo w gradiencie). Jeżeli i ten krok nie rozwiązuje naszych problemów, dodajemy 50%-100% dntp do wyjściowej reakcji. Możemy także zadziałać jednocześnie zwiększając magnez i dntp, a nawet wydłużając etap annealingu maksymalnie do minuty.
 Żel agaroza kopia Jeżeli szczęśliwie udało nam się otrzymać")
6 7. Może okazać się, że nie otrzymamy w ogóle produktu. Wówczas rezygnujemy z tej pary starterów lub próbujemy innego zestawu do reakcji PCR (czasami to wystarczy, żeby ruszyć oporną reakcję) Żel agaroza kopia Jeżeli szczęśliwie udało nam się otrzymać produkty, rozważmy dopracowanie reakcji poprzez manipulację stężeniami starterów, magnezu i stężenia matrycy. Multiplex-PCR powinien być niezawodny i powtarzalny. Dobrze działająca reakcja wynagrodzi nam trudy i czas spędzony podczas optymalizacji! Jak przygotować wodę wolną od RNAz (H2O-DEPC) protokół Woda wolna od RNAz to podstawa wszelkich manipulacji z RNA, podatnym na działanie wszędobylskich i bardzo stabilnych rybonukleaz. Wodę taką, najlepszej jakości, można zakupić u dystrybutorów większych firm sprzedających odczynniki dla biologii molekularnej. Gotowa woda ultraczysta jest niezbędna w sytuacji, gdy jakość każdej próbki jest niezwykle ważna (np. w diagnostyce medycznej), jednak w placówkach naukowych i przy zastosowaniach niekomercyjnych warto pomyśleć o
7 własnoręcznym przygotowaniu wody do izolacji RNA. Jest to tak zwana woda DEPC, nazywana tak od odczynnika, który jest używany do degradacji RNAz. Dlaczego akurat DEPC? Dietylopirowęglan to silny środek alkilujący, któremu nie straszne są żadne białka, także bardzo stabilne i powszechne w środowisku RNAzy. Zaletą DEPC jest za to jego termolabilność i rozpad do neutralnych związków chemicznych pod wpływem autoklawowania. DEPC rozpada się na CO2, H2O oraz etanol, który w niewielkim stężeniu nie zagraża RNA. W temperaturze pokojowej przy dłuższym przechowywaniu lub przy zbyt wysokim stężeniu dietylopirowęglan ulega przekształceniu do niższych estrów, dlatego jeżeli trąci on owocowym, estrowym zapachem, nie nadaje się już do neutralizacji nukleaz. Przygotowanie wody wolnej od rybonukleaz: 1. Do 1 litra wody destylowanej (najlepiej zaś poddanej odwrotnej osmozie) dodaj 1ml dietylopirowęglanu (DEPC), by otrzymać roztwór 0.1% (v/v). 2. Dobrze wymieszaj, pozwól roztworowi odstać się w temperaturze pokojowej ok minut 3. Autoklawuj w standardowych warunkach (121, 15 minut, 1 Bar) 4. Voila! Woda wolna od RNAz gotowa. Przed użyciem pozwól jej ostygnąć do temperatury pokojowej. Dobrze jest przepipetować część świeżo przygotowanej wody na małe alikwoty np. do zakręcanych próbówek i zamrozić. Uwagi: DEPC jest silnie toksyczny i kancerogenny (odczynnik alkilujący!), należy obchodzić się z nim ostrożnie. Dietylopirowęglan jest niestabilny rozcieńczony w wodzie i buforach, dlatego roztwór do autoklawowania przygotowuje się na świeżo, a przechowuje się już po sterylizacji jako czystą wodę DEPC. Marzena Pieronkiewicz

8 Żel agaroza kopia Labowe know-how bakterie kompetentne i transformacja plazmidowym DNA Czy zdarzyło Ci się niepowodzenie w tak podstawowej metodzie laboratoryjnej, jaką jest transformacja bakterii obcym DNA? Ta względnie mało skomplikowana metoda potrafi czasem być na tyle kapryśna, by spędzić nam sen z powiek. Wystarczy użyć starych lub nie do końca kompetentnych bakterii lub zastosować zbyt wysokie ilości DNA, aby na drugi dzień zamiast pięknych kolonii zobaczyć czyste szalki. Zaoszczędź sobie tej frustracji i użyj sprawdzonego przepisu, który przedstawiam poniżej: Przygotowanie chemicznie kompetentnych E. coli
9 (przed rozpoczęciem procedury przygotuj około 25 ml sterylnego 50 mm CaCl2 i schłódź go w lodówce) zaszczep 1 ml pożywki (LB lub BHI) odpowiednim szczepem E.coli (np. DH5α, TOP10) hoduj przez noc w 37 st. z wytrząsaniem (~ 16 h) zaszczep 40 ml pożywki 1 ml kultury nocnej i hoduj w 37 st. z wytrząsaniem (sprawdzaj regularnie OD600) gdy OD600 = 0,2 0,25 zakończ hodowlę i przetrzymaj bakterie na lodzie przez 15 min zwiruj hodowlę (5 000 rpm, 10 min, 4 st.) rozpuść pelet bakteryjny w 20 ml zimnego CaCl2 (50 mm) inkubuj 20 min na lodzie zwiruj hodowlę (5 000 rpm, 10 min, 4 st.) rozpuść pelet bakteryjny w 1 ml zimnego CaCl2 (50 mm) inkubuj około min na lodzie po tym czasie bakterie są gotowe do użycia Uwaga! Bakterie pozostaną kompetentne przez następne 2-3 dni jeśli przechowasz je w lodówce (nie dłużej!) Możesz również przechować je przez dłuższy czas (2-3 miesiące) w -80 st Transformacja bakterii kompetentnych metodą heat shock przygotuj porcję chemicznie kompetentnych bakterii ( µl) zmieszaj bakterie z wektorem, który chcesz wprowadzić użyj około ng plazmidowego DNA zawieszonego w wodzie inkubuj min na lodzie przełóż próbkę na 90 sek do 42 st. ( heat shock ) przełóż bakterie z powrotem na lód (5 min) dodaj 250 µl pożywki (LB, BHI lub SOC) możesz ją wcześniej ogrzać do

10 37 st. hoduj przez min w 37 st. z wytrząsaniem posiej około 100 µl zawiesiny na płytki selekcyjne hoduj przez noc w 37 st. (lub przez weekend w temperaturze pokojowej, jeśli doświadczenie wypadło na piątek..) uważaj, żeby nie doprowadzić do przerośniecia kolonii, gdyż w takim wypadku poszczególne klony mogą zrosnąć się ze sobą! wyrosłe kolonie powinny posiadać obce DNA; możesz sprawdzić to metodami Colony PCR, czy Rapid Colony Screening lub wyizolować plazmid z pojedynczej hodowli (po jej uprzednim rozhodowaniu) i zsekwencjonować. Uwaga! Pamiętaj, aby posiać również kontrolę negatywną (bakterie nietransformowane), dzięki której sprawdzisz, czy antybiotyk w podłożu nie uległ rozkładowi. Pamiętaj również, że podłoża stałe z antybiotykiem pomimo tego, że przechowywane są w lodówce nie nadają się do użycia po kilku miesiącach, gdyż antybiotyk ulega rozkładowi. Jeśli w Twoich bakteriach nie ma wprowadzonego DNA (pomimo obecnych transformantów) może być to wina starych szalek. AM
11 Oczyszczanie oligonukleotydów: odsalanie, elektroforeza czy HPLC? Gdy zamawiamy oligonukleotydy, stajemy przed wyborem metody ich oczyszczenia. Do wyboru zwykle są opcje odsalania i HPLC, chociaż niektóre firmy oferują także mniej wydajną od HPLC chromatografię odwróconej fazy lub sprawdzenie wydajności syntezy poprzez elektroforezę PAGE. Najtańsze jest odsalanie, czy jednak jest ono wystarczające? Którą opcję wobec tego wybrać? Oligonukleotydy są syntezowane automatycznie przez syntetyzery sterowane komputerowo. Nić DNA jest w nich produkowana w kierunku 3 5, każda zasada dołączana jest poprzez serię następujących po sobie reakcji chemicznych. Musimy wiedzieć, że jak to w chemii bywa, żadna synteza nie jest w 100% wydajna. Dlatego otrzymany oligonukleotyd o zadanej długości to tylko jeden z produktów końcowych- synteza powoduje też powstanie niedokończonych, krótszych oligo. Zwykle na każdym etapie przyłączania nukleotydu do rosnącego łańcucha ok. 1% reakcji przebiega nieprawidłowo. W związku z tym produkt syntezy 30-merowego oligonukleotydu w efekcie stanowi mieszaninę poprawnie zsyntezowanych łańcuchów (54%-74%), a resztę stanowią produkty uboczne reakcji : 29-merowe, 28-merowe itd. Im dłuższy łańcuch, tym mniejsza zawartość prawidłowego produktu syntezy. Produkty uboczne reakcji mogą współzawodniczyć z pełnymi oligonukleotydami o matrycę reakcji, obniżając wydajność amplifikacji lub skutkując niespecyficzną amplifikacją. Dlatego właśnie czasami niezbędne jest dobre oczyszczenie oligonukleotydu, jeżeli korzystamy np. z czułych metod allelospecyficznych, albo jeżeli potrzebne nam są sondy molekularne, lub długie oligo (>40pz). Istnieje pięć głównych metody oczyszczania: odsalanie, chromatografia odwróconej fazy ( Cartdridge ), wysokoprężna chromatografia cieczowa (HPLC), oczyszczanie żelowe na akrylamidzie (PAGE), oraz filtracja żelowa Podczas odsalania roztwór oligonukleotydu jest przepuszczany przez prostą kolumnę chromatograficzną, z której odpłukiwane są sole, natomiast produkty
12 uboczne syntezy nie są usuwane. Odsalanie jest standardowym wyborem dla starterów tradycyjnych PCR, oraz dla większości starterów do reakcji qpcr. Metoda chromatografii odwróconej fazy w kardridżu wymywa oprócz soli także część produktów niespecyficznych, pozwalając na otrzymanie frakcji ok % żądanego oligonukleotydu. Ustępuje skuteczności HPLC, zapewnia też niższy odzysk oligonukleotydu więc jest dobra tylko dla małych skal syntezy. Metoda nie ma zastosowania dla bardzo długich oligo (>50nt). Wspomniana już wyżej technika HPLC to najwydajniejsza metoda oczyszczania (ale też najdroższa). Pozwala ona na oczyszczenie nawet dużych ilości oligonukleotydów w najwyższej skali syntezy, o dużej czystości (90-97%). Metoda tak jak poprzednia, nie ma zastosowania dla bardzo długich oligo (>50nt). Rozdział żelowy PAGE jest jedynym skutecznym sposobem oczyszczania długich oligonukleotydów, którym nie podoła HPLC. Pozwala otrzymać bardzo czyste (do 99% czystości) oligonukleotydy, jednakże ma poważną wadę: niski odzysk z żelu znacząco zmniejsza ilość końcową czystego oligo. Tak oczyszcza się aptamery i krótkie konstrukty syntetyczne. Sączenie żelowe jest metodą z wyboru dla konstruktów zawierających fofotioniany (tzw. s-oligo) w badaniach nad antysensem. Ma także zastosowanie dla wszelkich oligonukleotydów podawanych do hodowli komórkowych, ponieważ zapewnia najlepsze odpłukanie śladowych zanieczyszczeń chemicznych. Filtracja żelowa pozwala podobnie jak PAGE na oczyszczenie dużych fragmentów DNA. Jest wykorzystywana np. podczas oczyszczania sond do techniki FISH, doskonale wymywając niezwiązany z sondą barwnik. Marzena Wojtaszewska Żel agaroza kopia Źródło:

13 Life Technologies SigmaAldrich Izolacja RNA metodą kolumienkową protokół Istnieją dwa główne sposoby izolacji RNA. Izolacja przez ekstrakcję odczynnikiem zawierającym fenol i izotiocyjanian guanidyny oraz izolacja wykorzystująca powinowactwo kwasu nukleinowego do odpowiednio modyfikowanych chemicznie złóż silikonowych. Ze względu na łatwość zastosowania, obecnie dominuje metoda kolumienkowa. Poniżej przedstawiamy szczegółowy protokół takiej izolacji. Cel Szybka, pozwalająca na automatyzację, minimalizująca ryzyko kontaminacji metoda izolacji RNA z tkanek, kultur komórkowych, płynów ustrojowych i innych świeżych materiałów biologicznych Konieczne zasoby 1. Mikrowirówka na próbówki typu eppendorf 1,5 i 2ml 2. Pipety o zmiennej obiętości 3. Zestawy komercyjne do izolacji RNA dostosowane do izolowanego materiału i warunków w naszym laboratorium
14 4. Odczynniki lub akcesoria niedodawane do zestawu, a często niezbędne: alkohol etylowy (co najmniej 96%) woda DEPC próbówki typu eppendorf do ostatecznej elucji rękawice lateksowe inne, opisane w poszczególnych zestawach (bufory, odczynniki organiczne, plastiki) Metodyka Każdy zestaw posiada odrębny protokół postępowania, wymaga specyficznej ilości wirowań, odpowiednich ich parametrów, dodatku kilku różnych buforów o nieznanym nam składzie (tajemnica producenta) itd. Poszczególne etapy mogą wyglądać inaczej, ale odczynniki i plastiki są zawsze podobne, bo idea jest niezmienna od lat. Każda izolacja kolumienkowa, czy to DNA czy RNA, opiera się na kilku stałych etapach, przedstawionych poniżej. 1. Homogenizacja tkanki i jej upłynnienie 2. Efekt chaotropowy: zniszczenie komórek i inaktywacja enzymów 3. Związanie ze złożem kwasów nukleinowych i ich wytrącenie 4. Odpłukanie ciał komórek 5. Wypłukanie związków organicznych innych niż kwasy nukleinowe 6. Odsolenie złoża kolumny, wypłukanie resztek poprzednich buforów 7. Wysuszenie złoża z alkoholu 8. Elucja złoża, czyli rozpuszczenie RNA i odwirowanie z wodą lub buforem do świeżej próbówki. Szczegółowa procedura 1-2. Izolacja i liza komórek odbywa się w agresywnym buforze z rozpuszczalnikami organicznymi i/lub enzymami. W przypadku RNA nie stosuje się za to podwyższonej temperatury. Czasami kultury zawiesinowe, krew lub inne tkanki płynne są od razu przepuszczane przez wstępną kolumnę szatkującą (np: Qiagen Shredder Column w zestawach QiaQuick), w innych etap wstępny przypomina ekstrakcję fenolową (np. mirvana z Ambionu). Jeszcze inne korzystają z homogenizacji z kulkami szklanymi lub silikonowymi czy też z sonikacji. Opcji
