(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
|
|
|
- Kacper Bednarski
- 5 lat temu
- Przeglądów:
Transkrypt
1 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: (97) O udzieleniu patentu europejskiego ogłoszono: Europejski Biuletyn Patentowy 2010/16 EP B1 (51) Int. Cl. C07K16/46 A61K39/395 A61P35/00 C12N15/13 C07K16/28 C12N1/21 ( ) ( ) ( ) ( ) ( ) ( ) (54) Tytuł wynalazku: Humanizowani antagoniści anty c-met (30) Pierwszeństwo: US P (43) Zgłoszenie ogłoszono: Europejski Biuletyn Patentowy 2007/16 (45) O złożeniu tłumaczenia patentu ogłoszono: Wiadomości Urzędu Patentowego 09/2010 (73) Uprawniony z patentu: Genentech, Inc., South San Francisco, US (72) Twórca (y) wynalazku: PL/EP T3 DENNIS Mark S., San Carlos, US BILLECI Karen, San Bruno, US YOUNG Judy, San Carlos, US ZHENG Zhong, Foster City, US (74) Pełnomocnik: Jan Wierzchoń&Partnerzy Biuro Patentów i Znaków Towarowych Sp.j. rzecz. pat. Twardowska Aleksandra Warszawa skr. poczt. 709 Uwaga: W ciągu dziewięciu miesięcy od publikacji informacji o udzieleniu patentu europejskiego, każda osoba może wnieść do Europejskiego Urzędu Patentowego sprzeciw dotyczący udzielonego patentu europejskiego. Sprzeciw wnosi się w formie uzasadnionego na piśmie oświadczenia. Uważa się go za wniesiony dopiero z chwilą wniesienia opłaty za sprzeciw (Art. 99 (1) Konwencji o udzielaniu patentów europejskich).
2 11079/10 EP B1 HUMANIZOWANI ANTAGONIŚCI ANTY C-MET Opis DZIEDZINA WYNALAZKU [0001] Niniejszy wynalazek dotyczy w ogólnym ujęciu dziedziny biologii molekularnej i regulacji czynników wzrostu. W dokładniejszym ujęciu, wynalazek dotyczy modulatorów szlaku sygnałowego HGF/c-met i zastosowań tych modulatorów. STAN TECHNIKI [0002] HGF jest mezenchymalnym czynnikiem plejotropowym posiadającym działanie mitogenne, motogenne i morfogenne w różnych rodzajach komórek. W działaniu HGF pośredniczy specyficzna kinaza tyrozynowa, c-met, a zaburzoną ekspresję HGF i c-met często obserwuje się w różnych guzach nowotworowych. Zob. np. Maulik et al., Cytokine & Growth Factor Reviews (2002), 13:41-59; Danilkovitch-Miagkova & Zbar, J. Clin. Invest. (2002), 109(7): Regulacja szlaku sygnałowego HGF/c-Met uczestniczy w progresji i powstawaniu przerzutów nowotworu. Zob. np. Trusolino & Comoglio, Nature Rev. (2002), 2: [0003] HGF wiąże się z zewnątrzkomórkową domeną receptorowej kinazy tyrozynowej (RTK) Met, regulując różne procesy biologiczne, takie jak rozpraszanie, proliferację i przeżycie komórek. Szlak sygnałowy HGF-Met jest kluczowy w prawidłowym rozwoju embrionalnym, zwłaszcza w migracji komórek progenitorowych mięśni oraz rozwoju wątroby i układu nerwowego (Bladt et al., 1995; Hamanoue et al., 1996; Maina et al., 1996; Schmidt et al., 1995; Uehara et al., 1995). Fenotypy rozwojowe myszy pozbawionych genów kodujących Met i HGF są podobne, co wskazuje, że HGF jest ligandem skojarzonym z receptorem Met (Schmidt et al., 1995; Uehara et al., 1995). Szlak HGF-Met odgrywa również rolę w procesach regeneracji wątroby, angiogenezy i gojenia ran (Bussolino et al., 1992; Matsumoto i Nakamura, 1993; Nusrat et al., 1994). Prekursor receptora Met ulega proteolizie z wytworzeniem zewnątrzkomórkowej podjednostki α i przezmembranowej podjednostki β, połączonych wiązaniami disiarczkowymi (Tempest et al., 1988). Podjednostka β zawiera cytoplazmatyczną domenę kinazy i posiada przy C-końcu wielosubstratowe miejsce wiążące, z którym wiążą się białka adaptorowe, inicjując kaskadę sygnałową (Bardelli et al., 1997; Nguyen et al., 1997; Pelicci et al., 1995; Ponzetto et al., 1994; Weidner et al., 1996). Aktywacja Met po związaniu HGF prowadzi do fosforylacji tyrozyny i dalszego przekazania sygnału poprzez aktywację PI3-kinazy i Ras/MAPK odpowiednio za pośrednictwem Gab1 i Grb2/Sos, któru napędza ruchliwość i proliferację komórek (Furge et al., 2000; Hartmann et al., 1994; Ponzetto et al., 1996; Royal i Park, 1995). [0004] Stwierdzono transformację Met w traktowanych środkiem karcynogennym komórkach z linii osteosarcoma (Cooper et al., 1984; Park et al., 1986). W wielu ludzkich nowotworach stwierdzono nadekspresję Met lub amplifikację odpowiedniego genu. Na przykład w raku odbytu i jelita grubego białko Met ulega przynajmniej pięciokrotnie zwiększonej ekspresji; ponadto, doniesiono o amplifikacji genu w przerzutach do wątroby (Di Renzo et al., 1995; Liu et al., 1992). Donoszono również o nadekspresji białka Met w płaskokomórkowych nowotworach jamy ustnej, nowotworach wątrobowokomórkowych, nowotworach nerkowokomórkowych, nowotworach piersi i płuca (Jin et al., 1997; Morello et al., 2001; Natali et al., 1996; Olivero et al., 1996; Suzuki et al., 1994). Ponadto w nowotworach wątrobowokomórkowych, nowotworach żołądka oraz nowotworach odbytu i jelita grubego stwierdzano nadekspresję mrna (Boix et al., 1994; Kuniyasu et al., 1993; Liu et al., 1992). [0005] W przypadku brodawczaka nerki stwierdzono istnienie pewnej liczby mutacji w kinazowej domenie Met, prowadzących do konstytutywnej aktywacji receptora (Olivero et al., 1999; Schmidt et al., 1997; Schmidt et al., 1999). Tego rodzaju aktywujące mutacje prowadzą do konstytutywnej fosforylacji reszt tyrozynowych przez Met, prowadząc do aktywacji MAPK, powstawania ognisk nowotworowych i tumorogenezy (Jeffers et al., 1997). Ponadto mutacje tego rodzaju zwiększają ruchliwość i inwazyjność komórek (Giordano et al., 2000; Lorenzato et al., 2002). HGF-zależna aktywacja Met w transformowanych komórkach pośredniczy w zwiększeniu ruchliwości, rozpraszania i migracji komórek, co w końcu prowadzi do inwazyjnego wzrostu nowotworu i przerzutów (Jeffers et al., 1996; Meiners et al., 1998). [0006] Wykazano, że Met oddziałuje z innymi białkami napędzającymi aktywację receptora, transformację komórek i inwazję nowotworu. Doniesiono, że w komórkach nowotworowych Met oddziałuje z integryną α6β4, receptorem dla składników macierzy zewnątrzkomórkowej, takich jak
3 2 lamininy, promując w ten sposób HGF-zależny inwazyjny wzrost nowotworu (Trusolino et al., 2001). Ponadto wykazano, że zewnątrzkomórkowa domena Met oddziałuje ze związkiem z rodziny semaforyn pleksyną B1 i nasila inwazyjny wzrost nowotworu (Giordano et al., 2002). Ponadto istnieją doniesienia o tworzeniu kompleksów z Met i HGF przez CD44v6, uczestniczący w tumorogenezie i powstawaniu przerzutów. Oddziaływanie to prowadzi do aktywacji receptora Met (Orian-Rousseau et al., 2002). [0007] Met jest członkiem podrodziny RTK obejmującej receptory Ron i Sea (Maulik et al., 2002). Przewidywana struktura domeny zewnątrzkomórkowej wskazuje na istnienie sekwencji homologicznych z semaforynami i pleksynami. N-koniec białka Met zawiera domenę Sema, złożoną z około 500 aminokwasów, zakonserwowaną we wszystkich semaforynach i pleksynach. Semaforyny i pleksyny należą do dużej grupy białek wydzielanych i wiązanych przez błony komórkowe opisanych po raz pierwszy w związku z ich rolą w rozwoju układu nerwowego (Van Vactor i Lorenz, 1999). W ostatnim czasie jednak nadekspresję semaforyny skorelowano jednak również z inwazją i przerzutami nowotworu. Bogata w reszty cysteinowe domena PSI (nazywana również domeną MRS [Met Related Sequence]), występująca w pleksynach, semaforynach i integrynach leży w sąsiedztwie domeny Sema, po której następują cztery powtórzenia IPT, będące immunoglobulinopodobnymi regionami występującymi w pleksynach i czynnikach transkrypcyjnych. Wyniki ostatnich badań wskazują, że domena Sema białka Met wystarcza do wiązania HGF i heparyny (Gherardi et al., 2003). Ponadto, Kong-Beltran et al. (Cancer Cell (2004), 6:61-73) donieśli, że domena Sema białka Met jest konieczna do dimeryzacji i aktywacji receptora. [0008] Opisano liczne cząsteczki docelowe szlaku sygnałowego HGF/c-met. Cząsteczki te obejmują przeciwciała takie, jak opisane w patencie nr US 5,686,292. Wykazano również, że antagonistyczne działanie względem szlaku HGF/c-met może mieć również część zewnątrzkomórkowej domeny Met. W świetle ważnej roli, jaką omawiany szlak odgrywa w etiologii różnych schorzeń oczywiste jest jednak ciągłe zapotrzebowanie na środki o optymalnych właściwościach klinicznych, umozliwiający ich rozwój w charakterze środków terapeutycznych. Wynalazek opisany w niniejszym zgłoszeniu spełnia to zapotrzebowanie, zapewniając jednocześnie dodatkowe korzyści. UJAWNIENIE WYNALAZKU [0009] Wynalazek częściowo opiera się na wskazaniu szeregu antagonistów szlaku biologicznego HGF/c-met, będącego procesem biologicznym/komórkowym, który przedstawia się jako ważny i korzystny docelowy obiekt terapii. Wynalazek ujawnia kompozycje i zastosowania medyczne opisane w zastrzeżeniach, opartych na zakłócaniu aktywacji HGF/c-met, między innymi poprzez zakłócanie wiązania HGF z zewnątrzkomórkową częścią c-met i multimeryzacji receptora. Antagoniści będący przedmiotem wynalazku dostarczają ważnych środków terapeutycznych i diagnostycznych do stosowania w leczeniu kierowanym na schorzenia związane z nieprawidłowym lub niepożądanym przekazywaniem sygnałów na szlaku HGF/c-met. Zgodnie z powyższym, niniejszy wynalazek ujawnia metody i zastosowania medyczne opisane w zastrzeżeniach i dotyczy zestawów i wyrobów związanych z regulacją szlaku HGF/c-met, w tym z modulacją wiązania liganda przez c-met, dimeryzacją i aktywacją c-met oraz innymi czynnościami biologicznymi/fizjologicznymi związanymi z sygnałowaniem na szlaku HGF/c-met. [0010] W jednym z aspektów wynalazek ujawnia środki lecznicze przeciwko HGF/c-met odpowiednich do zastosowań terapeutycznych i zdolnych do różnych stopni zakłócania szlaku sygnałowego HGF/cmet. W szczególności, wynalazek ujawnia humanizowane przeciwciało anty c-met w którym monowalencyjne powinowactwo przeciwciała do ludzkiego c-met (np. powinowactwo przeciwciała w postaci fragmentu Fab do ludzkiego c-met) jest większe, na przykład 3-, 5-, 7-, 10- lub 13-krotnie większe niż monowalencyjne powinowactwo przeciwciała mysiego (np. powinowactwo przeciwciała mysiego w postaci fragmentu Fab do ludzkiego c-met) zawierające, składające się lub zasadniczo składające się ze zmiennych domen łańcucha lekkiego i ciężkiego, przedstawionych na Fig. 7 (SEQ ID NO: 9 i 10), zgodnie z opisem w zastrzeżeniach. Zgodnie z wiedzą panującą w dziedzinie wynalazku, powinowactwo wiązania liganda do receptora można oznaczyć w szeregu testów i wyrazić poprzez szereg wartości ilościowych. Zgodnie z powyższym, w jednym z wariantów wynalazku, powinowactwo wiązania wyrażane jest wartościami Kd i oddaje samoistne powinowactwo wiązania (np. ze zminimalizowanymi efektami zachłanności). Z reguły i korzystnie powinowactwo wiązania mierzy się in vitro, czy to w warunkach pozakomórkowych, czy też komórkowych. Jak to zostało bardziej szczegółowo opisane w niniejszym zgłoszeniu, wielokrotna różnica w powinowactwie wiązania może być opisana ilościowo przy użyciu stosunku wartości powinowactwa monowalencyjnego wiązania humanizowanego przeciwciała (np. w postaci Fab) i powinowactwa monowalencyjnego wiązania przeciwciała referencyjnego/porównawczego (np. przeciwciała mysiego
4 3 posiadającego donorowe sekwencje regionów hiperzmiennych), przy czym wartości powinowactwa wiązania oznaczane są w podobnych warunkach. Tak więc, w jednym z wariantów wynalazku, wielokrotna różnica w powinowactwie wiązania oznaczana jest jako stosunek wartości Kd dla humanizowanego przeciwciała w postaci Fab i rzeczonego referencyjnego/porównawczego przeciwciała Fab. Na przykład, w jednym z wariantów, jeśli przeciwciało będące przedmiotem wynalazku (C) posiada powinowactwo trzykrotnie większe niż powinowactwo przeciwciała referencyjnego (R), wówczas wartość Kd dla C wynosi 1x, wartość Kd dla R wynosi 3x, zaś stosunek wartości Kd dla C do Kd dla R wynosi 1:3. Do pomiarów powinowactwa wiązania można wykorzystać dowolne z wielu znanych w dziedzinie oznaczeń, w tym opisanych w niniejszym zgłoszeniu, na przykład w oznaczeniu techniką Biacore, teście radioimmunologicznym (RIA) czy teście immunoenzymatycznym (ELISA). [0011] W jednym z aspektów antagonista HGF/c-met będący przedmiotem wynalazku ma postać przeciwciała anty c-met, zawierającego: (a) co najmniej jedną, dwie, trzy, cztery lub pięć sekwencji regionu hiperzmiennego (HVR), wybranych z grupy obejmującej: (i) HVR-L1 zawierający sekwencję A1-A17, gdzie sekwencja A1-A17 ma postać KSSQSLLYTSSQKNYLA (SEQ ID NO: 1) (ii) HVR-L2 zawierający sekwencję B1-B7, gdzie sekwencja B1-B7 ma postać WASTRES (SEQ ID NO: 2) (iii) HVR-L3 zawierający sekwencję C1-C9, gdzie sekwencja C1-C9 ma postać QQYYAYPWT (SEQ ID NO: 3) (iv) HVR-H1 zawierający sekwencję D1-D10, gdzie D1-D10 ma postać GYTFTSYWLH (SEQ ID NO: 4) (v) HVR-H2 zawierający sekwencję E1-E18, gdzie E1-E18 ma postać GMIDPSNSOTRFNPNFKD (SEQ ID NO: 5) a także (vi) wariant HVR-H3 zawierający sekwencję określoną w zastrzeżeniu 1. [0012] W jednym z wariantów region HVR-L1 przeciwciała będącego przedmiotem wynalazku zawiera sekwencję przedstawioną jako SEQ ID NO: 1. W jednym z wariantów region HVR-L3 przeciwciała będącego przedmiotem wynalazku zawiera sekwencję przedstawioną jako SEQ ID NO: 3. Przeciwciało będące przedmiotem wynalazku zawierające te sekwencje (w opisanych tu połączeniach) jest przeciwciałem humanizowanym. [0013] Wariantowe regiony HVR w przeciwciele będącym przedmiotem wynalazku mogą posiadać modyfikacje jednej lub większej liczby reszt w obszarze HVR. W jednym z wariantów wynalazku wariant HVR-L2 zawiera sekwencję jak określono w zastrzeżeniu 1 lub 3. W jednym z wariantów wynalazku wariant HVR-H2 zawiera sekwencję jak określono w zastrzeżeniu 1 lub 4. W niektórych wariantach wynalazku rzeczone przeciwciało zawierające wariant HVR-H3 zawiera ponadto HVR-L1, HVR-L2, HVR-L3, HVR-H1 i HVR-H2, gdzie każdy z wyżej wymienionych regionów zawiera odpowiednio sekwencje SEQ ID NO: 1, 2, 3, 4 i 5. W niektórych wariantach przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha ciężkiego z podgrupy III. W jednym z wariantów rzeczonych przeciwciał konsensusowa sekwencja zrębowa zawiera substytucje aminokwasów w pozycjach 71, 73 i/lub 78. W niektórych wariantach rzeczonych przeciwciał aminokwasem w pozycji 71 jest A, aminokwasem w pozycji 73 jest T i/lub aminokwasem w pozycji 78 jest A. W jednym z wariantów przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha lekkiego z podgrupy κi. [0014] W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera wariant HVR-L2, gdzie aminokwasem B6 jest V. W niektórych wariantach rzeczone przeciwciało zawierające wariant HVR-L2 zawiera ponadto HVR-L1, HVR-L3, HVR-H1 i HVR-H2, gdzie każdy z wyżej wymienionych regionów zawiera odpowiednio sekwencje przedstawione jako SEQ ID NO: 1, 3, 4 i 5 i określone w zastrzeżeniach. W niektórych wariantach przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha ciężkiego z podgrupy III. W jednym z wariantów rzeczonych przeciwciał konsensusowa sekwencja zrębowa zawiera substytucje aminokwasów w pozycjach 71, 73 i/lub 78. W niektórych wariantach rzeczonych przeciwciał aminokwasem w pozycji 71 jest A, aminokwasem w pozycji 73 jest T i/lub aminokwasem w pozycji 78 jest A. W jednym z wariantów przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha lekkiego z podgrupy κi.