15 jest wiele. 3. Kwasy nukleinowe nie rozpuszczają się w alkoholu, za to mają powinowactwo do silikonowych złóż modyfikowanych polikationami (DNA jest polianionem) w wysokiej sile jonowej buforu do izolacji. 4. Teraz wykonuje się pierwsze wirowanie. Kwasy nukleinowe zostają w złożu, bufor i zlizowane komórki lądują w przesączu na dole i są do wyrzucenia. 5. Na złożu mamy bardzo zanieczyszczone solami i związkami organicznymi RNA. Kolejne bufory, zawierajace spadajace stężenie soli i wysokie stężenie etanolu (kwasy nukleinowe się w nim nie rozpuszczają i zostają na kolumnie!) są nakładane na kolumnę i przepuszczane przez nią dzięki wirowaniu. Eluaty w dalszym ciągu wyrzucamy. 6. Ostatnie wirowania, bez dodatku buforu osuszają złoże. Kolumnę przekłada się następnie do próbówki typu eppendorf, chwilę przetrzymuje otwartą do dosuszenia, po czym dodaje się buforu bez etanolu, w celu uzyskania wodnego roztworu kwasu nukleinowego. Ten eluat jest celem naszych starań. Kolumnę wyrzucamy a na eluacie wykonujemy dalsze zabiegi po spektrofotometrycznym sprawdzeniu ilości i jakości RNA. Uwagi: Ważne podczas izolacji kolumienkowej jest przede wszystkim stosowanie się do instrukcji producenta. Zwróćmy uwagę na: dodatek etanolu do buforów, które tego wymagają [patrz punkt 1], uważne nakrapianie buforów i próbek na kolumnę tak, by nie zachlapać wewnętrznej powierzchni wieczka [patrz punkt 2], wysuszenie złoża przed ostateczną elucją [patrz punkt 3], ogólna czystość pracy z RNA [patrz punkt 4]. [1] Bardzo często popełniany błąd, zdarzający się zarówno początkującym, jak i weteranom. Może Was kosztować sporo cennych próbek i zmarnowany zestaw do izolacji! Zawsze sprawdzajmy, czy na pudełku świeżo otwartego KITu nie ma informacji o dodatku alkoholu. Gdy go dodajemy, to tylko w odpowiedniej objętości, tylko stężonego. Zawsze odhaczamy na pudełku informację o dodanym alkoholu, by współpracownicy wiedzieli, że bufor jest gotowy do użycia. [2] Zachlapanie wieczka pogorszy nam czystość RNA, ponieważ sole i
16 zanieczyszczenia organiczne mogą pozostać pod wieczkiem podczas wirowania aż do końcowego etapu elucji i razem z eluatem zostać wymyte. Nie jest to kwestia życia i śmierci, ale wpływa na jakość. [3] Resztki etanolu w izolacie zmniejszają hybrydyzację. Najczęściej nie są to wielkie uniemożliwiają jakąkolwiek amplifikację. przygotowywania mikromacierzy cdna czy wydajność reakcji opartych o PCR i straty, ale w skrajnych przypadkach Szczególnie ważne jest to podczas też w diagnostyce in vitro. [4] Mowa głównie o rękawiczkach, używaniu wody DEPC, ogólnej higienie pracy z łatwo degradującym materiałem. Troubleshooting, czyli kiedy coś idzie niezgodnie z planem 1. Po wymyciu kolumienki nie otrzymuję w ogóle RNA lub jest go tyle, co nic. sprawdź bufory. Czy dodałeś etanol do buforów przemywających (wash)? Czy nie pomyliłeś buforu do elucji (ang. elution buffer) z buforem do przemywania? Spróbuj użyć zamiast buforu do elucji czystej wody. czy używasz zestawu odpowiedniego do twojej tkanki? Tkanki lite mogą nie strawić się w buforach do lizy przystosowanych np. do krwi czy używasz odpowiedniej ilości tkanki? czy użyto rękawiczek podczas izolacji? Czy tkanka była świeża i odpowiednio przechowywana? 2. Kolumna się zapycha i nie da się jej zwirować. czy używasz zestawu odpowiedniego do twojej tkanki? Tkanki lite mogą nie strawić się w buforach do lizy przystosowanych np. do krwi i zatykają kolumnę czy bufor do lizy jest dodany w odpowiedniej objętości i inkubowany zgodnie z zaleceniami producenta? czy kolumny są nowe, nieużywane? 3. Kolumny się zniekształcają/wybuchają/kruszą się podczas wirowania. czy parametry wirowania są odpowiednie? czy kolumny są nowe, nieużywane i nie są przeterminowane? 4. RNA jest paskudnej jakości. Nie nadaje się do żadnych oznaczeń. czy bufory były uważnie nakładane na kolumnę, czy moczyły wieczko kolumny? czy użyto buforów w odpowiedniej kolejności?

17 czy bufor do elucji był niezanieczyszczony? Spróbuj elucji wodą DEPC. czy użyto odpowiedniej, nie za dużej ilości tkanki? czy zbiorniczek kolumny był opróżniany między wirowaniami? czy używano rękawiczek podczas izolacji? Czy tkanka była świeża i odpowiednio przechowywana? czy używasz zestawu odpowiedniego do Twojej tkanki? Żel agaroza kopia Jeżeli powyższe opracowanie nie dało odpowiedzi na Wasze problemy, najlepszym wyjściem będzie kontakt z firmą, która wyprodukowała dane kolumienki. Nikt nie powinien lepiej orientować się niż ona we własnościach swojego towaru. Zdarzają się także wadliwe partie, które producent powinien wymienić, jeżeli zaistnieje taka sytuacja. Piśmiennictwo: Własne doświadczenie zawodowe
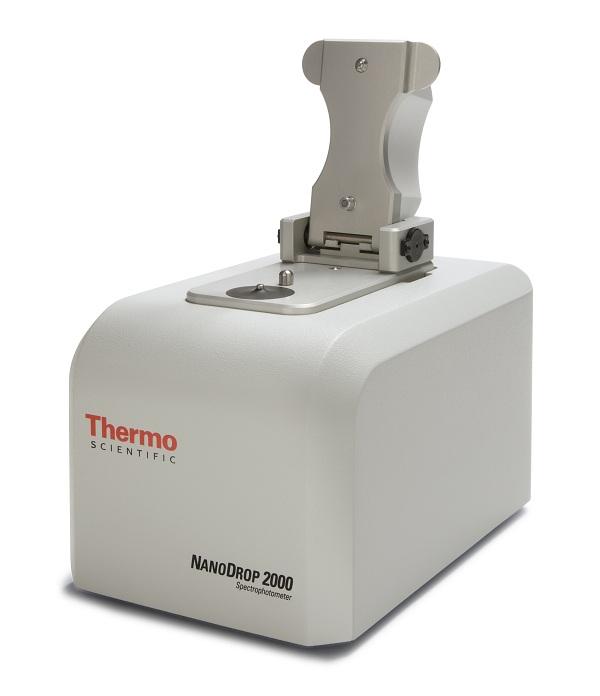
18 Spektrofotometryczna analiza jakościowa i ilościowa DNA i RNA za pomocą urządzenia NanoDrop protokół Wiele laboratoriów ma problem z izolacją RNA. Ten wstępny etap może zaważyć na powodzeniu lub klęsce całej serii badań, ponieważ RNA zanieczyszczone odczynnikami używanymi podczas ekstrakcji będzie nieefektywnie odwrotnie-transkrybowane. Dlatego niezwykle ważna jest kontrola wyizolowanego RNA przed jakąkolwiek obróbką enzymatyczną. Idealnym narzędziem do takiej kontroli jakości jest NanoDrop. Cel: określenie czystości DNA lub RNA po izolacji. Wprowadzenie: po skończonej izolacji kwasów nukleinowych musimy wiedzieć jakie jest stężenie i czystość materiału przed wykonaniem jakichkolwiek badań. Podczas ekstrakcji kwasów nukleinowych używa się rozpuszczalników organicznych (fenol, chloroform, alkohole) oraz soli (np. chlorek lub izotiocyjanian guanidyny), które mogą inhibować reakcje enzymatyczne takie jak PCR, RT-PCR, znakowanie rybonukleotydów i inne. RNA zanieczyszczone solami także szybciej degraduje (nawet przechowywany w -80 C!). Dlatego tak ważna jest poprawna analiza jakościowa i ilościowa każdej próbki. Wygodną formą takiej analizy jest pomiar absorbancji przy długości fali 230, 260 i 280nm. Obecnie większość laboratoriów dysponuje systemami typu NanoDrop (Thermo Scientific), które mierzą te parametry w kropli <1,5ul i automatycznie przeliczają absorbancję na stężenie i czystość białkową oraz organiczną. Wyznaczają także wykres absorbancji, bardzo pomocny przy ustalaniu źródła zanieczyszczeń kwasów nukleinowych. Wykonanie pomiaru: Próbkę DNA/ RNA przed pomiarem rozmrozić i trzymać na lodzie.
19 Bezpośrednio przed pomiarem zvortexować lub przepipetować góra-dół aby ją dobrze wymieszać. W menu urządzenia wybrać odpowiednia opcję (w przypadku NanoDrop a 1000 jest to Nucleic Acids ->ds DNA/ ssdna/ RNA. Opcje różnią się wartością jednostkowej absorbancji przyjmowaną w przeliczeniu na stężenie, należy o tym pamiętać!) Przygotować czystą wodę (do inicjalizacji) oraz bufor, w którym rozpuszczany był kwas nukleinowy (próba ślepa używana jako kalibrator). Inicjalizację i kalibrację wykonać nakrapiając każdorazowo 1,2-1,5µl płynu.za każdym razem zetrzeć szmatką okienko spektrofotometru. Nałożyć 1,2-1,5µl próbki na okienko, uważając na ewentualne bąbelki powietrza, które mogą zafałszować pomiar. Po wykonaniu pomiaru okienko przemyć wodą i przetrzeć szmatką. Jeżeli musimy zbierać próbkę z powrotem z okienka, dbajmy o jakość wody i dokładnie przemywajmy okienko pomiędzy pomiarami kolejnych próbek! Ryc. 1: Czyste RNA w zakresie stężeń od 1000 ng/µl (granatowa krzywa) do 3800 ng/µl (ceglasta). Widzimy, że stężenia powyżej 3000 ng/µl (żółta i ceglasta krzywa) są zbyt wysokie, by można było poprawnie oszacować A260. Charakterystyczne jest to, że w przypadku każdej z krzywych absorbancja A230 jest około 2x mniejsza niż A260 jest to wyznacznik dobrej jakości RNA
20 Analiza i interpretacja wyniku: Analiza ilościowa: Ilość kwasu nukleinowego jest określana jako stężenie w 1 µl. Zakres stężeń, w którym oznaczenie jest wiarygodne to ok ng/µl. Powyżej tej wartości DNA lub RNA jest zbyt stężone, by prawidłowo określić jego ilość i wyznaczyć krzywą absorbancji (ryc. 1). Poniżej wartości 10ul znacząca absorbancja tła powoduje przeszacowanie wyniku. Próbkę o zbyt wysokiej wartości absorbancji rozcieńczamy tym samym buforem, którego używaliśmy podczas izolacji i którym kalibrowaliśmy urządzenie. Przy zbyt niskiej ilości kwasu nukleinowego próbkę zagęszczamy lub przyjmujemy że ma ona stężenie niższe niż wyliczone przez urządzenie. Analiza jakościowa: maksimum absorbancji białek, częstego zanieczyszczenia DNA przy izolacji manualnej, wynosi 280 nm, zaś zanieczyszczenia organiczne posiadają najwyższą absorbancję poniżej 240 nm. Przyjęto więc arbitralnie 230nm jako długość fali dobrze określającą stopień zanieczyszczeń rozpuszczalnikami.organicznymi. Miarą stopnia czystości kwasów nukleinowych jest stosunek A260/230 oraz A260/280. Przyjmuje się, że czyste DNA lub RNA ma A260/280>1,8 i A260/230>1,8. Idealnie czyste RNA rozpuszczone w wodzie DEPC może mieć A260/230 nawet ok. 2,20.Na dokładną wartość tego parametru nieznacznie wpływa użyty bufor, ale generalnie parametry dobrze wyizolowanego DNA lub RNA oscylują wokół 1,8 2,05.Najprościej rzecz ujmując oznacza to, że absorbancja przy maksimum dla kwasów nukleinowych (A260) jest 2x większa niż absorbancja tła i zanieczyszczeń. Krytyczne dla reakcji enzymatycznych są wartości A260/280 i A260/230<1,4. Niższe wartości mogą spowodować bardzo kiepskie wyniki amplifikacji np. metodą Real-Time PCR czy też słabe znakowanie sond do mikromacierzy. Przy A260/230<1 nawet zwykły RT-PCR może okazać się problemem. Dlatego w przypadku kiepskiej czystości należy powtórzyć ekstrakcję alkoholem. Rodzaje zanieczyszczeń Zanieczyszczenie fenolem powodują wzrost absorbancji w zakresie nm i rozszerzenie krzywej w zakresie nm. Obraz (w przypadku ryciny 3 dotyczy próbek znaczonych strzałką, oprócz jasnoniebieskiej) jest
 Ryc.")
21 podobny jak w przypadku zanieczyszczenia solami chaotropowymi. Takie DNA/RNA nie nadaje się do zastosowań enzymatycznych. A260/230 i A260/280 mogą przyjmować zaskakujące wartości (>2. lub <1,4 w zależności od stężenia fenolu i towarzyszących zanieczyszczeń, np. chloroformu i alkoholu izoamylowego) Ryc. 2: Zanieczyszczenie fenolem (czerwona strzałka) oraz niepoprawna aplikacja próbki na okienko (oznaczone czarnymi strzałkami) Zanieczyszczenie izotiocyjanianem lub chlorkiem guanidyny widoczne na ryc. 2 (krzywa czarna i jasnozielona). Wydatny pik przy długości fali 230nm i towarzyszące mu przesunięcie absorbancji DNA/RNA w prawo w kierunku 270nm to cechy charakterystyczne tej kontaminacji. Taka próbka w żadnym wypadku nie nadaje się do zastosowań enzymatycznych, sole izotiocyjanianu są silnymi zwiazkami chaotropowymi i unieczynniają eznymy! Najczęściej oba parametry A260/A280 i A260/A230 <1,4.


22 Ryc. 3: Zanieczyszczenie solami chaotropowymi. Charakterystyczna skocznia mamucia na wykresie absorbancji. Zanieczyszczenie białkami powodują wzrost absorbancji w zakresie nm i rozszerzenie krzywej w zakresie nm. Przy niewielkim zanieczyszczeniu (<0,1% ) możemy nie obserwować górki z prawej strony wykresu, a jedynie niewielki spadek A260/280. Ryc 4. Wysokie zanieczyszczenie białkowe DNA we wszystkich próbkach. Zanieczyszczenie etanolem i/lub izopropanolem nie obserwuje się znaczącej zmiany absorbancji, szczególnie w roztworach zbuforowanych. W wodzie parametr A260/230 może być nieznacznie obniżony. Przy niewielkiej domieszce etanolu reakcje enzymatyczne nie są mocno zakłocóne, warto jednak
23 przywiązywać wagę do dokłądnego wysuszenia pelletu przd rozpuszczeniem, ponieważ zanieczyszczenia etanolem i izopropanolem są niewykrywalne spektrofotometrycznie! Zanieczyszczenie DNA przez RNA (i vice versa) oba kwasy nukleinowe są koekstragowane i zazwyczaj występują razem po izolacji. Mają także bardzo zbliżoną pochłanialność i emisję światła, więc są nirozróżnialne spektrofotometrycznie. W przypadku konieczności pozbycia się DNA z RNA musimy wykonać trawienie próbek DNAzą. Troubleshooting A260/230 jest >2,3 Powodem może być kalibracja urządzenia innym buform niż użyty do rozpuszczenia próbek (np.woda vs TRIS-HCl). Inna możliwość to nieprawidłowe nakropienie próbki na okienko i pomiar powietrza zamiast kropli przez spektrofotometr. Objawia się to poszarpaną krzywą (ryc. 2) Absorbancja A260 jest ujemna. Powodem może być kalibracja urządzenia innym buform niż użyty do rozpuszczenia próbek (np.woda vs TRIS-HCl). Inna możliwość to nieprawidłowe nakropienie próbki na okienko i pomiar powietrza zamiast kropli przez spektrofotometr. (ryc. 2) Urządzenie nie potrafi wyznaczyć absorbancji Próbka zawieszona jest w pieniącym się bądź lepkim buforze, nakropiono zbyt mało próbki bądź poza strefą pomiaru (okienko jest puste).
24
 Niejednokrotnie zdarza się, że projektujemy własną")

25 Przygotowanie liniowych standardów do reakcji Real-Time PCR (protokół) Niejednokrotnie zdarza się, że projektujemy własną metodykę opartą o technikę Real-Time PCR. Największym problemem przy optymalizacji gotowej metody, zarówno na etapie badań naukowych, jak i standaryzacji
26 oraz komercjalizacji, jest przygotowanie odpowiednich standardów. Jak zrobić to najprościej? Krzywe standardowe przygotowuje się zwykle na bazie plazmidów- kolistych lub linearyzowanych (poprzez cięcie restrykcyjne). Pokutuje teoria, że takie cząsteczki zachowują się podczas Real-Time PCRu tak samo jak mieszanina różnych cząsteczek we właściwej reakcji z cdna, podczas gdy reamplifikacja produktu PCR ma inną kinetykę niż właściwego cdna, przez co ilościowanie jest zafałszowane. Niewielu zdaje sobie sprawę, że jest to mit. Zamiast linearyzowanych cząsteczek plazmidu wystarczy użyć produktu PCR namnożonego na szerszych starterach, niż startery używane podczas właściwej reakcji ilościowej. Oto prosty przepis na standardy: Zaprojektuj specyficzny, dobrze działający PCR na starterach zewnętrznych względem starterów użytych do Real-Time u o ok nt z każdej strony (patrz rycina) Zamplifikuj dużą ilość matrycy na zewnętrznych starterach. Oczyść produkt na kolumienkach, pozbywając się zanieczyszczeń i buforu reakcyjnego. (KITy do oczyszczania produktu PCR oferowane są na polskim rynku przez wiele firm) Zmierz przy pomocy Nanodropa lub innego spektrofotometru stężenie matrycy wobec buforu użytego do elucji produktu PCR. Przelicz stężenie według poniższego wzoru: Liczba kopii w 1ul Lk =(x )/(długość matrycy w pz ), gdzie x to wartość stężenia wyrażona w ng/ul. Żeby zrobić to szybko i bez pomyłek, użyj gotowego kalkulatora Rozcieńcz standard w dowolny sposób. Pierwszego stężenia przygotuj tyle, żeby starczyło go na wszystkie eksperymenty, jeżeli zabraknie go, należy kolejny batch pierwszego standardu wywzorcować w Real-Time PCR z uwzglęnieniem standardu używanego wcześniej!
27 Przykład: Produkt Real-Time PCRu ma 223 pz. Zewnętrzne względem nich startery mają 580pz. Po amplifikacji i sprawdzeniu na żelu agarozowym jakości i wysokości prążka (silny, wąski prążek produktu o wielkości ~580pz) oczyszczamy próbkę na żelu. Zmierzone stężenie wynosi 26ng/ul. Wstawiamy wartości 26 i 580 w odpowiednie miejsca kalkulatora (lub liczymy ręcznie). Wyliczona wartość to 4,15 10^10
28 Żeby przygotować kolejne rozcieńczenia pipetujemy np. po 90ul H2O (lub buforu do elucji, zależy czym eluowaliśmy) do kolejnych próbówek, po czym do pierwszego rozcieńczenia dodajemy 10ul. Przygotowanego standardu, vortexujemy, następnie do 2-go standardu 10ul pierwszego itd. Podczas RealTime u definiujemy kolejne stężenia jako 4,15 10^10, 4,15 10^9, 4,15 10^8 itd. Uwagi dotyczące metody: Metoda obliczenia opiera się na założeniu, że średnio mol 1 pary zasad waży 650g (czyli 1 para zasad 650 Daltonów). Oczywiście można obliczyć dokładną wagę naszej matrycy, używając skryptu zliczającego ilość par GC i AT, mnożąc to następnie przez ich wagę, ale doświadczenie podpowiada, że jest to niepotrzebne. Stężenie oczyszczonego produktu PCR mierzone spektrofotometrycznie także nie jest bardzo dokładne, ale jeżeli amplifikacja poszła dobrze, taka dokładność wystarcza. Jeżeli zmierzone stężenie produktu jest mniejsze niż 10ng/ul (czyli mocno zawyżone z powodu bliskiego poziomu tła), polecam powtórzyć amplifikację, to na pewno lepsze niż powtarzanie całej procedury przygotowania standardów, jeżeli okazałoby się że ilościowy PCR nie chodzi przy 10 czy też 100 kopiach na reakcję. Żel agaroza kopia

29 Western poradnik. Blot. Praktyczny Western Blot jest techniką powszechnie stosowaną w biologii molekularnej w różnych laboratoriach na całym świecie. W przeciwieństwie do hybrydyzacji Southern i Northern, które służą identyfikacji odpowiednio DNA i RNA, Western Blot pozwala na jakościowe wykrycie konkretnego białka z bardzo kompleksowej mieszaniny białek izolowanych z ekstraktu komórkowego. Jak przebiega WB? Hybrydyzacja Western Blot, określana także jako immunoblotting, obejmuje trzy etapy. W pierwszej kolejności zdenaturowane białka rozdziela się na podstawie ich mas przy wykorzystaniu elektroforezy na żelu poliakrylamidowym. Następnie, białka są przenoszone na membranę (nitrocelulozową lub PVDF) na drodze elektrotransferu, aż w końcu następuje identyfikacja konkretnego białka poprzez inkubację membrany przeciwciałami pierwszo- i drugorzędowymi (immunodetekcja). Elektroforeza Rozdział białek podczas elektroforezy następuje na żelu składającym się z dwóch części: żelu zagęszczającego (górna warstwa) i żelu rozdzielającego (dolna warstwa). Próbki białka, uprzednio przygotowane w buforze o niskim przewodnictwie (ang. loading buffer), są aplikowane do studzienek żelu zagęszczającego. Zagęszczanie białek odbywa się w wyniku obustronnego kontaktu z buforami o wysokim przewodnictwie i następuje ono na granicy z żelem rozdzielającym [1].
30 Żele poliakrylamidowe używane w elektroforezie są polimerami akrylamidu i bisakrylamidu. Od proporcji i od ilości tego ostatniego składnika zależy usieciowanie żelu. Polimeryzacja katalizowana jest przez wolne rodniki, których źródłem jest nadsiarczan amonu (ang. ammonium persulfate, APS), a ich uwalnianie przyspiesza dodanie N,N,N -Tetramethylethylenediaminy (TEMED). Stężenie żelu poliakrylamidowego decyduje o rozdziale białek i jest ono odwrotnie proporcjonalnie do wielkości białka: im większe białko chcemy oznaczyć, tym mniejsze stężenie żelu powinno być wykonane. Elektrotransfer Elektrotransfer to przeniesienie rozdzielonych białek na membranę. Bardzo ważne jest ułożenie w odpowiedniej kolejności elementów tzw. kanapki, aby zapewnić prawidłowy transfer. Ujemnie naładowane białka migrują w kierunku dodatniej elektrody i zostają zatrzymane na membranie umieszczonej między żelem a anodą. W ten sposób białka w formie prążków znajdujących się na różnych poziomach są przeniesione na membranę i dają lustrzany obraz rozdzielonych w żelu polipeptydów [1]. Tutaj warto podać kilka informacji na temat membran. Istnieją dwa typy membran powszechnie stosowanych w Western Blot: nitrocelulozowa i poliwinylidenowa (ang. polyvinylidene fluoride, PVDF). Różnią się one zasadniczo. Membrana nitrocelulozowa cechuje się wysokim powinowactwem do białek. Jest ona jednak bardzo krucha w strukturze, a hybrydyzację na niej można wykonać tylko jeden raz. Z kolei membrana PVDF odznacza się bardzo dobrymi właściwościami mechanicznymi, jest bardziej wytrzymała, a hybrydyzację na niej można wykonać wielokrotnie. W przypadku membran PVDF bardzo istotne jest płukanie membrany przy technice Western Blot, aby ograniczyć sygnał tła [2]. Immunodetekcja Jednym z jej typów immunodetekcji jest inkubacja dwoma rodzajami przeciwciał: pierwszorzędowymi i drugorzędowymi. Pomiędzy tymi inkubacjami przeprowadza się etapy płukania membrany w celu usunięcia niezwiązanych przeciwciał. Przeciwciała pierwszorzędowe bezpośrednio reagują z antygenem (czyli białkiem, które chcemy wykryć). Z kolei przeciwciała drugorzędowe zazwyczaj są znakowane, radioaktywnie albo przyłączonym enzymem np. peroksydazy chrzanowej (ang. horseradish peroxidase, HRP). Membranę pokrywa się
31 substratem dla HRP, np. luminolem. Na rynku dostępnych jest kilka komercyjnych zestawów z przygotowanym roztworami substratów. HRP katalizuje utlenienie luminolu w obecności nadtlenku wodoru. Luminol, który przeszedł w stan wzbudzenia, emituje promieniowanie świetlne. Obraz doświadczenia uzyskuje się poprzez ekspozycję membrany na kliszę, następnie ją wywołując [3]. Przejdźmy teraz do praktycznego i technicznego spojrzenia na procedurę Western Blot. SDS PAGE przygotowanie i elektroforeza 1. Przygotowanie aparatury i żeli Do przeprowadzenia elektroforezy SDS PAGE niezbędnym elementem są żele poliakrylamidowe. Jeżeli nie możemy sobie pozwolić na zakup gotowych żeli, musimy je własnoręcznie przygotować. Przy dobrej organizacji nie jest to jednak żmudna i czasochłonna praca. Poniżej przedstawiona jest lista wszystkich odczynników i buforów potrzebnych do elektroforezy SDS PAGE i elektrotransferu: 3x Gel Buffer (bufor do przygotowania żeli poliakrylamidowych, przechowywać w temperaturze 4 st.c): 3M Tris-HCl 0.3% SDS ph 8,45 Bufory do elektroforezy: a) Bufor anodowy: 0,2 M Tris ph 8,9 b) Bufor katodowy: 0,1 M Tris 0,1 M Tricine 0,1 % SDS ph 8,25
 TBS-T (2 L) 40 ml 1M Tris ph 8 100 ml 3 M NaCl 2 ml TWEEN 20 (0.")
32 Bufor do elektrotransferu: 192 mm roztwór glicyny 25 mm Tris 20% metanol 2x bufor do przygotowania próbek białka: 100 mm Tris HCl ph 6,8 20% glicerol 5% SDS 0,02% błękit bromofenolowy (opcjonalnie 10% 2-merkaptoetanol) TBS-T (2 L) 40 ml 1M Tris ph ml 3 M NaCl 2 ml TWEEN 20 (0.1%) Zanim zaczniemy przygotowywać mieszaniny żeli, pierwszym krokiem jest przygotowanie całej aparatury zgodnie z wytycznymi producenta. Składamy kasetę żelową, umieszczamy w niej grzebień i zaznaczamy markerem miejsce ok cm poniżej końca ząbków grzebienia jest to górna granica żelu rozdzielającego. Przykładowa kaseta żelowa przedstawiona jest na Ryc. 1. Ryc. 1. Przykładowa kaseta żelowa. Źródło: Wikimedia Commons, autor: Lilly_M, licencja: CC BY-SA 2.5 Aby przygotować wystarczającą ilość żelu, obliczamy objętość kasety żelowej (Objętość = szerokość x wysokość x głębokość). Przykładowo, objętość kasety wynosi 8.2 cm3 (V = 6.8 cm x 8 cm x 0.15 cm) i z praktycznego punktu widzenia
 waha się zazwyczaj pomiędzy 4-5%, podczas gdy w żelu rozdzielającym (ang.")