5 4 [0015] W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera wariant HVR-H2, gdzie aminokwasem E14 jest T, aminokwasem E15 jest K, a aminokwasem E17 jest E. W niektórych wariantach rzeczone przeciwciało zawierające wariant HVR-H2 zawiera ponadto HVR-L1, HVR-L2, HVR-L3 i HVR-H1, gdzie każdy z wyżej wymienionych regionów zawiera odpowiednio sekwencje przedstawione jako SEQ ID NO: 1, 2, 3 i 4 i określone w zastrzeżeniach. W niektórych wariantach przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha ciężkiego z podgrupy III. W jednym z wariantów rzeczonych przeciwciał konsensusowa sekwencja zrębowa zawiera substytucje aminokwasów w pozycjach 71, 73 i/lub 78. W niektórych wariantach rzeczonych przeciwciał aminokwasem w pozycji 71 jest A, aminokwasem w pozycji 73 jest T i/lub aminokwasem w pozycji 78 jest A. W jednym z wariantów przeciwciała te zawierają ponadto ludzką konsensusową sekwencję zrębową łańcucha lekkiego z podgrupy κi. [0016] Przeciwciało może zawierać jeden, dwa, trzy, cztery, pięć lub wszystkie sekwencje HVR przedstawione na Fig. 2, 3 i/lub 4 (SEQ ID NO: ). [0017] W korzystnym wariancie, środek terapeutyczny przeznaczony do stosowania w organizmie gospodarza nie wywołuje u rzeczonego gospodarza odpowiedzi immunogennej lub wywołuje tę odpowiedź w niewielkim stopniu. Na przykład w niniejszym zgłoszeniu opisuje się humanizowane przeciwciało wywołujące i/lub mające wywoływać u gospodarza odpowiedź w postaci ludzkich przeciwciał przeciwmysich (HAMA) na znacznie niższym poziomie niż przeciwciało zawierające sekwencję przedstawioną jako SEQ ID NO: 9 i 10. W innym przykładzie opisywane jest humanizowane przeciwciało wywołujące i/lub mające wywoływać minimalną lub zerową odpowiedź w postaci ludzkich przeciwciał przeciwmysich (HAMA). W jednym przykładzie przeciwciało wywołuje odpowiedź w postaci ludzkich przeciwciał przeciwmysich na poziomie dopuszczalnym klinicznie lub niższym. [0018] Humanizowane przeciwciało będące przedmiotem wynalazku może zawierać w zmiennej domenie łańcucha ciężkiego i/lub lekkiego jedną lub większą liczbę ludzkich i/lub ludzkich konsensusowych sekwencji regionów niehiperzmiennych (np. zrębowych). W niektórych wariantach w ludzkich i/lub ludzkich konsensusowych sekwencjach regionów niehiperzmiennych występuje jedna lub większa liczba dodatkowych modyfikacji. W jednym z wariantów, zmienna domena łańcucha ciężkiego przeciwciała będącego przedmiotem wynalazku zawiera ludzką konsensusową sekwencję zrębową, która w jednym z wariantów jest konsensusową sekwencją zrębową z podgrupy III. W jednym z wariantów, przeciwciało będące przedmiotem wynalazku zawiera wariantową ludzką konsensusową sekwencję zrębową z podgrupy III, z modyfikacją aminokwasu w co najmniej jednej pozycji. Na przykład, w jednym z wariantów, wariantowa konsensusowa sekwencja zrębowa z podgrupy III może zawierać substytucje w jednej lub większej liczbie pozycji spośród pozycji 71, 73 i/lub 78. W jednym z wariantów wynalazku rzeczone substytucje mają postać R71A, N73T i/lub N78A lub kombinacji wyżej wymienionych. [0019] Zgodnie z wiedzą panującą w dziedzinie i z przedstawionym poniżej opisem, pozycja granica aminokwasu wyznaczającego hiperzmienny region przeciwciała może być zmienna w zależności od kontekstu i różnych definicji znanych w dziedzinie (zgodnie z opisem poniżej). Niektóre pozycje w ramach domeny zmiennej mogą być postrzegane jako hybrydowe pozycje hiperzmienne, które mogą być uznane z wchodzące w skład regionu hiperzmiennego przy określonym zestawie kryteriów lub za znajdujące się poza regionem hiperzmiennym przy innym zestawie kryteriów. Jedna lub większa liczba tego rodzaju pozycji może również znaleźć się w obszarze poszerzonych regionów hiperzmiennych (zgodnie z definicją podaną poniżej). Wynalazek ujawnia przeciwciała zawierające modyfikacje w rzeczonych hybrydowych pozycjach hiperzmiennych. Rzeczone hybrydowe pozycje hiperzmienne mogą obejmować jedną lub większą liczbę pozycji wśród pozycji 26-30, 33-35B, 47-49, 57-65, 93, 94 i 102 w zmiennej domenie łańcucha ciężkiego. Rzeczone hybrydowe pozycje hiperzmienne mogą obejmować jedną lub większą liczbę pozycji wśród pozycji 24-29, 35-36, 46-49, 56 i 97 w zmiennej domenie łańcucha lekkiego. Przeciwciało może zawierać wariantową ludzką konsensusową sekwencję zrębową z określonej podgrupy zmodyfikowaną w jednej lub większej liczbie hybrydowych pozycji hiperzmiennych. Przeciwciało może zawierać zmienną domenę łańcucha ciężkiego zawierającą wariantową ludzką konsensusową sekwencję zrębową z podgrupy III zmodyfikowaną w jednej lub większej liczbie pozycji 27-28, 30, 33-35, 49, 57-65, 94 i 102. W jednym z przykładów przeciwciało zawiera substytucję F27Y. Przeciwciało może zawierać substytucje T28N, P, L, S, A lub I, S30I, T lub Y, A33W, M34L lub M34V, S35H,T57I, Y58R, Y59F, A60N, D61P, T lub Q, S62N, D, K, T lub V, V63F lub V63L, K64E, H, N, D lub Q, G65D, Y, E lub H, R94T lub R94S lub Y102Q, S, H lub F. W jednym z przykładów, przeciwciało zawierające rzeczoną modyfikację R94T lub R94S zawiera ponadto jedną lub więcej modyfikacji w pozycjach 96 i/lub 100. W jednym z przypadków
6 5 rzeczone modyfikacje obejmują substytucje G96R i/lub S100T (np. w HVR-H3). Przeciwciało może zawierać zmienną domenę łańcucha lekkiego zawierającą wariantową ludzką konsensusową sekwencję zrębową kappa z podgrupy I zmodyfikowaną w jednej lub większej liczbie pozycji 24, 25, 29 i 56. W jednym z przykładów przeciwciało zawiera substytucję R24K. Przeciwciało może zawierać substytucje A25S, I29Q lub S56R, I, M lub G. [0020] Przeciwciało może zawierać zmienną domenę łańcucha ciężkiego zawierającą wariantową ludzką konsensusową sekwencję zrębową z podgrupy III zmodyfikowaną w 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 lub wszystkich pozycjach spośród pozycji 27-28, 30, 33-35, 49, 57-65, 94 i 102. W jednym z przykładów modyfikacja należy do grupy obejmującej F27Y i T28(N, P, L, S, A lub I), S30(I, T lub Y), A33W, M34(L, V), S35H, T57I, Y58R, Y59F, A60N, D61(P, T, Q), S62(N, D, K, T, V), V63(F, L), K64 (E, H, N, D, Q), G65(D, Y, E, H), R94(T, S) i Y102(Q, S, H, F). W jednym z przykładów, przeciwciało zawierające rzeczoną modyfikację R94T lub R94S zawiera ponadto jedną lub więcej modyfikacji w pozycjach 96 i/lub 100. W jednym z przypadków rzeczone modyfikacje obejmują substytucje G96R i/lub S100T (np. w HVR-H3). [0021] Przeciwciało może zawierać zmienną domenę łańcucha lekkiego zawierającą wariantową ludzką konsensusową sekwencję zrębową kappa z podgrupy I zmodyfikowaną w 1, 2, 3 lub wszystkich pozycjach spośród pozycji 24, 25, 29 i 56. W jednym z przykładów modyfikacja należy do grupy obejmującej R24K, A25S, I29Q i S56(R, I, M, G). [0022] Przeciwciało będące przedmiotem wynalazku może zawierać dowolne odpowiednie ludzkie lub ludzkie konsensusowe sekwencje zrębowe łańcucha lekkiego, pod warunkiem wykazywania pożądanych cech biologicznych (np. pożądanego powinowactwa wiązania). W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera co najmniej część (lub całość) ludzkiej sekwencji zrębowej κ łańcucha lekkiego. W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera co najmniej część (lub całość) ludzkiej sekwencji zrębowej κ z podgrupy I. [0023] W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera zmienną domenę łańcucha ciężkiego i/lub lekkiego, przedstawioną jako SEQ ID NO: 13 i/lub 16 (FIGURA 1), pod warunkiem, że w pozycji 94 łańcucha ciężkiego nie znajduje się R (zaś korzystnie, choć nie koniecznie, S lub T). [0024] W jednym z aspektów przeciwciało będące przedmiotem wynalazku jest humanizowanym przeciwciałem anty c-met, hamującym wiązanie ludzkiego czynnika wzrostu hepatocytów przez właściwy receptor lepiej niż referencyjne przeciwciało w postaci chimerycznego przeciwciała anty c- met zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione na Fig. 7 jako SEQ ID NO: 9 i 10). Na przykład, w jednym z wariantów wynalazku, przeciwciało będące przedmiotem wynalazku hamuje wiązanie receptora z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. W jednym z wariantów wynalazku, wartość IC50 dla przeciwciała będącego przedmiotem