33 potrzeba w tym przypadku 6-7 ml żelu rozdzielającego, jednak lepiej przygotować mały zapas np. 10 ml dla jednego żelu. Stężenia akrylamidu w żelach różnią się w zależności od wykonywanego rozdziału. Stężenie akrylamidu w żelu zagęszczającym (ang. stacking gel) waha się zazwyczaj pomiędzy 4-5%, podczas gdy w żelu rozdzielającym (ang. separating gel) jego stężenie różni się w zależności od masy cząsteczkowej danego białka. Odpowiednie stężenia dla obydwóch żeli dla białka o znanej masie cząsteczkowej, które poddajemy elektroforezie, można znaleźć na stronach różnych producentów. Poniżej przedstawiony skład można użyć do przygotowania kilku żeli (około 3) do rozdziału białek, o masie cząsteczkowej w przedziale kda. Tabela 1. Skład żelu zagęszczającego i rozdzielającego dla elektroforezy białek o masie cząsteczkowej kda. Źródło: własne Żele przygotowujemy pod wyciągiem, ponieważ niespolimeryzowany akrylamid jest neurotoksyczny. Odpowiednio wcześnie organizujemy wszystkie potrzebne narzędzia i odczynniki. W 50 ml probówkach typu Falcon mieszamy wszystkie składniki żelu rozdzielającego, pamiętając o tym, że TEMED dodajemy na sam koniec, gdyż inicjuje on polimeryzację żelu. Tak przygotowany roztwór przenosimy pipetą do kasety żelowej do wcześniej zaznaczonej linii cm (wcześniej wyjmujemy grzebyk). Nałożony żel rozdzielający pokrywamy cienką warstwą n-butanolu (około 500 µl) i upewniamy się, że jest on równomiernie rozmieszczony na górnej linii żelu. Pozostawiamy tak przygotowany żel na minut, aby spolimeryzował. Proces ten możemy także obserwować na pozostałościach roztworu w 50 ml tubce Falcon. Poliakrylamid (który uległ polimeryzacji) nie jest już silnie neurotoksyczny. Gdy żel rozdzielający ulegnie polimeryzacji, usuwamy n-butanol z powierzchni żelu, a jego nadmiar osuszamy delikatnie bibułą. Następnie przygotowujemy żel zagęszczający poprzez zmieszanie komponentów z APS, dodając na koniec TEMED. Nakładamy pipetą mieszaninę, szybko umieszczając grzebień pomiędzy

34 krawędzie płytek szklanych w ten sposób, aby nie powstały pęcherzyki powietrza. Pozostawiamy żel do polimeryzacji. Warto pamiętać, że jeżeli planujemy przeprowadzenie dużej ilości elektroforez w krótkim czasie, można za jednym razem przygotować więcej żeli i przechować je kasetach żelowych z umieszczonym grzebykiem, owinięte w nasączony w buforze katodowym kawałek ręcznika papierowego w temperaturze 4 stc. 2. Elektroforeza na żelu poliakrylamidowym W przypadku, gdy elektroforezę przeprowadzamy tego samego dnia, ostrożnie wyjmujemy grzebień z żelu i przemywamy studzienki wodą destylowaną. Przygotowaną kasetę z żelem umieszczamy w module i mocujemy za pomocą uszczelek do ramki mocującej. Następnie cały moduł umieszczamy wewnątrz zbiornika elektroforezy. Górną komorę wypełniamy buforem katodowym do krawędzi górnej, upewniając się, że cały żel jest zanurzony w buforze. Dolną komorę wypełniamy buforem anodowym. Przykładowy dobrze zmontowany moduł do elektroforezy jest przedstawiony na Ryc. 2. Ryc. 2. Zmontowany moduł do elektroforezy. Źródło: Wikimedia Commons, autor: Jean-Eitenne Poirrier, licencja: CC BY-SA 2.0 Następnie zamykamy pokrywą zbiornik i podłączamy do zasilacza. Przy żelach własnoręcznie wylewanych ważne jest przeprowadzenie tzw. pre-run w celu usunięcia zanieczyszczeń z żelu. Proces Pre-run przeprowadzamy przez 20 minut przy napięciu V podczas przygotowywania próbek na rozdział. Oznaką dobrze zmontowanej aparatury do elektroforezy i poprawnie działającego systemu jest pojawienie się piany przy górnej krawędzi żelu. Ryc. 3 poniżej przedstawia przykładowy zestaw do elektroforezy na żelu poliakrylamidowym.
 i inkubujemy przez 5 minut w temperaturze 95 stc w celu denaturacji białka.")
35 Ryc. 3. SDS PAGE. Źródło: FLICKR, autor: Jean-Eitenne Poirrier, licencja: CC BYSA 2.0 Równocześnie z pre-run przygotowujemy próbki białka, których stężenie oznaczyliśmy wcześniej np. metodą Bradforda. Odpowiednią obliczoną ilość próbki mieszamy z 2x buforem do próbek (ang. loading buffer) i inkubujemy przez 5 minut w temperaturze 95 stc w celu denaturacji białka. Następnie odpowiednią ilość próbki nanosimy do poszczególnych studzienek. Pamiętamy także o zaaplikowaniu 5 µl markera. Elektroforezę przeprowadzamy w następujących warunkach. Przy napięciu 80 V załadowane próbki zagęszczają się, następnie przy wyższym napięciu V następuje ich rozdział. Elektroforezę prowadzimy do momentu, gdy linia wędrujących w żelu prążków znajdzie się przy dolnej krawędzi żelu. Po zakończeniu elektroforezy bufor katodowy zlewamy, natomiast bufor anodowy możemy użyć do następnej elektroforezy. Kasetę żelową wyjmujemy ostrożnie z modułu, następnie rozdzielamy za pomocą szpatułki płytki, pomiędzy którymi znajduje się żel. Skalpelem lub żyletką odcinamy żel zagęszczający i tak przygotowany żel umieszczamy w buforze do elektrotransferu. Transfer półsuchy W trakcie prowadzenia elektroforezy przygotowujemy wszystkie potrzebne czynniki do transferu rozdzielonych białek na membranę. Wycinamy membranę PVDF w rozmiarze odpowiadającym żelowi rozdzielającemu i aktywujemy ją (zmniejszamy jej hydrofobowość) w 100% metanolu przez 10 sekund, następnie obmywając membranę 5 minut w wodzie MQ i przenosząc do buforu do elektrotransferu (ang. Western buffer). W ten sam sposób traktujemy złożone po 3 bibuły Whatmann.


36 Składamy kanapkę w kasecie do elektrotransferu w następującej kolejności: 3 x nasączone bibuły Whatmann aktywowana membrana PVDF żel nasączony także buforem do elektrotransferu 3 x nasączone bibuły Whatmann. Przedstawiona poniżej Ryc. 4 przedstawia schemat układania kanapki do Western Blot. Ryc. 4. Elektrotransfer. Źródło: Wikimedia Commons, autor: Bensaccount, licencja: CC-BY-SA 3.0 Praktyczna rada: Dla ułatwienia orientacji membrany i żelu odcinamy żyletką mały górny lewy róg na membranie i żelu. Upewniamy się także, czy żadne pęcherzyki powietrza nie dostały się pomiędzy warstwy. Poprzez delikatne rolowanie na powierzchni kanapki probówką typu Falcon możemy zapewnić dokładne przyleganie poszczególnych warstw. Następnie, nakładamy górną pokrywę kasety Western Blot i przeprowadzamy elektrotransfer przez 1 h i 10 minut. Natężenie potrzebnego prądu obliczamy z następującego wzoru: I = S x 1,1 x N [ma] N ilość membran jaką poddajemy Western Blot S powierzchnia membrany. Napięcie ustawiamy w granicach V. O sukcesie przeprowadzonego elektrotransferu upewniamy się, jeżeli zaobserwujemy przeniesiony marker na membranę.

37 Przykładowa kaseta do Western Blot przedstawiona jest na Ryc. 5. Ryc. 5. Kaseta do elektrotransferu. Źródło: Wikimedia Commons, autor: Lilly_M, licencja: CC BY-SA 3.0 W trakcie trwania elektrotransferu przygotowujemy roztwór blokujący: 4% mleka odtłuszczonego w proszku, 1% BSA w 100 ml roztworu TBS-T. Po wykonanym elektrotransferze membranę zanurzamy w przygotowanym roztworze blokującym i pozostawiamy w temperaturze 4 stc przez noc na wolnym kołysaniu. Roztwór ten ma na celu zablokować niespecyficzne wiązania. Immunodetekcja Inkubacja membrany przeciwciałami pierwszorzędowymi Wykonujemy odpowiednie rozcieńczenie przeciwciał pierwszorzędowych w TBS-T, zgodnie z zaleceniami producenta. Rozcieńczenia zazwyczaj wahają się w granicach 1:500 1:5000. Dla przykładu przeciwciała anty-cd4 zgodnie ze wskazówkami powinny być rozcieńczone 1:500. W związku z tym, 8 µl z próbki przeciwciał rozcieńczamy w 4 ml TBS-T. Jest to wystarczająca objętość na inkubację przeciwciałami pierwszorzędowymi. Praktycznym sposobem na inkubację jest umieszczenie membrany w 50 ml probówce typu Falcon (do wewnątrz stroną, na której znajdują się białka) tak, aby strony membrany na siebie się nie nakładały. Następnie układamy 50 ml probówkę poziomo na urządzeniu rotującym, tak aby zapewnić równomiernie rozłożenie roztworu z przeciwciałami na całą membranę. Inkubację prowadzimy przez 1 godzinę w temperaturze pokojowej. Płukanie membrany. Membranę płuczemy w TBS-T w celu usunięcia niezwiązanych przeciwciał. Proces
38 ten prowadzimy 5 razy przez 5 minut upewniając się, aby za każdym razem cała membrana była zanurzona w roztworze. Inkubacja przeciwciałami drugorzędowymi Prowadzimy ją w ten sam sposób jak w przypadku inkubacji przeciwciałami pierwszorzędowymi. Rozcieńczenie przeciwciał w tym wypadku jest zazwyczaj wyższe, np. 1: (rozcieńczenie wykonujemy zgodnie ze wskazówkami producenta.) Płukanie membrany Pięciokrotnie płuczemy membranę w roztworze TBS-T. Ostatnie płukanie wykonujemy w roztworze TBS bez Tween, aby wypłukać i usunąć detergent. Nałożenie czynnika detekcji Po drugim płukaniu membranę lekko osuszamy z nadmiaru TBS poprzez dotknięcie jednego jej końca o papierowy ręcznik, który wsiąknie nadmiar buforu. Następnie pokrywamy membranę czynnikiem detekcji, który wiąże się z drugorzędowymi przeciwciałami. Nakładamy reagent równomiernie pipetą na powierzchnię membrany i inkubujemy przez 1 minutę. Usuwamy nadmiar substratu i umieszczamy membranę w kasecie X ray. Membrana jest dodatkowo zabezpieczona, tj. włożona w zwykłą folię biurową. Białka do detekcji znajdują się po wierzchniej stronie. Przed wystawieniem membrany na ekspozycję, upewniamy się, że w pomieszczeniu, gdzie będziemy wywoływać film jest ciemno, a jedynym dozwolonym światłem jest światło czerwone. Następnie umieszczamy jeden film na powierzchni folii, zamykamy X ray kasetę i poddajemy ekspozycji w różnym czasie. Ekspozycja może trwać, np. 10 sec, 1 min 10 minut, ale czasem potrzeba i paru godzin [4]. Interpretacja wyniku. Przykładowy wynik Western Blot przedstawiony jest na Ryc. 6.

39 Ryc. 6. Przykładowy wynik Western Blot. Pierwszy rząd z lewej strony reprezentuje marker ze zdefiniowanymi rozmiarami białka, które miało być zidentyfikowane. Na górnej rycinie obserwujemy wyraźnie zarysowane prążki pochodzące od białka beta-actin o masie cząsteczkowej 42 kda. Na dolnej rycinie zostało zidentyfikowane białko PCP4 o masie cząsteczkowej 7.6 kda. Źródło: Wikimedia Commons, autor: Herizora, licencja: CC BY-SA 3.0 Żel agaroza kopia Western Blot jest techniką popularną w biologii molekularnej. Ze względu na swoją czułość i jednoznaczność wyniku jest jedną z najczęściej wybieranych przez naukowców metod. Pomimo faktu, iż trwa ona aż dwa dni, to przy optymalizacji testu oraz doborze odpowiednich przeciwciał mamy pewność, że otrzymamy wyniki wysokiej jakości. Piśmiennictwo: 1. Opiela J. Analiza ekspresji genów na poziomie białek przy użyciu techniki western-blot. Wiadomości Zootechniczne, 2006; R. XLIV; 1: Mahmood T., Yang P-C. Western Blot: Technique, Theory, and Trouble Shooting. N Am J Med Sci, 2012; 4(9): Kurien B., Scofield R. Western blotting. Methods; 2006; 38: Doświadczenia własne.

40 Zapisywanie mutacji polimorfizmów (II) DNA i Jak zapisać koordynaty sekwencji nukleotydowej? Jak zawrzeć informację o długości i charakterze delecji lub insercji tak, by inni badacze poprawnie ją zinterpretowali? Przedstawiamy drugi z serii artykułów o poprawnej nomenklaturze mutacji i polimorfizmów. Dziś zajmiemy się sekwencją DNA. Zapisywanie zmian na poziomie DNA substytucje oznaczamy zawsze nawiasem trójkątnym, skierowanym w prawo, t.j. > Kierunek zapisu biegnie od nukleotydu pierwotnie znajdującego się w sekwencji referencyjnej, do nukleotydu zmienionego. np. 76A>C oznacza zmianę w pozycji 76 nukleotydu z adeniny na cytozynę. 88+1G>T (lub w innym zapisie IVS2+1G>T) opisuje substytucję G na T w pierwszym nukleotydzie drugiego intronu, intron znajduje się w naszym przykładzie pomiędzy nukleotydami 88 i 89 względem sekwencji cdna. W przypadku, gdy dochodzi do alternatywnego splicingu zawsze zaznaczajmy, którego wariantu transkryptu dotyczy zapis. 89-2A>C (lub IVS2-2A>C) analogicznie jak w poprzednim przykładzie symbolizuje substytucje A>C w drugim od końca kodonie intronu drugiego, znajdującego się pomiędzy 88 a 89 nukleotydem cdna.