wynalazku wynosi około 0,1; 0,2; 0,3 lub 0,4 wartości właściwej dla przeciwciała chimerycznego. Porównanie zdolności hamowania wiązania HGF z receptorem można przeprowadzić przy użyciu różnych znanych w dziedzinie metod, w tym zgodnie z opisem podanym w Przykładach poniżej. W jednym z wariantów wartości IC50 oznaczane są w zakresie stężeń przeciwciała od około 0,01 nm do około 1000 nm. [0025] W jednym z aspektów przeciwciało będące przedmiotem wynalazku jest humanizowanym przeciwciałem anty c-met, hamującym aktywację receptora ludzkiego czynnika wzrostu hepatocytów (HGF) lepiej niż referencyjne przeciwciało w postaci chimerycznego przeciwciała anty c-met zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione na Fig. 7 jako SEQ ID NO: 9 i 10). Na przykład, w jednym z wariantów wynalazku, przeciwciało będące przedmiotem wynalazku hamuje aktywację receptora z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. W jednym z wariantów wynalazku, wartość IC50 dla przeciwciała będącego przedmiotem wynalazku wynosi około 0,1; 0,2; 0,3 lub 0,4 wartości właściwej dla przeciwciała chimerycznego. Porównanie zdolności aktywacji receptora HGF można przeprowadzić przy użyciu różnych znanych w dziedzinie metod, w tym zgodnie z opisem podanym w Przykładach poniżej. W jednym z wariantów wartości IC50 oznaczane są w zakresie stężeń przeciwciała od około 0,1 nm do około 100 nm. [0026] W jednym z aspektów przeciwciało będące przedmiotem wynalazku jest humanizowanym przeciwciałem anty c-met, hamującym c-met zależną proliferacje komórek lepiej niż referencyjne przeciwciało w postaci chimerycznego przeciwciała anty c-met zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione na Fig. 7 (SEQ ID NO: 9 i 10), z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. W jednym z
7 6 wariantów wynalazku, wartość IC50 dla przeciwciała będącego przedmiotem wynalazku wynosi około 0,1; 0,2; 0,3 lub 0,4 wartości właściwej dla przeciwciała chimerycznego. Porównanie zdolności hamowania proliferacji komórek można przeprowadzić przy użyciu różnych znanych w dziedzinie metod, w tym zgodnie z opisem podanym w Przykładach poniżej. W jednym z wariantów wartości IC50 oznaczane są w zakresie stężeń przeciwciała od około 0,01 nm do około 100 nm. [0027] W jednym z wariantów wynalazku zarówno przeciwciało humanizowane, jak i przeciwciało chimeryczne są przeciwciałami monowalencyjnymi. W jednym z wariantów wynalazku zarówno przeciwciało humanizowane, jak i przeciwciało chimeryczne zawierają pojedynczy region Fab połączony z regionem Fc. W jednym z wariantów referencyjne przeciwciało chimeryczne zawiera sekwencje domeny zmiennej przedstawione na Fig. 7 (SEQ ID NO: 9 i 10), połączone z ludzkim regionem Fc. W jednym z wariantów ludzki region Fc jest regionem właściwym dla IgG (np. IgG1, 2, 3 lub 4). [0028] W jednym z aspektów wynalazek dotyczy przeciwciała zawierającego zmienną domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3-HC, przedstawione na Figurze 13. W jednym z wariantów wynalazku domena zmienna zawiera sekwencje FR1-HC, FR2-HC, FR3-HC i FR4-HC, przedstawione na Figurze 13. W jednym z wariantów wynalazku przeciwciało zawiera sekwencje CH1 i/lub Fc, przedstawione na Figurze 13. W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera zmienną domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3-HC oraz sekwencje FR1-HC, FR2-HC, FR3-HC i FR4-HC, przedstawione na Figurze 13. W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera zmienną domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3- HC, a także sekwencje CH1 i/lub Fc, przedstawione na Figurze 13. W jednym z wariantów przeciwciało stanowiące przedmiot wynalazku zawiera zmienna domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3-HC oraz sekwencje FR1-HC, FR2-HC, FR3-HC i FR4-HC, przedstawione na Figurze 13, a także sekwencje CH1 i/lub Fc przedstawione na Figurze 13. W jednym z wariantów region Fc przeciwciała będącego przedmiotem wynalazku stanowi kompleks polipeptydu zawierającego sekwencję Fc przedstawioną na Figurze 13 i polipeptydu zawierającego sekwencję Fc przedstawioną na Figurze 14. [0029] W jednym z aspektów wynalazek dotyczy przeciwciała zawierającego zmienną domenę łańcucha lekkiego, zawierającą sekwencje HVR1-LC, HVR2-LC i HVR3-LC, przedstawione na Figurze 13. W jednym z wariantów wynalazku domena zmienna zawiera sekwencje FR1-LC, FR2-LC, FR3-LC i FR4-LC, przedstawione na Figurze 13. W jednym z wariantów wynalazku przeciwciało zawiera sekwencję CL1, przedstawioną na Figurze 13. [0030] W jednym z wariantów przeciwciało będące przedmiotem wynalazku zawiera zmienne domeny łańcucha lekkiego i ciężkiego zgodne z opisem zamieszczonym w poprzednich dwóch akapitach. W jednym z wariantów wynalazku przeciwciało jest monowalencyjne i zawiera region Fc. W jednym z wariantów wynalazku region Fc zawiera co najmniej jedno wybrzuszenie ( knob ) i co najmniej jedno zagłębienie ( hole ), przy czym obecność wybrzuszenia i zagłębienia wspomaga tworzenie kompleksu pomiędzy polipeptydem Fc zawierającym wybrzuszenie a polipeptydem Fc zawierającym zagłębienie, na przykład zgodnie z opisem w patencie nr WO W jednym z wariantów region Fc przeciwciała będącego przedmiotem wynalazku zawiera pierwszy i drugi polipeptyd Fc, gdzie zarówno pierwszy, jak i drugi polipeptyd zawiera jedną lub większą liczbę mutacji ludzkiej sekwencji Fc dzikiego typu. W jednym z wariantów wynalazku, mutacje zagłębienia obejmują T366S, L368A i/lub Y407V. W jednym z wariantów wynalazku, mutacją wybrzuszenia jest T366W. W jednym z wariantów pierwszy polipeptyd zawiera sekwencję Fc przedstawioną na Figurze 13, zaś drugi polipeptyd zawiera sekwencję Fc przedstawioną na Figurze 14. [0031] Antagoniści będący przedmiotem wynalazku mogą być stosowani w modulowaniu jednego lub większej liczby aspektów związanych z sygnałowaniem HGF/c-met, między innymi z aktywacją c-met, sygnalizacją cząsteczkową w dół szlaku (np. z fosforylacją kinazy białkowej aktywowanej mitogenami [MAPK]), proliferacja komórek, migracją komórek, przeżyciem komórek, morfogenezą komórek i angiogenezą. Efekty te mogą być modulowane poprzez dowolny mający zastosowanie mechanizm biologiczny, w tym poprzez zakłócenie wiązania liganda (np. HGF) z c-met, fosforylacji c-met i/lub multimeryzacji c/met. Zgodnie z powyższym, w jednym z wariantów wynalazek ujawnia przeciwciało antagonistyczne względem c-met hamującego wiązanie HGF z c-met. W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne względem c-met zakłóca multimeryzację (np. dimeryzację) c-met. W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne względem c-met zakłóca dimeryzacyjną aktywność domeny Sema białka c-met. W jednym z przykładów przeciwciało antagonistyczne względem c-met zakłóca zdolność domeny Sema
8 7 białka c-met do dimeryzowania białka c-met. Zakłócanie to może mieć charakter bezpośredni lub pośredni. Na przykład przeciwciało antagonistyczne względem c-met może wiązać się z sekwencją w obrębie domeny Sema białka c-met, tym samym hamując interakcje rzeczonej związanej domeny z właściwym partnerem wiązania (np. z inną cząsteczką c-met). W innym przykładzie, przeciwciało antagonistyczne względem c-met może wiązać się z sekwencją poza obrębem domeny Sema białka c-met, przy czym rzeczone wiązanie hamuje interakcje domeny Sema białka c-met z właściwym partnerem wiązania (np. z inną cząsteczką c-met). W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne wiąże c-met (np. domenę zewnątrzkomórkową) w taki sposób, że zakłócona zostaje dimeryzacja c-met. W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne wiąże c-met w taki sposób, że zakłócona zostaje zdolność domeny Sema białka c-met do wywoływania dimeryzacji c-met. Na przykład, w jednym z wariantów, wynalazek ujawnia antagonistyczne przeciwciało, które wiążąc się z cząsteczkę C-met hamuje dimeryzację rzeczonej cząsteczki. W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne specyficznie wiąże sekwencję w obrębie domeny Sema białka c-met. [0032] W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne względem c-met zakłóca dimeryzację c-met, w tym homodimeryzację c-met. W