Projektowanie reakcji multiplex- PCR
 Projektowanie reakcji multiplex- PCR Dzięki nowoczesnym, wydajnym zestawom do PCR, amplifikacja DNA nie jest już takim wyzwaniem, jak w latach 80-tych i 90-tych. Wraz z poprawą metodyki, pojawiły się ciekawe
Projektowanie reakcji multiplex- PCR Dzięki nowoczesnym, wydajnym zestawom do PCR, amplifikacja DNA nie jest już takim wyzwaniem, jak w latach 80-tych i 90-tych. Wraz z poprawą metodyki, pojawiły się ciekawe
Spektrofotometryczna analiza jakościowa i ilościowa DNA i RNA za pomocą urządzenia NanoDrop protokół
 Spektrofotometryczna analiza jakościowa i ilościowa DNA i RNA za pomocą urządzenia NanoDrop protokół Wiele laboratoriów ma problem z izolacją RNA. Ten wstępny etap może zaważyć na powodzeniu lub klęsce
Spektrofotometryczna analiza jakościowa i ilościowa DNA i RNA za pomocą urządzenia NanoDrop protokół Wiele laboratoriów ma problem z izolacją RNA. Ten wstępny etap może zaważyć na powodzeniu lub klęsce
Izolacja RNA metodą kolumienkową protokół
 Izolacja RNA metodą kolumienkową protokół Istnieją dwa główne sposoby izolacji RNA. Izolacja przez ekstrakcję odczynnikiem zawierającym fenol i izotiocyjanian guanidyny oraz izolacja wykorzystująca powinowactwo
Izolacja RNA metodą kolumienkową protokół Istnieją dwa główne sposoby izolacji RNA. Izolacja przez ekstrakcję odczynnikiem zawierającym fenol i izotiocyjanian guanidyny oraz izolacja wykorzystująca powinowactwo
Western Blot. Praktyczny poradnik.
 Western Blot. Praktyczny poradnik. Western Blot jest techniką powszechnie stosowaną w biologii molekularnej w różnych laboratoriach na całym świecie. W przeciwieństwie do hybrydyzacji Southern i Northern,
Western Blot. Praktyczny poradnik. Western Blot jest techniką powszechnie stosowaną w biologii molekularnej w różnych laboratoriach na całym świecie. W przeciwieństwie do hybrydyzacji Southern i Northern,
Metody badania ekspresji genów
 Metody badania ekspresji genów dr Katarzyna Knapczyk-Stwora Warunki wstępne: Proszę zapoznać się z tematem Metody badania ekspresji genów zamieszczonym w skrypcie pod reakcją A. Lityńskiej i M. Lewandowskiego
Metody badania ekspresji genów dr Katarzyna Knapczyk-Stwora Warunki wstępne: Proszę zapoznać się z tematem Metody badania ekspresji genów zamieszczonym w skrypcie pod reakcją A. Lityńskiej i M. Lewandowskiego
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa tel. 22 572 0735, 606448502
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa tel. 22 572 0735, 606448502
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa IZOLACJA DNA Z HODOWLI KOMÓRKOWEJ.
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa IZOLACJA DNA Z HODOWLI KOMÓRKOWEJ.
TaqNova-RED. Polimeraza DNA RP20R, RP100R
 TaqNova-RED Polimeraza DNA RP20R, RP100R RP20R, RP100R TaqNova-RED Polimeraza DNA Rekombinowana termostabilna polimeraza DNA Taq zawierająca czerwony barwnik, izolowana z Thermus aquaticus, o przybliżonej
TaqNova-RED Polimeraza DNA RP20R, RP100R RP20R, RP100R TaqNova-RED Polimeraza DNA Rekombinowana termostabilna polimeraza DNA Taq zawierająca czerwony barwnik, izolowana z Thermus aquaticus, o przybliżonej
AmpliTest Babesia spp. (PCR)
") AmpliTest Babesia spp. (PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla pierwotniaków z rodzaju Babesia techniką PCR Nr kat.: BAC21-100 Wielkość zestawu: 100 oznaczeń Objętość pojedynczej reakcji:
AmpliTest Babesia spp. (PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla pierwotniaków z rodzaju Babesia techniką PCR Nr kat.: BAC21-100 Wielkość zestawu: 100 oznaczeń Objętość pojedynczej reakcji:
E.coli Transformer Kit
 E.coli Transformer Kit zestaw do przygotowywania i transformacji komórek kompetentnych Escherichia coli. Metoda chemiczna. wersja 1117 6 x 40 transformacji Nr kat. 4020-240 Zestaw zawiera komplet odczynników
E.coli Transformer Kit zestaw do przygotowywania i transformacji komórek kompetentnych Escherichia coli. Metoda chemiczna. wersja 1117 6 x 40 transformacji Nr kat. 4020-240 Zestaw zawiera komplet odczynników
TaqNovaHS. Polimeraza DNA RP902A, RP905A, RP910A, RP925A RP902, RP905, RP910, RP925
 TaqNovaHS RP902A, RP905A, RP910A, RP925A RP902, RP905, RP910, RP925 RP902A, RP905A, RP910A, RP925A RP902, RP905, RP910, RP925 TaqNovaHS Polimeraza TaqNovaHS jest mieszaniną termostabilnej polimerazy DNA
TaqNovaHS RP902A, RP905A, RP910A, RP925A RP902, RP905, RP910, RP925 RP902A, RP905A, RP910A, RP925A RP902, RP905, RP910, RP925 TaqNovaHS Polimeraza TaqNovaHS jest mieszaniną termostabilnej polimerazy DNA
Techniki molekularne ćw. 1 1 z 6
 Techniki molekularne ćw. 1 1 z 6 Instrukcja do ćwiczeń Nr 1. Temat: Izolacja całkowitego DNA z tkanki ssaczej metodą wiązania DNA do kolumny krzemionkowej oraz spektrofotometryczna ocena jego czystości
Techniki molekularne ćw. 1 1 z 6 Instrukcja do ćwiczeń Nr 1. Temat: Izolacja całkowitego DNA z tkanki ssaczej metodą wiązania DNA do kolumny krzemionkowej oraz spektrofotometryczna ocena jego czystości
RT31-020, RT , MgCl 2. , random heksamerów X 6
 RT31-020, RT31-100 RT31-020, RT31-100 Zestaw TRANSCRIPTME RNA zawiera wszystkie niezbędne składniki do przeprowadzenia syntezy pierwszej nici cdna na matrycy mrna lub całkowitego RNA. Uzyskany jednoniciowy
RT31-020, RT31-100 RT31-020, RT31-100 Zestaw TRANSCRIPTME RNA zawiera wszystkie niezbędne składniki do przeprowadzenia syntezy pierwszej nici cdna na matrycy mrna lub całkowitego RNA. Uzyskany jednoniciowy
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa tel. 22 572 0735, 606448502
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa tel. 22 572 0735, 606448502
Zestaw do wykrywania Chlamydia trachomatis w moczu lub w kulturach komórkowych
 Nr kat. PK15 Wersja zestawu: 1.2016 Zestaw do wykrywania w moczu lub w kulturach komórkowych na 50 reakcji PCR (50µl), włączając w to kontrole Detekcja oparta jest na amplifikacji fragmentu genu crp (cysteine
Nr kat. PK15 Wersja zestawu: 1.2016 Zestaw do wykrywania w moczu lub w kulturach komórkowych na 50 reakcji PCR (50µl), włączając w to kontrole Detekcja oparta jest na amplifikacji fragmentu genu crp (cysteine
Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, Warszawa. Zakład Biologii Molekularnej
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa 1 Ćwiczenie 1 Izolacja oraz
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa 1 Ćwiczenie 1 Izolacja oraz
Zestaw do wykrywania Babesia spp. i Theileria spp. w kleszczach, krwi i hodowlach komórkowych
 Nr kat. PK25N Wersja zestawu: 1.2012 Zestaw do wykrywania spp. i Theileria spp. w kleszczach, krwi i hodowlach komórkowych dwie oddzielne reakcje PCR 2x50 reakcji PCR (50 µl), włączając w to kontrole Zestaw
Nr kat. PK25N Wersja zestawu: 1.2012 Zestaw do wykrywania spp. i Theileria spp. w kleszczach, krwi i hodowlach komórkowych dwie oddzielne reakcje PCR 2x50 reakcji PCR (50 µl), włączając w to kontrole Zestaw
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa Ćwiczenie 3 Izolacja i rozdział
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa Ćwiczenie 3 Izolacja i rozdział
Syngen Gel/PCR Mini Kit
 Syngen Gel/PCR Mini Kit Syngen Gel/PCR ME Mini Kit Zestawy do oczyszczania DNA: - z żelu agarozowego - po reakcji PCR i innych reakcjach enzymatycznych - z żelu agarozowego z małą objętością elucji - po
Syngen Gel/PCR Mini Kit Syngen Gel/PCR ME Mini Kit Zestawy do oczyszczania DNA: - z żelu agarozowego - po reakcji PCR i innych reakcjach enzymatycznych - z żelu agarozowego z małą objętością elucji - po
E.coli Transformer Zestaw do przygotowywania i transformacji komórek kompetentnych Escherichia coli
 E.coli Transformer Zestaw do przygotowywania i transformacji komórek kompetentnych Escherichia coli Wersja 0211 6x40 transformacji Nr kat. 4020-240 Zestaw zawiera komplet odczynników do przygotowania sześciu
E.coli Transformer Zestaw do przygotowywania i transformacji komórek kompetentnych Escherichia coli Wersja 0211 6x40 transformacji Nr kat. 4020-240 Zestaw zawiera komplet odczynników do przygotowania sześciu
Saccharomyces Transformer Kit zestaw do przygotowywania i transformacji komórek kompetentnych Saccharomyces cerevisiae. Metoda chemiczna.
 Saccharomyces Transformer Kit zestaw do przygotowywania i transformacji komórek kompetentnych Saccharomyces cerevisiae. Metoda chemiczna. wersja 0916 6 x 20 transformacji Nr kat. 4010-120 Zestaw zawiera
Saccharomyces Transformer Kit zestaw do przygotowywania i transformacji komórek kompetentnych Saccharomyces cerevisiae. Metoda chemiczna. wersja 0916 6 x 20 transformacji Nr kat. 4010-120 Zestaw zawiera
AmpliTest Salmonella spp. (Real Time PCR)
") AmpliTest Salmonella spp. (Real Time PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla bakterii z rodzaju Salmonella techniką Real Time PCR Nr kat.: BAC01-50 Wielkość zestawu: 50 oznaczeń Objętość
AmpliTest Salmonella spp. (Real Time PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla bakterii z rodzaju Salmonella techniką Real Time PCR Nr kat.: BAC01-50 Wielkość zestawu: 50 oznaczeń Objętość
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Farmacja 2016
 Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Farmacja 2016 Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa farmacjamolekularna@wum.edu.pl
Zakład Biologii Molekularnej Materiały do ćwiczeń z przedmiotu: BIOLOGIA MOLEKULARNA Farmacja 2016 Zakład Biologii Molekularnej Wydział Farmaceutyczny, WUM ul. Banacha 1, 02-097 Warszawa farmacjamolekularna@wum.edu.pl
Gel-Out. 50 izolacji, 250 izolacji. Nr kat , Zestaw do izolacji DNA z żelu agarozowego. wersja 0617
 Gel-Out Zestaw do izolacji DNA z żelu agarozowego. wersja 0617 50 izolacji, 250 izolacji Nr kat. 023-50, 023-250 Pojemność kolumny do izolacji DNA - do 20 µg DNA, minimalna pojemność - 2 µg DNA (przy zawartości
Gel-Out Zestaw do izolacji DNA z żelu agarozowego. wersja 0617 50 izolacji, 250 izolacji Nr kat. 023-50, 023-250 Pojemność kolumny do izolacji DNA - do 20 µg DNA, minimalna pojemność - 2 µg DNA (przy zawartości
PathogenFree DNA Isolation Kit Zestaw do izolacji DNA Instrukcja użytkownika
 PathogenFree DNA Isolation Kit Zestaw do izolacji DNA Instrukcja użytkownika Spis treści 1. Zawartość 2 1.1 Składniki zestawu 2 2. Opis produktu 2 2.1 Założenia metody 2 2.2 Instrukcja 2 2.3 Specyfikacja
PathogenFree DNA Isolation Kit Zestaw do izolacji DNA Instrukcja użytkownika Spis treści 1. Zawartość 2 1.1 Składniki zestawu 2 2. Opis produktu 2 2.1 Założenia metody 2 2.2 Instrukcja 2 2.3 Specyfikacja
Novabeads Food DNA Kit
 Novabeads Food DNA Kit Novabeads Food DNA Kit jest nowej generacji narzędziem w technikach biologii molekularnej, umożliwiającym izolację DNA z produktów spożywczych wysoko przetworzonych. Metoda oparta
Novabeads Food DNA Kit Novabeads Food DNA Kit jest nowej generacji narzędziem w technikach biologii molekularnej, umożliwiającym izolację DNA z produktów spożywczych wysoko przetworzonych. Metoda oparta
Zestaw do wykrywania Anaplasma phagocytophilum w kleszczach, krwi i hodowlach komórkowych
 Nr kat. PK24N Wersja zestawu: 1.2016 Zestaw do wykrywania phagocytophilum w kleszczach, krwi i hodowlach komórkowych dwie oddzielne reakcje PCR 2x50 reakcji PCR (50 µl), włączając w to kontrole Detekcja
Nr kat. PK24N Wersja zestawu: 1.2016 Zestaw do wykrywania phagocytophilum w kleszczach, krwi i hodowlach komórkowych dwie oddzielne reakcje PCR 2x50 reakcji PCR (50 µl), włączając w to kontrole Detekcja
PCR bez izolacji testujemy Direct PCR Kits od ThermoFisher Scientific
 PCR bez izolacji testujemy Direct PCR Kits od ThermoFisher Scientific Specjalnie dla Was przetestowaliśmy w naszym laboratorium odczynniki firmy Thermo Scientific umożliwiające przeprowadzanie reakcji
PCR bez izolacji testujemy Direct PCR Kits od ThermoFisher Scientific Specjalnie dla Was przetestowaliśmy w naszym laboratorium odczynniki firmy Thermo Scientific umożliwiające przeprowadzanie reakcji
AmpliTest GMO screening-nos (Real Time PCR)
") AmpliTest GMO screening-nos (Real Time PCR) Zestaw do wykrywania sekwencji DNA terminatora NOS techniką Real Time PCR Nr kat.: GMO03-50 GMO03-100 Wielkość zestawu: 50 reakcji 100 reakcji Objętość pojedynczej
AmpliTest GMO screening-nos (Real Time PCR) Zestaw do wykrywania sekwencji DNA terminatora NOS techniką Real Time PCR Nr kat.: GMO03-50 GMO03-100 Wielkość zestawu: 50 reakcji 100 reakcji Objętość pojedynczej
CEL ĆWICZENIA: Zapoznanie się z przykładową procedurą odsalania oczyszczanych preparatów enzymatycznych w procesie klasycznej filtracji żelowej.