jednym z wariantów, będące przedmiotem wynalazku przeciwciało antagonistyczne zakłóca dimeryzację c-met, w tym heterodimeryzację c-met (tj. dimeryzację c-met z cząsteczką inną niż c-met). [0033] W niektórych przypadkach korzystne może być posiadanie przeciwciała antagonistycznego względem c-met niezakłócającego wiązania liganda (np. HGF) z c-met. Zgodnie z powyższym opisuje się przeciwciało niewiążące miejsca wiązania HGF w cząsteczce c-met. Przeciwciało nie może zasadniczo hamować wiązania HGF przez c-met lub nie może zasadniczo konkurować z HGF o wiązanie z c-met. W jednym z przykładów, przeciwciało antagonistyczne będące przedmiotem wynalazku może być stosowane w połączeniu z jednym lub większą liczbą antagonistów, gdzie rzeczeni antagoniści są obiektami docelowymi w różnych procesach i/lub funkcjach w osi HGF/c-met. Tak więc, w jednym z wariantów, przeciwciało antagonistyczne względem c-met, będące przedmiotem wynalazku, wiąże epitop cząsteczki c-met inny niż epitop wiązany przez innego antagonistę c-met (na przykład fragment Fab monoklonalnego przeciwciała wytwarzanego przez linię komórek hybrydoma złożoną w kolekcji komórek ATCC pod numerem dostępu HB (hybrydoma 1A3.3.13)). W innym wariancie, przeciwciało antagonistyczne względem c-met, będące przedmiotem wynalazku jest różne od fragmentu Fab monoklonalnego przeciwciała wytwarzanego przez linię komórek hybrydoma złożoną w kolekcji komórek ATCC pod numerem dostępu HB (hybrydoma 1A3.3.13) (tj. nie jest tożsame z ww. fragmentem). [0034] W jednym z wariantów wynalazek ujawnia przeciwciało antagonistyczne względem c-met hamującego zarówno multimeryzację c-met, jak i wiązanie ligandów przez c-met. Na przykład przeciwciało antagonistyczne będące przedmiotem wynalazku i hamujące multimeryzację (np. dimeryzację) c-met może ponadto posiadać zdolność konkurowania z HGF o wiązanie c-met. [0035] W jednym z wariantów będącego przedmiotem wynalazku przeciwciała antagonistycznego względem c-met, wiązanie antagonisty przez c-met hamuje aktywację c-met przez HGF. W innym z wariantów będącego przedmiotem wynalazku przeciwciała antagonistycznego względem c-met, wiązanie antagonisty przez c-met w komórce hamuje proliferację, przeżycie, rozpraszanie, morfogenezę i/lub ruchliwość komórki. [0036] Przeciwciało specyficzne względem c-met może specyficznie wiązać co najmniej część domeny Sema białka c-met lub jej wariantu. W jednym z przykładów antagonistyczne przeciwciało specyficznie wiąże co najmniej jedną z sekwencji wybranych z grupy obejmującej LDAQT (SEQ ID NO: 15) (np. reszty białka c-met), LTEKRKKRS (SEQ ID NO: 16) (np. reszty białka c-met), KPDSAEPM (SEQ ID NO: 17) (np. reszty białka c-met) i NVRCLQHF (SEQ ID NO: 18) (np. reszty białka c-met). W jednym z przykładów antagonistyczne przeciwciało specyficznie wiąże konformacyjny epitop utworzony przez część lub całość co najmniej jednej z sekwencji wybranych z grupy obejmującej LDAQT (np. reszty białka c-met), LTEKRKKRS (np. reszty białka c-met), KPDSAEPM (np. reszty białka c-met) i NVRCLQHF (np. reszty białka c-met). W jednym z przykładów przeciwciało antagonistyczne specyficznie wiąże sekwencje aminokwasów charakteryzującą się sekwencją identyczną lub podobną w co najmniej 50%, 60%, 70%, 80%, 90%, 95%, 98% z sekwencją LDAQT, LTEKRKKRS, KPDSAEPM l/lub NVRCLQHF. [0037] W jednym z wariantów, przeciwciało będące przedmiotem wynalazku specyficznie wiąże receptor HGF pierwszego gatunku zwierzęcego i nie wiąże specyficznie receptora HGF drugiego
9 8 gatunku zwierzęcego. W jednym z wariantów pierwszym gatunkiem zwierzęcym jest człowiek i/lub gatunek z rzędu naczelnych (np. makak), zaś drugim gatunkiem zwierzęcym jest gatunek należący do rodziny myszowatych (np. mysz) i/lub psowatych. W jednym z wariantów pierwszym gatunkiem zwierzęcym jest człowiek. W jednym z wariantów pierwszym gatunkiem zwierzęcym jest gatunek z rzędu naczelnych, na przykład makak. W jednym z wariantów drugim gatunkiem zwierzęcym jest gatunek należy do rodziny myszowatych, np. mysz. W jednym z wariantów drugim gatunkiem zwierzęcym jest gatunek należy do rodziny psowatych, np. pies. [0038] W jednym z aspektów, niniejszy wynalazek ujawnia kompozycje zawierające jedno lub większą liczbę będących przedmiotem wynalazku przeciwciał antagonistycznych oraz substancję nośną. W jednym z wariantów substancja nośna jest dopuszczalna farmaceutycznie. [0039] W jednym z wariantów wynalazek ujawnia kwasy nukleinowe kodujące będące przedmiotem wynalazku przeciwciało antagonistyczne względem c-met. [0040] W jednym z aspektów, wynalazek ujawnia wektory zawierające kwas nukleinowy będący przedmiotem wynalazku. [0041] W jednym z aspektów, wynalazek ujawnia komórki gospodarza zawierające kwas nukleinowy lub wektor będący przedmiotem wynalazku. Wektor może być wektorem dowolnego rodzaju, na przykład wektorem rekombinacyjnym takim, jak wektor ekspresyjny. Możliwe jest zastosowanie dowolnej z wielu komórek gospodarza. W jednym z wariantów komórka gospodarza jest komórką prokariotyczną, na przykład E. coli. W jednym z wariantów komórka gospodarza jest komórką eukariotyczną, komórką ssaczą taką, jak na przykład komórka jajowa chomika chińskiego (CHO). [0042] Opisuje się również metody wytwarzania przeciwciał antagonistycznych będących przedmiotem wynalazku. Na przykład, opisuje się metodę wytwarzania przeciwciała antagonistycznego względem c-met (które zgodnie ze stosowaną tu definicją obejmuje przeciwciało o pełnej długości i jego fragmenty), przy czym rzeczona metoda obejmuje ekspresję będącego przedmiotem wynalazku rekombinowanego wektora kodującego rzeczone przeciwciało (lub jego fragment) w odpowiedniej komórce gospodarza i wyodrębnianie rzeczonego przeciwciała. [0043] Opisuje się również wyrób przemysłowy obejmujący pojemnik oraz kompozycję zawartą w pojemniku, przy czym kompozycja zawiera jedno lub większą liczbę przeciwciał antagonistycznych względem c-met, będących przedmiotem wynalazku. W jednym z przykładów, kompozycja zawiera kwas nukleinowy będący przedmiotem wynalazku. W jednym z przykładów, kompozycja zawierająca przeciwciało antagonistyczne zawiera ponadto substancję nośną, która w niektórych wariantach jest substancją dopuszczalną farmaceutycznie. W jednym z przykładów, będący przedmiotem wynalazku wyrób przemysłowy zawiera ponadto instrukcje podawania kompozycji (np. przeciwciała antagonistycznego) pacjentowi. [0044] Opisuje się również zestaw obejmujący pierwszy pojemnik wyrób przemysłowy obejmujący pojemnik oraz kompozycję zawierającą jedno lub większą liczbę przeciwciał antagonistycznych względem c-met, będących przedmiotem wynalazku oraz drugi pojemnik zawierający bufor. W jednym z przykładów bufor jest dopuszczalny farmaceutycznie. W jednym z przykładów, kompozycja zawierająca przeciwciało antagonistyczne zawiera ponadto substancję nośną, która w niektórych przypadkach jest substancją dopuszczalną farmaceutycznie. W jednym z przykładów, rzeczony zestaw zawiera ponadto instrukcje podawania kompozycji (np. przeciwciała antagonistycznego) pacjentowi. [0045] W jednym z aspektów wynalazek ujawnia wykorzystanie przeciwciała antagonistycznego względem c-met do produkcji substancji leczniczej przeznaczonej do terapeutycznego i/lub profilaktycznego leczenia chorób takich jak rak, nowotwór, choroba proliferacyjna, schorzenie immunologiczne (na przykład autoimmunologiczne) i/lub schorzenie związane z angiogenezą. [0046] W jednym z aspektów wynalazek dotyczy wykorzystania kwasu nukleinowego będącego przedmiotem wynalazku do produkcji substancji leczniczej przeznaczonej do terapeutycznego i/lub profilaktycznego leczenia chorób takich jak rak, nowotwór, choroba proliferacyjna, schorzenie immunologiczne (na przykład autoimmunologiczne) i/lub schorzenie związane z angiogenezą. [0047] W jednym z aspektów wynalazek dotyczy wykorzystania wektora ekspresyjnego będącego przedmiotem wynalazku do produkcji substancji leczniczej przeznaczonej do terapeutycznego i/lub profilaktycznego leczenia chorób takich jak rak, nowotwór, choroba proliferacyjna, schorzenie immunologiczne (na przykład autoimmunologiczne) i/lub schorzenie związane z angiogenezą.
10 9 [0048] W jednym z aspektów wynalazek dotyczy wykorzystania komórki gospodarza będącej przedmiotem wynalazku do produkcji substancji leczniczej przeznaczonej do terapeutycznego i/lub profilaktycznego leczenia chorób takich jak rak, nowotwór, choroba proliferacyjna, schorzenie immunologiczne (na przykład autoimmunologiczne) i/lub schorzenie związane z angiogenezą. [0049] Wynalazek ujawnia zastosowania i kompozycje medyczne użyteczne w modulowaniu stanów chorobowych związanych z deregulacją osi sygnalizacyjnej HGF/c-met. Szlak sygnałowy HGF/c-met uczestniczy w wielu czynnościach biologicznych i fizjologicznych, na przykład w proliferacji komórek i angiogenezie. W związku z tym, w jednym z aspektów niniejszy wynalazek ujawnia medyczne zastosowanie przeciwciała będącego przedmiotem wynalazku w metodzie obejmującej podanie rzeczonego przeciwciała, na przykład metodę hamowania proliferacji komórek aktywowanej przez c-mat, przy czym rzeczona metoda obejmuje zetknięcie komórki lub tkanki ze skuteczną ilością przeciwciała będącego przedmiotem wynalazku, w wyniku czego hamowana jest proliferacja komórek związana z aktywacją c-met, metodę leczenia stanu chorobowego związanego z deregulacją aktywacji c-met u pacjenta, przy czym rzeczona metoda obejmuje podanie pacjentowi skutecznej ilości przeciwciała będącego przedmiotem wynalazku, w wyniku czego następuje leczenie rzeczonego stanu chorobowego, metodę hamowania wzrostu komórki, w której zachodzi ekspresja c-met, czynnika wzrostu hepatocytów lub obu tych związków, przy czym rzeczona metoda obejmuje zetknięcie rzeczonej komórki z przeciwciałem będącym przedmiotem wynalazku, w wyniku czego następuje zahamowanie wzrostu rzeczonych komórek. W jednym z wariantów komórka wchodzi w kontakt z HGF podlegającym ekspresji w innej komórce (np. w mechanizmie parakrynnym), metodę terapeutycznego leczenia ssaka z guzem nowotworowym zawierającym komórkę, w której zachodzi ekspresja c-met, czynnika wzrostu hepatocytów lub obu tych związków, przy czym rzeczona metoda obejmuje podanie rzeczonemu ssakowi skutecznej ilości przeciwciała będącego przedmiotem wynalazku, a tym samym skuteczne leczenie rzeczonego ssaka, gdzie w jednym z przykładów komórka wchodzi w kontakt z HGF podlegającym ekspresji w innej komórce (np. w mechanizmie parakrynnym), metodę leczenia lub zapobiegania chorobie proliferacyjnej związanej z podwyższoną ekspresją lub aktywnością c-met, czynnika wzrostu hepatocytów lub obu tych związków, przy czym rzeczona metoda obejmuje podanie pacjentowi wymagającemu takiego leczenia skutecznej ilości przeciwciała będącego przedmiotem wynalazku, a tym samym skuteczne leczenie rzeczonej choroby proliferacyjnej, gdzie rzeczoną chorobą proliferacyjną może być choroba nowotworowa, metodę hamowania wzrostu komórki, gdzie wzrost rzeczonej komórki przynajmniej w części zależy od wzmacniającego wzrost działania c-met, czynnika wzrostu hepatocytów lub obu tych związków, przy czym rzeczona metoda obejmuje zetknięcie rzeczonej komórki ze skuteczną ilością przeciwciała będącego przedmiotem wynalazku, a tym samym zahamowanie wzrostu rzeczonej komórki, gdzie w jednym z przykładów komórka wchodzi w kontakt z HGF podlegającym ekspresji w innej komórce (np. w mechanizmie parakrynnym), lub metodę terapeutycznego leczenia guza nowotworowego u ssaka, gdzie rzeczony guz nowotworowy jest przynajmniej w części zależy od wzmacniającego wzrost działania c-met, czynnika wzrostu hepatocytów lub obu tych związków, przy czym rzeczona metoda obejmuje zetknięcie rzeczonej komórki ze skuteczną ilością przeciwciała będącego przedmiotem wynalazku, a tym samym zahamowanie wzrostu rzeczonej komórki, gdzie w jednym z przykładów komórka wchodzi w kontakt z HGF podlegającym ekspresji w innej komórce (np. w mechanizmie parakrynnym). [0050] Powyższe metody mogą być stosowane w leczeniu dowolnego odpowiedniego stanu chorobowego, na przykład komórek i/lub tkanek związanych z deregulację szlaku sygnałowego HGF/c-met. W jednym z przykładów docelową komórką jest komórka rakowa. Na przykład, komórka rakowa może należeć do grupy obejmującej komórkę raka piersi, komórkę raka odbytu i jelita grubego, komórkę raka płuca, komórkę brodawczaka (np. tarczycy), komórkę raka jelita grubego, komórkę raka jajnika, komórkę raka szyjki macicy, komórkę raka ośrodkowego układu nerwowego, komórkę mięsaka osteogennego, komórkę raka nerki, komórkę raka wątrobowokomórkowego, komórkę raka pęcherza, komórkę raka żołądka, komórką raka płaskokomórkowego szyi i głowy, komórki czerniaka i komórki białaczki. W jednym z przykładów docelową komórką metody leczniczej jest komórka hiperproliferacyjna i/lub hiperplastyczna. W jednym z przykładów docelową komórką
11 10 metody leczniczej jest komórka dysplastyczna. W jeszcze innym przykładzie docelową komórką metody leczniczej jest komórka metastatyczna. [0051] Tego rodzaju metody mogą ponadto obejmować dodatkowe etapy lecznicze. Na przykład, w jednym z przykładów, metoda obejmuje ponadto etap, w którym komórka i/lub tkanka docelowa (np. komórka rakowa) poddawana jest radioterapii lub chemioterapii. [0052] Zgodnie z niniejszym opisem, aktywacja c-met jest ważnym procesem biologicznym, którego deregulacja prowadzi do licznych stanów chorobowych. I tak, w jednej z przykładowych opisanych tu metod, docelowa komórka (np. komórka rakowa) jest komórką, w której aktywacja c-met jest nasilona w porównaniu z normalną komórką pochodzącą z tej samej tkanki. W jednym z przykładów rzeczona metoda powoduje śmierć docelowej komórki. Na przykład zetknięcie z antagonistą będącym przedmiotem wynalazku może skutkować utratą zdolności komórki do przesyłania sygnałów na szlaku c-met, co prowadzi do śmierci komórki. [0053] Deregulacja aktywacji c-met (a tym samym szlaku sygnałowego) może wynikać z wielu zmian komórkowych, w tym na przykład z nadekspresji HGF (liganda skojarzonego z c-met) i/lub samego c- met. Zgodnie z powyższym, w niektórych przykładach metoda obejmuje działanie skierowane przeciwko komórce, w której c-met, czynnik wzrostu hepatocytów lub oba te związki ulegają w rzeczonej komórce (np. komórce rakowej) liczniejszej ekspresji niż w normalnej komórce pochodzącej z tej samej tkanki. Komórka, w której zachodzi ekspresja c-met może być regulowana przez HGF pochodzący z wielu źródeł, np. w mechanizmie autokrynnym lub parakrynnym. Na przykład, docelowa komórka styka się/jest wiązana przez czynnik wzrostu hepatocytów ulegający ekspresji w innej komórce (np. w mechanizmie parakrynnym). Rzeczona inna komórka może być komórką pochodzącą z tej samej lub z innej tkanki. W jednym z przykładów docelowa komórka styka się/jest wiązana przez HGF ulegający ekspresji w tej samej komórce (np. w mechanizmie autokrynnym/pętli autokrynnej). Aktywacja c-met i/lub przekazanie sygnału może również zachodzić niezależnie od liganda. I tak, w jednym z przykładów, aktywacja c-met w docelowej komórce zachodzi niezależnie od liganda. SKRÓCONY OPIS RYSUNKÓW [0054] FIG. 1 przedstawia ułożenie następujących sekwencji w ramach zmiennych domen łańcucha lekkiego i ciężkiego dla: ludzka konsensusowa sekwencja zrębowa łańcucha lekkiego z podgrupy I, ludzka konsensusowa sekwencja zrębowa łańcucha ciężkiego z podgrupy III, mysie przeciwciało 5D5 anty c- met i humanizowane przeciwciało z wszczepionym przeciwciałem 5D5. FIG. 2 przedstawia różne sekwencje HVR wybranych przeciwciał o dojrzałym powinowactwie, pochodzących z bibliotek przeciwciał o poszczególnie różnicowanych regionach HVR. FIG. 3 przedstawia sekwencje HVR-H3 wybranych przeciwciał o dojrzałym powinowactwie, pochodzących z puli złożonej z 6 bibliotek obejmujących wszystkie sześć regionów HVR, gdzie każda z bibliotek zawiera przeciwciała różniące się w ramach pojedynczego HVR. FIG. 4 przedstawia wyniki analizy wybranych przeciwciał anty c-met techniką Biacore. FIG. 5A, B oraz 6A, B przedstawiają przykładowe ludzkie akceptorowe konsensusowe sekwencje zrębowe przeznaczone do stosowania w niniejszym wynalazku z następującymi numerami identyfikacyjnymi sekwencji: Konsensusowe sekwencje zrębowe zmiennej domeny łańcucha ciężkiego (VH) (FIG. 5A, B) ludzka konsensusowa sekwencja zrębowa VH z podgrupy I minus CDR Kabata (SEQ ID NO: 19) ludzka konsensusowa sekwencja zrębowa VH z podgrupy I minus rozszerzone regiony hiperzmienne (SEQ ID NO: 20-22) ludzka konsensusowa sekwencja zrębowa VH z podgrupy II minus CDR Kabata (SEQ ID NO: 23) ludzka konsensusowa sekwencja zrębowa VH z podgrupy II minus rozszerzone regiony hiperzmienne (SEQ ID NO: 24-26) ludzka konsensusowa sekwencja zrębowa VH z podgrupy III minus CDR Kabata (SEQ ID NO: 27) ludzka konsensusowa sekwencja zrębowa VH z podgrupy III minus rozszerzone regiony hiperzmienne (SEQ ID NO: 28-30) ludzka akceptorowa sekwencja zrębowa VH minus CDR Kabata (SEQ ID NO: 31)