 LABORATORIUM 3 Filtracja żelowa preparatu oksydazy polifenolowej (PPO) oczyszczanego w procesie wysalania siarczanem amonu z wykorzystaniem złoża Sephadex G-50 CEL ĆWICZENIA: Zapoznanie się z przykładową
LABORATORIUM 3 Filtracja żelowa preparatu oksydazy polifenolowej (PPO) oczyszczanego w procesie wysalania siarczanem amonu z wykorzystaniem złoża Sephadex G-50 CEL ĆWICZENIA: Zapoznanie się z przykładową
Polimeraza Taq (1U/ l) 1-2 U 1 polimeraza Taq jako ostatni składniki mieszaniny końcowa objętość
 1-2 U 1 polimeraza Taq jako ostatni składniki mieszaniny końcowa objętość") Ćwiczenie 6 Technika PCR Celem ćwiczenia jest zastosowanie techniki PCR do amplifikacji fragmentu DNA z bakterii R. leguminosarum bv. trifolii TA1 (RtTA1). Studenci przygotowują reakcję PCR wykorzystując
Ćwiczenie 6 Technika PCR Celem ćwiczenia jest zastosowanie techniki PCR do amplifikacji fragmentu DNA z bakterii R. leguminosarum bv. trifolii TA1 (RtTA1). Studenci przygotowują reakcję PCR wykorzystując
AmpliTest Chlamydia/Chlamydophila (Real Time PCR)
") AmpliTest Chlamydia/Chlamydophila (Real Time PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla bakterii z rodzajów Chlamydia i Chlamydophila techniką Real Time PCR Nr kat.: BAC18-50 BAC18-100 Wielkość
AmpliTest Chlamydia/Chlamydophila (Real Time PCR) Zestaw do wykrywania sekwencji DNA specyficznych dla bakterii z rodzajów Chlamydia i Chlamydophila techniką Real Time PCR Nr kat.: BAC18-50 BAC18-100 Wielkość
Odczynnik do izolacji RNA z tkanek zwierzęcych, roślinnych oraz linii komórkowych
 Nr kat.: EM30-100, EM30-200 Wersja zestawu: 1.2015 Odczynnik do izolacji RNA z tkanek zwierzęcych, roślinnych oraz linii komórkowych EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. I. PRZEZNACZENIE
Nr kat.: EM30-100, EM30-200 Wersja zestawu: 1.2015 Odczynnik do izolacji RNA z tkanek zwierzęcych, roślinnych oraz linii komórkowych EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. I. PRZEZNACZENIE
fix RNA Roztwór do przechowywania i ochrony przed degradacją próbek przeznaczonych do izolacji RNA kat. nr. E0280 Sierpień 2018
 Sierpień 2018 fix RNA Roztwór do przechowywania i ochrony przed degradacją próbek przeznaczonych do izolacji RNA kat. nr. E0280 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow 3, NIP 957-07-05-191 KRS
Sierpień 2018 fix RNA Roztwór do przechowywania i ochrony przed degradacją próbek przeznaczonych do izolacji RNA kat. nr. E0280 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow 3, NIP 957-07-05-191 KRS
Syngen Gel/PCR Mini Kit
 Syngen Gel/PCR Mini Kit Syngen Gel/PCR ME Mini Kit Zestawy do oczyszczania DNA: - z żelu agarozowego - po reakcji PCR i innych reakcjach enzymatycznych - z żelu agarozowego z małą objętością elucji - po
Syngen Gel/PCR Mini Kit Syngen Gel/PCR ME Mini Kit Zestawy do oczyszczania DNA: - z żelu agarozowego - po reakcji PCR i innych reakcjach enzymatycznych - z żelu agarozowego z małą objętością elucji - po
Instrukcje do ćwiczeń oraz zakres materiału realizowanego na wykładach z przedmiotu Inżynieria bioprocesowa na kierunku biotechnologia
 1 Zakład Mikrobiologii UJK Instrukcje do ćwiczeń oraz zakres materiału realizowanego na wykładach z przedmiotu Inżynieria bioprocesowa na kierunku biotechnologia 2 Zakład Mikrobiologii UJK Zakres materiału
1 Zakład Mikrobiologii UJK Instrukcje do ćwiczeń oraz zakres materiału realizowanego na wykładach z przedmiotu Inżynieria bioprocesowa na kierunku biotechnologia 2 Zakład Mikrobiologii UJK Zakres materiału
Instrukcja ćwiczeń Biologia molekularna dla II roku Analityki Medycznej. Reakcja odwrotnej transkrypcji. Projektowanie starterów do reakcji PCR.
 INSTRUKCJA Ćwiczenie nr 5 Odczynniki: Sprzęt: 1. 5xNG cdna Buffer 2. 50 µm oligo(dt)20 3. NG dart RT mix l 4. Probówki typu Eppendorf 0,2 ml 5. Woda wolna od RNaz 6. Lód 1. Miniwirówka 2. Termocykler 3.
INSTRUKCJA Ćwiczenie nr 5 Odczynniki: Sprzęt: 1. 5xNG cdna Buffer 2. 50 µm oligo(dt)20 3. NG dart RT mix l 4. Probówki typu Eppendorf 0,2 ml 5. Woda wolna od RNaz 6. Lód 1. Miniwirówka 2. Termocykler 3.
Zestaw do izolacji DNA z żeli agarozowych. Nr kat. EM08 Wersja zestawu:
 Zestaw do izolacji DNA z żeli agarozowych Nr kat. EM08 Wersja zestawu: 1.2014 www.dnagdansk.com 2 Nr kat. EM08 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA GEL OUT przeznaczony jest do szybkiej i wydajnej
Zestaw do izolacji DNA z żeli agarozowych Nr kat. EM08 Wersja zestawu: 1.2014 www.dnagdansk.com 2 Nr kat. EM08 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA GEL OUT przeznaczony jest do szybkiej i wydajnej
Klonowanie molekularne Kurs doskonalący. Zakład Geriatrii i Gerontologii CMKP
 Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Ćwiczenie 5 Klonowanie DNA w wektorach plazmidowych
 Ćwiczenie 5 Klonowanie DNA w wektorach plazmidowych Celem ćwiczenia jest sklonowanie fragmentu DNA (powielonego techniką PCR i wyizolowanego z żelu agarozowego na poprzednich zajęciach) do wektora plazmidowego
Ćwiczenie 5 Klonowanie DNA w wektorach plazmidowych Celem ćwiczenia jest sklonowanie fragmentu DNA (powielonego techniką PCR i wyizolowanego z żelu agarozowego na poprzednich zajęciach) do wektora plazmidowego
Zestawy do izolacji DNA i RNA
 Syngen kolumienki.pl Gotowe zestawy do izolacji i oczyszczania kwasów nukleinowych Zestawy do izolacji DNA i RNA Katalog produktów 2012-13 kolumienki.pl Syngen Biotech Sp. z o.o., 54-116 Wrocław, ul. Ostródzka
Syngen kolumienki.pl Gotowe zestawy do izolacji i oczyszczania kwasów nukleinowych Zestawy do izolacji DNA i RNA Katalog produktów 2012-13 kolumienki.pl Syngen Biotech Sp. z o.o., 54-116 Wrocław, ul. Ostródzka
Zestaw do oczyszczania DNA po reakcjach enzymatycznych. Nr kat. EM03 Wersja zestawu:
 Zestaw do oczyszczania DNA po reakcjach enzymatycznych Nr kat. EM03 Wersja zestawu: 1.2012 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA CLEAN-UP przeznaczony jest do szybkiego i wydajnego oczyszczania
Zestaw do oczyszczania DNA po reakcjach enzymatycznych Nr kat. EM03 Wersja zestawu: 1.2012 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA CLEAN-UP przeznaczony jest do szybkiego i wydajnego oczyszczania
Zestaw do izolacji DNA z żeli agarozowych. Nr kat. EM08 Wersja zestawu:
 Zestaw do izolacji DNA z żeli agarozowych Nr kat. EM08 Wersja zestawu: 1.2012 www.dnagdansk.com Nr kat. EM08 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA GEL OUT przeznaczony jest do szybkiej i wydajnej
Zestaw do izolacji DNA z żeli agarozowych Nr kat. EM08 Wersja zestawu: 1.2012 www.dnagdansk.com Nr kat. EM08 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME DNA GEL OUT przeznaczony jest do szybkiej i wydajnej
Biologia Molekularna ĆWICZENIE I PREPARATYKA RNA
 Biologia Molekularna ĆWICZENIE I PREPARATYKA RNA ĆWICZENIE I (RNA) W komórkach występują trzy główne rodzaje RNA: mrna, trna, rrna. Największą pulę RNA stanowi rrna kodujące podjednostki rybosomów (u Eukariota
Biologia Molekularna ĆWICZENIE I PREPARATYKA RNA ĆWICZENIE I (RNA) W komórkach występują trzy główne rodzaje RNA: mrna, trna, rrna. Największą pulę RNA stanowi rrna kodujące podjednostki rybosomów (u Eukariota
Spis treści. Aparatura
 Spis treści Aparatura I. Podstawowe wyposażenie laboratoryjne... 13 I.I. Probówki i naczynia laboratoryjne... 13 I.II. Pipety... 17 I.II.I. Rodzaje pipet automatycznych... 17 I.II.II. Techniki pipetowania...
Spis treści Aparatura I. Podstawowe wyposażenie laboratoryjne... 13 I.I. Probówki i naczynia laboratoryjne... 13 I.II. Pipety... 17 I.II.I. Rodzaje pipet automatycznych... 17 I.II.II. Techniki pipetowania...
Izolowanie DNA i RNA z komórek hodowlanych. Ocena jakości uzyskanego materiału
 II ROK KIERUNEK WETERYNARIA UJCM INSTRUKCJA DO ĆWICZEŃ LABORATORYJNYCH VIII Izolowanie DNA i RNA z komórek hodowlanych. Ocena jakości uzyskanego materiału 2013/2014 2 ĆWICZENIE VIII Preparatyka całkowitego
II ROK KIERUNEK WETERYNARIA UJCM INSTRUKCJA DO ĆWICZEŃ LABORATORYJNYCH VIII Izolowanie DNA i RNA z komórek hodowlanych. Ocena jakości uzyskanego materiału 2013/2014 2 ĆWICZENIE VIII Preparatyka całkowitego
GeneMATRIX Agarose-Out DNA Purification Kit
 Wersja zestawu 7.1 Kwiecień 2019 GeneMATRIX Agarose-Out DNA Purification Kit Uniwersalny zestaw do izolacji DNA z żeli agarozowych kat. nr. E3540 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow 3, NIP
Wersja zestawu 7.1 Kwiecień 2019 GeneMATRIX Agarose-Out DNA Purification Kit Uniwersalny zestaw do izolacji DNA z żeli agarozowych kat. nr. E3540 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow 3, NIP
Total RNA Zol-Out. 25 izolacji, 100 izolacji
 Total RNA Zol-Out Zestaw do szybkiej izolacji ultraczystego, całkowitego RNA z odczynników opartych na mieszaninie fenolu oraz rodanku lub chlorowodorku guanidyny (TRIzol, TRI Reagent, RNAzol, QIAzol,
Total RNA Zol-Out Zestaw do szybkiej izolacji ultraczystego, całkowitego RNA z odczynników opartych na mieszaninie fenolu oraz rodanku lub chlorowodorku guanidyny (TRIzol, TRI Reagent, RNAzol, QIAzol,
Zestaw przeznaczony jest do całkowitej izolacji RNA z bakterii, drożdży, hodowli komórkowych, tkanek oraz krwi świeżej (nie mrożonej).
.") Total RNA Mini Plus Zestaw do izolacji całkowitego RNA. Procedura izolacji nie wymaga użycia chloroformu wersja 0517 25 izolacji, 100 izolacji Nr kat. 036-25, 036-100 Zestaw przeznaczony jest do całkowitej
Total RNA Mini Plus Zestaw do izolacji całkowitego RNA. Procedura izolacji nie wymaga użycia chloroformu wersja 0517 25 izolacji, 100 izolacji Nr kat. 036-25, 036-100 Zestaw przeznaczony jest do całkowitej
Zestaw do oczyszczania DNA po reakcjach enzymatycznych
 Cat. No. EM07.1 Wersja: 1.2017 NOWA WERSJA Zestaw do oczyszczania DNA po reakcjach enzymatycznych EXTRACTME jest zarejestrowanym znakiem towarowym BLIRT S.A. www.blirt.eu Nr kat. EM07.1 I. PRZEZNACZENIE
Cat. No. EM07.1 Wersja: 1.2017 NOWA WERSJA Zestaw do oczyszczania DNA po reakcjach enzymatycznych EXTRACTME jest zarejestrowanym znakiem towarowym BLIRT S.A. www.blirt.eu Nr kat. EM07.1 I. PRZEZNACZENIE
SKUTECZNOŚĆ IZOLACJI JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI? JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI?