12 11 ludzka akceptorowa sekwencja zrębowa VH minus rozszerzone regiony hiperzmienne (SEQ ID NO: 32-33) ludzka akceptorowa sekwencja zrębowa VH nr 2 minus CDR Kabata (SEQ ID NO: 34) ludzka akceptorowa sekwencja zrębowa VH nr 2 minus rozszerzone regiony hiperzmienne (SEQ ID NO: 35-37) Konsensusowe sekwencje zrębowe zmiennej domeny łańcucha lekkiego (VL) (FIG. 6A, B) ludzka konsensusowa sekwencja zrębowa VL kappa z podgrupy I (SEQ ID NO: 38) ludzka konsensusowa sekwencja zrębowa VL kappa z podgrupy II (SEQ ID NO: 39) ludzka konsensusowa sekwencja zrębowa VL kappa z podgrupy III (SEQ ID NO: 40) ludzka konsensusowa sekwencja zrębowa VL kappa z podgrupy IV (SEQ ID NO: 41) FIG. 7 przedstawia donorowe (pochodzące z mysiego przeciwciała 5D5) sekwencje zmiennych domen łańcucha lekkiego i ciężkiego. FIG. 8 przedstawia graficznie dane dotyczące blokowania wiązania HGF z receptorem przez przeciwciało będące przedmiotem wynalazku. FIG. 9 przedstawia graficznie dane dotyczące hamowania aktywacji receptora HGF przez przeciwciało będące przedmiotem wynalazku. FIG. 10 przedstawia graficznie dane dotyczące hamowania proliferacji komórek przez przeciwciało będące przedmiotem wynalazku. rchoa5d5 HGF oznacza chimeryczne jednoramienne przeciwciało 5D5 plus HGF; rhuoa5d5v2 HGF oznacza OA5D5.v2 plus HGF; rhuoa5d5v1 HGF oznacza OA5D5.v1 plus HGF. rchoa5d5 Control oznacza chimeryczne jednoramienne przeciwciało 5D5 bez HGF; rhuoa5d5v2 Control oznacza OASD5.v2 bez HGF; rhuoa5d5v1 Control oznacza OA5D5.v1 bez HGF. FIG. 11A, B przedstawia dane dotyczące hamowania fosforylacji receptora w obecności przeciwciała będącego przedmiotem wynalazku. FIG. 11A przedstawia fosforylację receptora w komórkach H358. FIG. 11B przedstawia fosforylację receptora w komórkach H358 transfekowanych HGF. FIG. 12 przedstawia graficznie dane dotyczące skuteczności przeciwciała będącego przedmiotem wynalazku w warunkach in vivo. TI oznacza zachorowalność na nowotwór. TI=8/10 oznacza wystąpienie nowotworu u 8 na 10 myszy. TI=2/8 oznacza wystąpienie nowotworu u 2 na 8 myszy. FIG. 13 przedstawia kolejność aminokwasów w sekwencji zrębowej (FR), regionie hiperzmiennym (HVR), pierwszej domenie stałej (CL lub CH1) i regionie Fc (Fc) w ramach jednego z wariantów przeciwciała będącego przedmiotem wynalazku (5D5.v2). Przedstawiona sekwencja Fc zawiera mutacje tworzące zagłębienie ( hole ) T366S, L368A i Y407V, zgodnie z opisem w patencie nr WO 2005/ FIG. 14 przedstawia sekwencję polipeptydu Fc zawierającą mutację tworzącą wybrzuszenie ( knob ) T366W, zgodnie z opisem w patencie nr WO 2005/ W jednym z wariantów wynalazku polipeptyd Fc zawierający tę sekwencję tworzy kompleks z polipeptydem Fc zawierającym sekwencję Fc przedstawiona n a Fig. 13 z wytworzeniem regionu Fc przeciwciała będącego przedmiotem wynalazku. TRYBY REALIZACJI WYNALAZKU [0055] Wynalazek ujawnia zastosowania i kompozycje medyczne; opisuje się również zestawy i wyroby przemysłowe przeznaczone do identyfikacji i/lub wykorzystania inhibitorów szlaku sygnałowego HGF/c-met. [0056] Niniejszym przedstawia się szczegółowe informacje na temat rzeczonych metod, kompozycji, zestawów i wyrobów przemysłowych. Techniki ogólne [0057] Praktyka niniejszego wynalazku wykorzystuje o ile nie zaznaczono inaczej konwencjonalne techniki biologii molekularnej (w tym techniki rekombinacyjne), mikrobiologii, biologii komórkowej, biochemii i immunologii, znane osobom wyszkolonym w dziedzinie wynalazku. Techniki takie są dokładnie opisane w literaturze, np. w pozycjach Molecular Cloning: A Laboratory Manual, wydanie drugie (Sambrook et al., 1989); Oligonucleotide Synthesis (M. J. Gait, ed., 1984); Animal Cell
13 12 Culture (R. I. Freshney (red.), 1987); Methods in Enzymology (Academic Press, Inc.); Current Protocols in Molecular Biology (F. M. Ausubel et al. (red.), 1987, z okresowymi aktualizacjami); PCR: The Polymerase Chain Reaction, (Mullis et al. (red.), 1994); A Practical Guide to Molecular Cloning (Perbal Bernard V., 1988); Phage Display: A Laboratory Manual" (Barbas et al., 2001). Definicje [0058] Modyfikacja reszty/pozycji aminokwasowej w rozumieniu niniejszego opisu oznacza zmianę pierwszorzędowej sekwencji aminokwasów w porównaniu z wyjściową sekwencją aminokwasów, gdzie rzeczona zmiana wynika z modyfikacji sekwencji w rzeczonej reszcie/pozycji aminokwasowej. Na przykład, typowe modyfikacje obejmują substytucję reszty (lub w rzeczonej pozycji) innym aminokwasem (np. substytucje konserwatywną lub niekonserwatywną), insercję jednego lub większej liczby (z reguły mniejszej niż 5 lub 3) aminokwasów w sąsiedztwie rzeczonej reszty/pozycji oraz delecję rzeczonej reszty/pozycji. Termin substytucja aminokwasu lub jego wariacje dotyczą zamiany istniejącej reszty aminokwasowej w określonej z góry (wyjściowej) sekwencji aminokwasów przez inna resztę aminokwasową. Z reguły i korzystnie modyfikacja prowadzi do zmiany co najmniej jednej cechy fizykobiochemicznej wariantowego polipeptydu w porównaniu z polipeptydem zawierającym wyjściową ( dzikiego typu ) sekwencje wyjściową. Na przykład, w przypadku przeciwciał cechą fizykobiochemiczną ulegającą zmienia może być powinowactwo wiązania, zdolność wiązania i/lub wpływ wiązania na cząsteczkę docelową. [0059] Przeciwciało izolowane to przeciwciało zidentyfikowane i oddzielone i/lub wyodrębnione z elementów jego naturalnego środowiska. Zanieczyszczającymi elementami środowiska przeciwciała są substancje mogące zakłócać diagnostyczne lub terapeutyczne zastosowania przeciwciała, które to substancje mogą obejmować enzymy, hormony oraz inne soluty białkowe lub niebiałkowe. W korzystnych wariantach przeciwciało jest oczyszczone (1) do zawartości ponad 95% wagowych przeciwciała, oznaczonej metoda Lowry ego, najkorzystniej ponad 99% wagowych, (2) do stopnia wystarczającego na uzyskanie co najmniej 15 reszt N-końcowej lub wewnętrznej sekwencji aminokwasów przy użyciu aparatu do sekwencjonowania z naczynkiem lub (3) do homogeniczności oznaczonej metodą SDS-PAGE w warunkach nieredukujących z barwieniem metodą Coomassie lub korzystnie barwieniem srebrowym. Izolowane przeciwciało obejmuje przeciwciało in situ w komórkach rekombinowanych, ponieważ nie będzie w nich obecny co najmniej jeden element naturalnego środowiska przeciwciała. Zazwyczaj jednak izolowane przeciwciało będzie uzyskiwane w co najmniej jednym etapie oczyszczania. [0060] Termin numeracja reszt domeny zmiennej wg Kabata lub numeracja pozycji aminokwasowych wg Kabata oraz wariacje tych terminów dotyczą systemu numeracji stosowanego dla zmiennych domen łańcucha ciężkiego lub zmiennych domen łańcucha lekkiego kompilowanych przeciwciał zawartego w pracy Kabat et al., Sequences of Proteins of Immunological Interest, wyd. 5, Public Health Service, National Institutes of Health, Bethesda, MD, USA. (1991). W oparciu o rzeczony system numerowania faktyczna liniowa sekwencja aminokwasów może zawierać mniejszą liczbę aminokwasów lub dodatkowe aminokwasy, co odpowiada skróceniu lub insercji w regionie FR lub CDR zmiennej domeny. Na przykład, zmienna domena łańcucha ciężkiego może zawierać pojedynczy insert aminokwasowy (reszta 52a w systemie Kabata) za resztą 52 w H2 oraz insertowane reszty (np. reszty 82a, 82b, 82c itd. w systemie Kabata) za resztą 82 w regionie FR łańcucha ciężkiego. Numerację reszt wg Kabata można dla danego przeciwciała ustalić poprzez zestawienie regionów charakteryzujących się homologicznością sekwencji przeciwciała ze standardową, ponumerowaną, sekwencją Kabata. [0061] Sformułowanie zasadniczo podobny lub zasadniczo taki sam w rozumieniu niniejszego opisu oznacza stopień podobieństwa pomiędzy dwiema wartościami numerycznymi (zazwyczaj jedną związana z przeciwciałem będącym przedmiotem wynalazku, a drugą związaną z przeciwciałem referencyjnym/porównawczym) wystarczająco duży, by osoba biegła w dziedzinie uznała różnicę pomiędzy obiema wartościami za posiadającą niewielkie bądź żadne znaczenie biologiczne i/lub statystyczne w kontekście cech biologicznych mierzonych rzeczonym i wartościami (np. wartościami Kd). Różnica pomiędzy rzeczonymi dwiema wartościami wynosi korzystnie mniej niż około 50%, korzystnie mniej niż około 40%, korzystnie mniej niż około 30%, korzystnie mniej niż około 20%, korzystnie mniej niż około 10% wartości dla przeciwciała referencyjnego/ porównawczego. [0062] Termin powinowactwo wiązania z reguły oznacza siłę sumarycznych oddziaływań niekowalencyjnych pomiędzy pojedynczym miejscem wiązania cząsteczki (np. przeciwciała) i partnera wiązania (np. antygenu). O ile nie zaznaczono inaczej, w rozumieniu niniejszego opisu termin powinowactwo wiązania dotyczy wewnętrznego powinowactwa wiązania oddającego interakcję 1:1