 SKUTECZNOŚĆ IZOLACJI Wydajność izolacji- ilość otrzymanego kwasu nukleinowego Efektywność izolacji- jakość otrzymanego kwasu nukleinowego w stosunku do ilości Powtarzalność izolacji- zoptymalizowanie procedury
SKUTECZNOŚĆ IZOLACJI Wydajność izolacji- ilość otrzymanego kwasu nukleinowego Efektywność izolacji- jakość otrzymanego kwasu nukleinowego w stosunku do ilości Powtarzalność izolacji- zoptymalizowanie procedury
Ćwiczenie 7 Klonowanie DNA w wektorach plazmidowych
 Ćwiczenie 7 Klonowanie DNA w wektorach plazmidowych Celem ćwiczenia jest sklonowanie fragmentu DNA (powielonego techniką PCR i wyizolowanego z żelu agarozowego na poprzednich zajęciach) do wektora plazmidowego
Ćwiczenie 7 Klonowanie DNA w wektorach plazmidowych Celem ćwiczenia jest sklonowanie fragmentu DNA (powielonego techniką PCR i wyizolowanego z żelu agarozowego na poprzednich zajęciach) do wektora plazmidowego
Inżynieria Genetyczna ćw. 3
 Materiały do ćwiczeń z przedmiotu Genetyka z inżynierią genetyczną D - blok Inżynieria Genetyczna ćw. 3 Instytut Genetyki i Biotechnologii, Wydział Biologii, Uniwersytet Warszawski, rok akad. 2018/2019
Materiały do ćwiczeń z przedmiotu Genetyka z inżynierią genetyczną D - blok Inżynieria Genetyczna ćw. 3 Instytut Genetyki i Biotechnologii, Wydział Biologii, Uniwersytet Warszawski, rok akad. 2018/2019
ĆWICZENIE 3 DGGE- ELEKTROFOREZA W ŻELU Z GRADIENTEM CZYNNIKA DENATURUJĄCEGO
 ĆWICZENIE 3 DGGE- ELEKTROFOREZA W ŻELU Z GRADIENTEM CZYNNIKA DENATURUJĄCEGO CZĘŚĆ TEORETYCZNA Metody badań i cechy w oparciu o które przeprowadzana jest klasyfikacja i identyfikacja mikroorganizmówh Struktura
ĆWICZENIE 3 DGGE- ELEKTROFOREZA W ŻELU Z GRADIENTEM CZYNNIKA DENATURUJĄCEGO CZĘŚĆ TEORETYCZNA Metody badań i cechy w oparciu o które przeprowadzana jest klasyfikacja i identyfikacja mikroorganizmówh Struktura
Genomic Midi AX Direct zestaw do izolacji genomowego DNA (procedura bez precypitacji) wersja 1215
 wersja 1215") Genomic Midi AX Direct zestaw do izolacji genomowego DNA (procedura bez precypitacji) wersja 1215 20 izolacji Nr kat. 895-20D Pojemność kolumny do oczyszczania DNA wynosi 100 µg 1 Skład zestawu Składnik
Genomic Midi AX Direct zestaw do izolacji genomowego DNA (procedura bez precypitacji) wersja 1215 20 izolacji Nr kat. 895-20D Pojemność kolumny do oczyszczania DNA wynosi 100 µg 1 Skład zestawu Składnik
Genomic Maxi AX Direct
 Genomic Maxi AX Direct Uniwersalny zestaw o zwiększonej wydajności do izolacji genomowego DNA z różnych materiałów. Procedura bez etapu precypitacji. wersja 0517 10 izolacji Nr kat. 995-10D Pojemność kolumny
Genomic Maxi AX Direct Uniwersalny zestaw o zwiększonej wydajności do izolacji genomowego DNA z różnych materiałów. Procedura bez etapu precypitacji. wersja 0517 10 izolacji Nr kat. 995-10D Pojemność kolumny
ĆWICZENIE 5 MECHANIZMY PROMUJĄCE WZROST ROŚLIN
 ĆWICZENIE 5 MECHANIZMY PROMUJĄCE WZROST ROŚLIN CZĘŚĆ TEORETYCZNA Mechanizmy promujące wzrost rośli (PGP) Metody badań PGP CZĘŚĆ PRAKTYCZNA 1. Mechanizmy promujące wzrost roślin. Odczyt. a) Wytwarzanie
ĆWICZENIE 5 MECHANIZMY PROMUJĄCE WZROST ROŚLIN CZĘŚĆ TEORETYCZNA Mechanizmy promujące wzrost rośli (PGP) Metody badań PGP CZĘŚĆ PRAKTYCZNA 1. Mechanizmy promujące wzrost roślin. Odczyt. a) Wytwarzanie
PathogenFree RNA Isolation Kit Zestaw do izolacji RNA
 PathogenFree RNA Isolation Kit Zestaw do izolacji RNA Instrukcja użytkownika Spis treści 1. Zawartość 2 1.1 Składniki zestawu 2 2. Opis produktu 2 2.1 Założenia metody 2 2.2 Specyfikacja zestawu 2 2.3
PathogenFree RNA Isolation Kit Zestaw do izolacji RNA Instrukcja użytkownika Spis treści 1. Zawartość 2 1.1 Składniki zestawu 2 2. Opis produktu 2 2.1 Założenia metody 2 2.2 Specyfikacja zestawu 2 2.3
AmpliTest TBEV (Real Time PCR)
") AmpliTest TBEV (Real Time PCR) Zestaw do wykrywania sekwencji RNA specyficznych dla TBEV (Tick-borne encephalitis virus) techniką Real Time PCR Nr kat.: RV03-50 Wielkość zestawu: 50 oznaczeń Objętość pojedynczej
AmpliTest TBEV (Real Time PCR) Zestaw do wykrywania sekwencji RNA specyficznych dla TBEV (Tick-borne encephalitis virus) techniką Real Time PCR Nr kat.: RV03-50 Wielkość zestawu: 50 oznaczeń Objętość pojedynczej
Ćwiczenie numer 6. Analiza próbek spożywczych na obecność markerów GMO
 Ćwiczenie numer 6 Analiza próbek spożywczych na obecność markerów GMO 1. Informacje wstępne -screening GMO -metoda CTAB -qpcr 2. Izolacja DNA z soi metodą CTAB 3. Oznaczenie ilościowe i jakościowe DNA
Ćwiczenie numer 6 Analiza próbek spożywczych na obecność markerów GMO 1. Informacje wstępne -screening GMO -metoda CTAB -qpcr 2. Izolacja DNA z soi metodą CTAB 3. Oznaczenie ilościowe i jakościowe DNA
Zestaw do oczyszczania DNA po reakcjach enzymatycznych
 Nr kat. EM07 Wersja zestawu: 1.2014 Zestaw do oczyszczania DNA po reakcjach enzymatycznych EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. www.dnagdansk.com Nr kat. EM07 I. PRZEZNACZENIE
Nr kat. EM07 Wersja zestawu: 1.2014 Zestaw do oczyszczania DNA po reakcjach enzymatycznych EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. www.dnagdansk.com Nr kat. EM07 I. PRZEZNACZENIE
Genomic Midi AX. 20 izolacji
 Genomic Midi AX Uniwersalny zestaw o zwiększonej wydajności do izolacji genomowego DNA z różnych materiałów. Procedura z precypitacją DNA. wersja 0517 20 izolacji Nr kat. 895-20 Pojemność kolumny do oczyszczania
Genomic Midi AX Uniwersalny zestaw o zwiększonej wydajności do izolacji genomowego DNA z różnych materiałów. Procedura z precypitacją DNA. wersja 0517 20 izolacji Nr kat. 895-20 Pojemność kolumny do oczyszczania
bi ~ Zestaw dydal<tyczny umożliwiający przeprowadzenie izolacji genomowego DNA z bakterii EasyLation Genomie DNA INSTRUI<CJA DLA STUDENTÓW
 Zestaw dydaktyczny EasyLation Genomie DNA Nr kat. DY80 Wersja zestawu: 1.2014 Zestaw dydal
Zestaw dydaktyczny EasyLation Genomie DNA Nr kat. DY80 Wersja zestawu: 1.2014 Zestaw dydal
JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI? JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI?
 Podstawowe miary masy i objętości stosowane przy oznaczaniu ilości kwasów nukleinowych : 1g (1) 1l (1) 1mg (1g x 10-3 ) 1ml (1l x 10-3 ) 1μg (1g x 10-6 ) 1μl (1l x 10-6 ) 1ng (1g x 10-9 ) 1pg (1g x 10-12
Podstawowe miary masy i objętości stosowane przy oznaczaniu ilości kwasów nukleinowych : 1g (1) 1l (1) 1mg (1g x 10-3 ) 1ml (1l x 10-3 ) 1μg (1g x 10-6 ) 1μl (1l x 10-6 ) 1ng (1g x 10-9 ) 1pg (1g x 10-12
Techniki immunoenzymatycznego oznaczania poziomu hormonów (EIA)
") Techniki immunoenzymatycznego oznaczania poziomu hormonów (EIA) Wstęp: Test ELISA (ang. Enzyme-Linked Immunosorbent Assay), czyli test immunoenzymatyczny (ang. Enzyme Immunoassay - EIA) jest obecnie szeroko
Techniki immunoenzymatycznego oznaczania poziomu hormonów (EIA) Wstęp: Test ELISA (ang. Enzyme-Linked Immunosorbent Assay), czyli test immunoenzymatyczny (ang. Enzyme Immunoassay - EIA) jest obecnie szeroko
EKSTRAHOWANIE KWASÓW NUKLEINOWYCH JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI? JAK ZMIERZYĆ ILOŚĆ KWASÓW NUKLEINOWYCH PO IZOLACJI?
 EKSTRAHOWANIE KWASÓW NUKLEINOWYCH Wytrącanie etanolem Rozpuszczenie kwasu nukleinowego w fazie wodnej (met. fenol/chloroform) Wiązanie ze złożem krzemionkowym za pomocą substancji chaotropowych: jodek
EKSTRAHOWANIE KWASÓW NUKLEINOWYCH Wytrącanie etanolem Rozpuszczenie kwasu nukleinowego w fazie wodnej (met. fenol/chloroform) Wiązanie ze złożem krzemionkowym za pomocą substancji chaotropowych: jodek
Genomic Maxi AX zestaw do izolacji genomowego DNA wersja 0616
 Genomic Maxi AX zestaw do izolacji genomowego DNA wersja 0616 10 izolacji Nr kat. 995-10 Pojemność kolumny do oczyszczania DNA wynosi 500 µg 1 Skład zestawu Składnik Ilość Temp. Przechowywania Kolumny
Genomic Maxi AX zestaw do izolacji genomowego DNA wersja 0616 10 izolacji Nr kat. 995-10 Pojemność kolumny do oczyszczania DNA wynosi 500 µg 1 Skład zestawu Składnik Ilość Temp. Przechowywania Kolumny
ĆWICZENIE 5 Barwniki roślinne. Ekstrakcja barwników asymilacyjnych. Rozpuszczalność chlorofilu
 ĆWICZENIE 5 Barwniki roślinne Ekstrakcja barwników asymilacyjnych 400 mg - zhomogenizowany w ciekłym azocie proszek z natki pietruszki 6 ml - etanol 96% 2x probówki plastikowe typu Falcon na 15 ml 5x probówki
ĆWICZENIE 5 Barwniki roślinne Ekstrakcja barwników asymilacyjnych 400 mg - zhomogenizowany w ciekłym azocie proszek z natki pietruszki 6 ml - etanol 96% 2x probówki plastikowe typu Falcon na 15 ml 5x probówki
Syngen Viral Mini Kit. Syngen Viral
 Syngen Viral Mini Kit Syngen Viral Mini Kit PLUS Zestawy do izolacji materiału genetycznego wirusów (DNA i RNA) z próbek: - osocza - surowicy - płynów ustrojowych pozbawionych komórek - pożywki znad hodowli
Syngen Viral Mini Kit Syngen Viral Mini Kit PLUS Zestawy do izolacji materiału genetycznego wirusów (DNA i RNA) z próbek: - osocza - surowicy - płynów ustrojowych pozbawionych komórek - pożywki znad hodowli
Biochemia: Ćw. 11 Metoda RT-PCR
 Ćwiczenie 11 METODA RT-PCR Wyciąg z kart charakterystyki substancji niebezpiecznych: bromek etydyny T+ EDTA Xi etanol, 96% F kwas octowy, 96% C -merkaptoetanol N, T Tris Xi UWAGI WSTĘPNE Praca z kwasami
Ćwiczenie 11 METODA RT-PCR Wyciąg z kart charakterystyki substancji niebezpiecznych: bromek etydyny T+ EDTA Xi etanol, 96% F kwas octowy, 96% C -merkaptoetanol N, T Tris Xi UWAGI WSTĘPNE Praca z kwasami
Zestaw do izolacji plazmidowego DNA
 Nr kat. EM01 Wersja zestawu: 1.2014 Zestaw do izolacji plazmidowego DNA EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. www.dnagdansk.com Nr kat. EM01 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME
Nr kat. EM01 Wersja zestawu: 1.2014 Zestaw do izolacji plazmidowego DNA EXTRACTME jest zastrzeżonym znakiem towarowym firmy BLIRT S.A. www.dnagdansk.com Nr kat. EM01 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME
Genomic Mini AX Bacteria+ Spin
 Genomic Mini AX Bacteria+ Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z bakterii Gram-dodatnich. wersja 1017 100 izolacji Nr kat. 060-100MS Pojemność kolumny do oczyszczania DNA wynosi
Genomic Mini AX Bacteria+ Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z bakterii Gram-dodatnich. wersja 1017 100 izolacji Nr kat. 060-100MS Pojemność kolumny do oczyszczania DNA wynosi
Genomic Micro AX Blood Gravity 96-well
 Genomic Micro AX Blood Gravity 96-well Zestaw do izolacji DNA z krwi w formacie płytek na 96 studzienek dedykowany do pracy z pipetą wielokanałową lub automatem typu liquid handler. 960 izolacji Nr kat.
Genomic Micro AX Blood Gravity 96-well Zestaw do izolacji DNA z krwi w formacie płytek na 96 studzienek dedykowany do pracy z pipetą wielokanałową lub automatem typu liquid handler. 960 izolacji Nr kat.
ĆWICZENIE II. PRZYGOTOWANIE ŻELU POLIAKRYLAMIDOWEGO DO ELEKTROFOREZY BIAŁEK W WARUNKACH DENATURUJĄCYCH (SDS-PAGE)
") ĆWICZENIE II. ELEKTROFOREZA W ŻELU POLIAKRYLAMIDOWYM (ANG. POLYACRYLAMIDE GEL ELECTROPHORESIS - PAGE) PRZYGOTOWANIE ŻELU POLIAKRYLAMIDOWEGO DO ELEKTROFOREZY BIAŁEK W WARUNKACH DENATURUJĄCYCH (SDS-PAGE)
ĆWICZENIE II. ELEKTROFOREZA W ŻELU POLIAKRYLAMIDOWYM (ANG. POLYACRYLAMIDE GEL ELECTROPHORESIS - PAGE) PRZYGOTOWANIE ŻELU POLIAKRYLAMIDOWEGO DO ELEKTROFOREZY BIAŁEK W WARUNKACH DENATURUJĄCYCH (SDS-PAGE)
SPECYFIKACJA TECHNICZNA AGAROZ
 SPECYFIKACJA TECHNICZNA AGAROZ D1 LOW EEO i D1 MEDIUM EEO, D1 HIGH EEO D1 LOW EEO GQT; (Genetic Quality Tested) D2 HIGH GELLING TEMPERATURE D5 HIGH STRENGTH GEL Agaroza LM Agaroza LM GQT; (Genetic Quality
SPECYFIKACJA TECHNICZNA AGAROZ D1 LOW EEO i D1 MEDIUM EEO, D1 HIGH EEO D1 LOW EEO GQT; (Genetic Quality Tested) D2 HIGH GELLING TEMPERATURE D5 HIGH STRENGTH GEL Agaroza LM Agaroza LM GQT; (Genetic Quality
ĆWICZENIE 1 i 2 Modyfikacja geu wołowej beta-laktoglobuliny przy użyciu metody Overlap Extension PCR (wydłużania nakładających się odcinków)
") ĆWICZENIE 1 i 2 Modyfikacja geu wołowej beta-laktoglobuliny przy użyciu metody Overlap Extension PCR (wydłużania nakładających się odcinków) Celem ćwiczenia jest wprowadzenie mutacji punktowej do genu
ĆWICZENIE 1 i 2 Modyfikacja geu wołowej beta-laktoglobuliny przy użyciu metody Overlap Extension PCR (wydłużania nakładających się odcinków) Celem ćwiczenia jest wprowadzenie mutacji punktowej do genu
Elektroforeza kwasów nukleinowych
 Elektroforeza kwasów nukleinowych Elektroforeza jest podstawową techniką wizualizacji kwasów nukleinowych pozwala bezpośrednio identyfikować cząsteczki DNA ze stosunkowo wysoką czułością (po odpowiednim
Elektroforeza kwasów nukleinowych Elektroforeza jest podstawową techniką wizualizacji kwasów nukleinowych pozwala bezpośrednio identyfikować cząsteczki DNA ze stosunkowo wysoką czułością (po odpowiednim
Laboratorium 5. Wpływ temperatury na aktywność enzymów. Inaktywacja termiczna
 Laboratorium 5 Wpływ temperatury na aktywność enzymów. Inaktywacja termiczna Prowadzący: dr inż. Karolina Labus 1. CZĘŚĆ TEORETYCZNA Szybkość reakcji enzymatycznej zależy przede wszystkim od stężenia substratu
Laboratorium 5 Wpływ temperatury na aktywność enzymów. Inaktywacja termiczna Prowadzący: dr inż. Karolina Labus 1. CZĘŚĆ TEORETYCZNA Szybkość reakcji enzymatycznej zależy przede wszystkim od stężenia substratu
Na tej pracowni wyjątkowo odpowiedzi na zadania należy udzielić na osobnym arkuszu odpowiedzi.
 Pracownia biochemiczna arkusz zadań Czas: 90 min. Łączna liczba punktów do zdobycia: 30 Na tej pracowni wyjątkowo odpowiedzi na zadania należy udzielić na osobnym arkuszu odpowiedzi. Drodzy uczestnicy!
Pracownia biochemiczna arkusz zadań Czas: 90 min. Łączna liczba punktów do zdobycia: 30 Na tej pracowni wyjątkowo odpowiedzi na zadania należy udzielić na osobnym arkuszu odpowiedzi. Drodzy uczestnicy!
Ćwiczenie 1. Izolacja genomowego DNA różnymi metodami
 Ćwiczenie 1 Izolacja genomowego DNA różnymi metodami Praca laboratoryjna z użyciem materiału biologicznego wymaga szczególnej dbałości o prawidłowe wykonanie każdego etapu pracy, dlatego też tylko zastosowanie
Ćwiczenie 1 Izolacja genomowego DNA różnymi metodami Praca laboratoryjna z użyciem materiału biologicznego wymaga szczególnej dbałości o prawidłowe wykonanie każdego etapu pracy, dlatego też tylko zastosowanie
PROJEKT WSPÓŁFINANSOWANY PRZEZ UNIĘ EUROPEJSKĄ Z EUROPEJSKIEGO FUNDUSZU ROZWOJU REGIONALNEGO 1 z 7
 Poznań, dnia 28.04.2014 r. BioVentures Institute Spółka z ograniczoną odpowiedzialnością ul. Promienista 83 60 141 Poznań Zapytanie ofertowe nr 01/2014 Projekt Nowa technologia wytwarzania szczepionek
Poznań, dnia 28.04.2014 r. BioVentures Institute Spółka z ograniczoną odpowiedzialnością ul. Promienista 83 60 141 Poznań Zapytanie ofertowe nr 01/2014 Projekt Nowa technologia wytwarzania szczepionek
Syngen Fungi DNA Mini Kit
 Syngen Fungi DNA Mini Kit Zestaw do izolacji całkowitego DNA z próbek: - kultur tkankowych grzybów - kultur tkankowych drożdży Instrukcja dla użytkownika, w. 2016.1 Instrukcja dla użytkownika strona 1
Syngen Fungi DNA Mini Kit Zestaw do izolacji całkowitego DNA z próbek: - kultur tkankowych grzybów - kultur tkankowych drożdży Instrukcja dla użytkownika, w. 2016.1 Instrukcja dla użytkownika strona 1
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów:
 Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Plan ćwiczeń wykonywanych w ramach Pracowni Biologii Molekularnej, część II Ekspresja i oczyszczanie białek.
 Pracownia Biologii Molekularnej Część II Plan ćwiczeń wykonywanych w ramach Pracowni Biologii Molekularnej, część II Ekspresja i oczyszczanie białek. Spis treści 1 I spotkanie: Ekspresja białka w komórkach
Pracownia Biologii Molekularnej Część II Plan ćwiczeń wykonywanych w ramach Pracowni Biologii Molekularnej, część II Ekspresja i oczyszczanie białek. Spis treści 1 I spotkanie: Ekspresja białka w komórkach
Adres strony internetowej, na której Zamawiający udostępnia Specyfikację Istotnych Warunków Zamówienia: www.imp.sosnowiec.pl
 Strona 1 z 7 Adres strony internetowej, na której Zamawiający udostępnia Specyfikację Istotnych Warunków Zamówienia: www.imp.sosnowiec.pl Sosnowiec: Sukcesywna dostawa odczynników dla Instytutu Medycyny
Strona 1 z 7 Adres strony internetowej, na której Zamawiający udostępnia Specyfikację Istotnych Warunków Zamówienia: www.imp.sosnowiec.pl Sosnowiec: Sukcesywna dostawa odczynników dla Instytutu Medycyny
PARAMETRY TECHNICZNE I ILOŚCIOWE PRZEDMIOTU NINIEJSZEGO POSTĘPOWANIA
 Znak sprawy: 13/G/2016 Załącznik nr 2 PARAMETRY TECHNICZNE I ILOŚCIOWE PRZEDMIOTU NINIEJSZEGO POSTĘPOWANIA 1. [APARAT DO AUTOMATYCZNEJ IZOLACJI KWASÓW NUKLEINOWYCH] MagCore HF16Plus szt.1 / NIE 1 Automatyczna
Znak sprawy: 13/G/2016 Załącznik nr 2 PARAMETRY TECHNICZNE I ILOŚCIOWE PRZEDMIOTU NINIEJSZEGO POSTĘPOWANIA 1. [APARAT DO AUTOMATYCZNEJ IZOLACJI KWASÓW NUKLEINOWYCH] MagCore HF16Plus szt.1 / NIE 1 Automatyczna
GeneMATRIX Cell Culture DNA Purification Kit
 Wersja zestawu 3.3 Kwiecień 2019 GeneMATRIX Cell Culture DNA Purification Kit Zestaw do izolacji DNA z kultur komórek ludzkich i zwierzęcych kat. nr. E3555 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow
Wersja zestawu 3.3 Kwiecień 2019 GeneMATRIX Cell Culture DNA Purification Kit Zestaw do izolacji DNA z kultur komórek ludzkich i zwierzęcych kat. nr. E3555 EURx Ltd. 80-297 Gdansk Poland ul. Przyrodnikow
Elektroforeza kwasów nukleinowych
 Elektroforeza kwasów nukleinowych Elektroforeza jest podstawową techniką wizualizacji kwasów nukleinowych pozwala bezpośrednio identyfikować cząsteczki DNA i jest stosunkowo czuła (po odpowiednim wybarwieniu
Elektroforeza kwasów nukleinowych Elektroforeza jest podstawową techniką wizualizacji kwasów nukleinowych pozwala bezpośrednio identyfikować cząsteczki DNA i jest stosunkowo czuła (po odpowiednim wybarwieniu
data ĆWICZENIE 12 BIOCHEMIA MOCZU Doświadczenie 1
 Imię i nazwisko Uzyskane punkty Nr albumu data /3 podpis asystenta ĆWICZENIE 12 BIOCHEMIA MOCZU Doświadczenie 1 Cel: Wyznaczanie klirensu endogennej kreatyniny. Miarą zdolności nerek do usuwania i wydalania
Imię i nazwisko Uzyskane punkty Nr albumu data /3 podpis asystenta ĆWICZENIE 12 BIOCHEMIA MOCZU Doświadczenie 1 Cel: Wyznaczanie klirensu endogennej kreatyniny. Miarą zdolności nerek do usuwania i wydalania
Badanie próbek żywności na obecność Genetycznie Zmodyfikowanych Organizmów. Rozdział 9
 Badanie próbek żywności na obecność Genetycznie Zmodyfikowanych Organizmów Rozdział 9 Wykrywanie jakościowe kukurydzy MON810, kukurydzy Bt-176 i soi Roundup Ready metodą PCR M. Querci, M. Maretti, M. Mazzara
Badanie próbek żywności na obecność Genetycznie Zmodyfikowanych Organizmów Rozdział 9 Wykrywanie jakościowe kukurydzy MON810, kukurydzy Bt-176 i soi Roundup Ready metodą PCR M. Querci, M. Maretti, M. Mazzara
Genetyczne modyfikowanie organizmów Kierunek OCHRONA ŚRODOWISKA, II rok semestr letni 2015/16
 Genetyczne modyfikowanie organizmów Kierunek OCHRONA ŚRODOWISKA, II rok semestr letni 2015/16 Ćwiczenie 3 Identyfikacja genetycznie modyfikowanych roślin w produktach spożywczych - jakościowe badanie obecności
Genetyczne modyfikowanie organizmów Kierunek OCHRONA ŚRODOWISKA, II rok semestr letni 2015/16 Ćwiczenie 3 Identyfikacja genetycznie modyfikowanych roślin w produktach spożywczych - jakościowe badanie obecności
Zestaw do izolacji plazmidowego DNA. Nr kat. EM01 Wersja zestawu:
 Zestaw do izolacji plazmidowego DNA Nr kat. EM01 Wersja zestawu: 1.2012 www.dnagdansk.com Nr kat. EM01 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME PLASMID DNA przeznaczony jest do szybkiej i wydajnej izolacji
Zestaw do izolacji plazmidowego DNA Nr kat. EM01 Wersja zestawu: 1.2012 www.dnagdansk.com Nr kat. EM01 I. PRZEZNACZENIE ZESTAWU Zestaw EXTRACTME PLASMID DNA przeznaczony jest do szybkiej i wydajnej izolacji
Genomic Mini AX Milk Spin
 Genomic Mini AX Milk Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z próbek mleka. wersja 1017 100 izolacji Nr kat. 059-100S Pojemność kolumny do oczyszczania DNA wynosi 15 μg. Produkt
Genomic Mini AX Milk Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z próbek mleka. wersja 1017 100 izolacji Nr kat. 059-100S Pojemność kolumny do oczyszczania DNA wynosi 15 μg. Produkt
Syngen Plasmid Mini Kit
 Syngen Plasmid Mini Kit Syngen Plasmid Screening Kit Zestawy do izolacji DNA: - plazmidowego z hodowli bakteryjnej - plazmidów niksokopijnych Instrukcja dla użytkownika, w. 2016.1 Instrukcja dla użytkownika
Syngen Plasmid Mini Kit Syngen Plasmid Screening Kit Zestawy do izolacji DNA: - plazmidowego z hodowli bakteryjnej - plazmidów niksokopijnych Instrukcja dla użytkownika, w. 2016.1 Instrukcja dla użytkownika
ĆWICZENIE I - BIAŁKA. Celem ćwiczenia jest zapoznanie się z właściwościami fizykochemicznymi białek i ich reakcjami charakterystycznymi.
 ĆWICZENIE I - BIAŁKA Celem ćwiczenia jest zapoznanie się z właściwościami fizykochemicznymi białek i ich reakcjami charakterystycznymi. Odczynniki: - wodny 1% roztwór siarczanu(vi) miedzi(ii), - 10% wodny
ĆWICZENIE I - BIAŁKA Celem ćwiczenia jest zapoznanie się z właściwościami fizykochemicznymi białek i ich reakcjami charakterystycznymi. Odczynniki: - wodny 1% roztwór siarczanu(vi) miedzi(ii), - 10% wodny
Uniwersalny zestaw do izolacji DNA (GeDI)
") Wersja zestawu 1.0 Listopad 2017 Uniwersalny zestaw do izolacji DNA (GeDI) Uniwersalny odczynnik do izolacji całkowitego DNA (GeDI) w zestawie z kolumienkami krzemionkowymi oraz buforami pozwalającymi
Wersja zestawu 1.0 Listopad 2017 Uniwersalny zestaw do izolacji DNA (GeDI) Uniwersalny odczynnik do izolacji całkowitego DNA (GeDI) w zestawie z kolumienkami krzemionkowymi oraz buforami pozwalającymi
Technika radioimmunologicznego oznaczania poziomu hormonów (RIA) dr Katarzyna Knapczyk-Stwora
 dr Katarzyna Knapczyk-Stwora") Technika radioimmunologicznego oznaczania poziomu hormonów (RIA) dr Katarzyna Knapczyk-Stwora Warunki wstępne: Proszę zapoznać się z tematem Radioimmunologiczne metody oznaczania hormonów steroidowych
Technika radioimmunologicznego oznaczania poziomu hormonów (RIA) dr Katarzyna Knapczyk-Stwora Warunki wstępne: Proszę zapoznać się z tematem Radioimmunologiczne metody oznaczania hormonów steroidowych
Genomic Mini AX Plant Spin
 Genomic Mini AX Plant Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z materiału roślinnego. wersja 1017 100 izolacji Nr kat. 050-100S Pojemność kolumny do oczyszczania DNA wynosi 15 μg.
Genomic Mini AX Plant Spin Zestaw o zwiększonej wydajności do izolacji genomowego DNA z materiału roślinnego. wersja 1017 100 izolacji Nr kat. 050-100S Pojemność kolumny do oczyszczania DNA wynosi 15 μg.