14 13 pomiędzy składnikami pary wiązanej (np. przeciwciała i antygenu). Powinowactwo cząsteczki X do partnera Y można z reguły przedstawić poprzez stałą dysocjacji (Kd). Powinowactwo może być mierzone typowymi metodami znanymi w dziedzinie, w tym metodami tu opisanymi. Przeciwciała o niskim powinowactwie z reguły wiążą antygen powoli, podczas gdy przeciwciała o wysokim powinowactwie z reguły wiążą antygen szybciej i pozostają związane przez dłuższy czas. W dziedzinie znanych jest wiele metod mierzenia powinowactwa wiązania, z których każda może być wykorzystana do celów niniejszego wynalazku. W dalszej części przedstawiono konkretne warianty o charakterze ilustracyjnym. [0063] W jednym z wariantów Kd lub wartość Kd według niniejszego wynalazku mierzy się w teście radioimmunologicznym (RIA) wykonywanym z wykorzystaniem wersji Fab badanego przeciwciała i jego antygenu zgodnie z poniższym opisem badania, mierzącego powinowactwo wiązania antygenu przez Fab w roztworach poprzez osiągnięcie stanu równowagi dla Fab w obecności minimalnego stężenia antygenu znakowanego izotopem 125 I w obecności serii rozcieńczeń nieznakowanego antygenu, a następnie wychwytywanie związanego antygenu na płytce powleczonej przeciwciałem anty Fab (Chen, et al., (1999) J. Mol Biol 293: ). W celu stworzenia warunków do przeprowadzenia badania płytki mikrotitracyjne (Dynex) powleka się przeciwciałem wychwytującym anty Fab (Cappel Labs) w stężeniu 5 µg/ml w 50 mm węglanie sodu (ph 9,6), pozostawia na noc, a następnie blokuje 2% (wag./obj.) roztworem albuminy surowicy bydlęcej w PBS w temperaturze pokojowej (około 23 C) przez dwie do pięciu godzin. Na nieadsorbującej płytce (Nunc #269620), miesza się 100 pm lub 26 pm [ 125 I]-antygenu z serią rozcieńczeń badanego Fab (np. zgodnie z oznaczeniem przeciwciała anty VEGF, Fab-12, w pracy Presta et al., (1997) Cancer Res. 57: ). Badany region Fab poddaje się inkubacji przez noc; czas inkubacji może być jednak dłuższy (np. 65 godzin) w celu zapewnienia osiągnięcia stanu równowagi. Następnie mieszaniny przenosi się na płytkę wychwytującą w celu przeprowadzenia inkubacji w temperaturze pokojowej (na przykład przez jedną godzinę). Następnie usuwa się roztwór, a płytką przemywa osiem razy 0,1% roztworem Tween-20 w PBS. Po wyschnięciu płytek dodaje się płyn scyntylacyjny (MicroScint-20; Packard) w ilości 150 µl/dołek, po czym impulsy promieniowania gamma zlicza się przy użyciu licznika Topcount (Packard) przez dziesięć minut. Do badań konkurencyjnego wiązania antygenu wybiera się stężenia każdego Fab zapewniające nie więcej niż 20% maksymalnego stopnia związania antygenu. W innym wariancie wynalazku Kd lub wartość Kd mierzy się techniką rezonansu plazmonów powierzchniowych przy użyciu aparatu BIAcore lub BIAcore (BIAcore, Inc., Piscataway, NJ) w temperaturze 25 C z chipami CM5 z immobilizowanym antygenem na poziomie ~10 jednostek odpowiedzi (RU). W skrócie, chipy biosensorowe z karboksymetylowanego dekstranu (CM5, BIAcore Inc.) aktywuje się chlorowodorkiem N-etylo-N'-(3-dimetyloaminopropylo)-karbodiimidu (EDC) i N- hydroksysukcynoimidem (NHS) zgodnie z instrukcjami dostawcy. Antygen rozcieńcza się 10mM octanem sodu o ph 4,8 do stężenia 5 µg/ml (~0,2 µm) przed wstrzyknięciem z prędkością 5 µl/minutę do momentu uzyskania około 10 jednostek odpowiedzi (RU) sprzężonego białka. Po wstrzyknięciu antygenu do mieszaniny wstrzykuje się 1M roztwór etanoloaminy w celu zablokowania nieprzereagowanych grup. W ramach pomiarów kinetyki, serie dwukrotnych rozcieńczeń Fab (0,78 nm do 500 nm) wstrzykuje się do PBS z zawartością 0,05% Tween 20 (PBST) w temperaturze 25 C, z prędkością około 25 µl/min. Stałe asocjacji (k on ) i dysocjacji (k off ) oblicza się w oparciu o prosty model wiązania jeden na jeden Langmuira (oprogramowanie BIAcore Evaluation Software, wersja 3.2) poprzez równoległe dopasowanie sensorogramów asocjacji i dysocjacji. Równowagową stałą dysocjacji (Kd) oblicza się jako stosunek k off /k on. Zob. np. Chen, Y. et al., (1999) J. Mol Biol 293: Jeśli stała asocjacji zmierzona w powyższym oznaczeniu metodą rezonansu plazmonów powierzchniowych przekracza 10 6 M -1 S -1, może zostać zmierzona techniką gaszenia fluorescencji, mierzącą przy użyciu spektrometru, na przykład spektrometru z zatrzymaniem przepływu (Aviv Instruments) lub spektrometrem SLM- Aminco serii 8000 wyposażonym w kuwetę z mieszaniem, wzrost lub spadek intensywności emisji fluorescencji (częstotliwość ekscytacji = 295 nm; częstotliwość emisji = 340 nm, filtr środkowoprzepustowy 16 nm) w temperaturze 25 C dla 20nM przeciwciała antagonistycznego względem antygenu (postać Fab) w PBS, ph 7,2, w obecności rosnących stężeń antygenu. [0064] Stałą asocjacji lub k on według niniejszego wynalazku można również oznaczyć tą samą opisaną powyżej techniką rezonansu plazmonów powierzchniowych przy użyciu aparatu BIAcore lub BIAcore (BIAcore, Inc., Piscataway, NJ) w temperaturze 25 C z chipami CM5 z immobilizowanym antygenem na poziomie ~10 jednostek odpowiedzi (RU). W skrócie, chipy biosensorowe z karboksymetylowanego dekstranu (CM5, BIAcore Inc.) aktywuje się chlorowodorkiem N-etylo-N'-(3-dimetyloaminopropylo)-karbodiimidu (EDC) i N-N-hydroksysukcynoimidem (NHS) zgodnie z instrukcjami dostawcy. Antygen rozcieńcza się 10mM octanem sodu o ph 4,8 do stężenia 5
15 14 µg/ml (~0,2 µm) przed wstrzyknięciem z prędkością 5 µl/minutę do momentu uzyskania około 10 jednostek odpowiedzi (RU) sprzężonego białka. Po wstrzyknięciu antygenu do mieszaniny wstrzykuje się 1M roztwór etanoloaminy w celu zablokowania nieprzereagowanych grup. W ramach pomiarów kinetyki, serie dwukrotnych rozcieńczeń Fab (0,78 nm do 500 nm) wstrzykuje się do PBS z zawartością 0,05% Tween 20 (PBST) w temperaturze 25 C, z prędkością około 25 µl/min. Stałe asocjacji (k on ) i dysocjacji (k off ) oblicza się w oparciu o prosty model wiązania jeden na jeden Langmuira (oprogramowanie BIAcore Evaluation Software, wersja 3.2) poprzez równoległe dopasowanie sensorogramów asocjacji i dysocjacji. Równowagową stałą dysocjacji (Kd) obliczano jako stosunek k off /k on. Zob. np. Chen, Y. et al., (1999) J. Mol Biol 293: Jeżeli jednak stała asocjacji mierzona powyższą techniką rezonansu plazmonów powierzchniowych przekracza 10 6 M -1 S - 1 stałą asocjacji korzystnie oznacza się techniką gaszenia fluorescencji, mierzącą przy użyciu spektrometru, na przykład spektrometru z zatrzymaniem przepływu (Aviv Instruments) lub spektrometrem SLM-Aminco serii 8000 wyposażonym w kuwetę z mieszaniem, wzrost lub spadek intensywności emisji fluorescencji (częstotliwość ekscytacji = 295 nm; częstotliwość emisji = 340 nm, filtr środkowoprzepustowy 16 nm) w temperaturze 25 C dla 20nM przeciwciała antagonistycznego względem antygenu (postać Fab ) w PBS, ph 7,2, w obecności rosnących stężeń antygenu. W jednym z wariantów Kd lub wartość Kd według niniejszego wynalazku mierzy się w teście radioimmunologicznym (RIA) wykonywanym z wykorzystaniem wersji Fab badanego przeciwciała i jego antygenu zgodnie z poniższym opisem badania, mierzącego powinowactwo wiązania antygenu przez Fab w roztworach poprzez osiągnięcie stanu równowagi dla Fab w obecności minimalnego stężenia antygenu znakowanego izotopem 125 I w obecności serii rozcieńczeń nieznakowanego antygenu, a następnie wychwytywanie związanego antygenu na płytce powleczonej przeciwciałem anty Fab (Chen, et al., (1999) J. Mol Biol 293: ) W celu stworzenia warunków do przeprowadzenia badania płytki mikrotitracyjne (Dynex) powleka się przeciwciałem wychwytującym anty Fab (Cappel Labs) w stężeniu 5 µg/ml w 50 mm węglanie sodu (ph 9,6), pozostawia na noc, a następnie blokuje 2% (wag./obj.) roztworem albuminy surowicy bydlęcej w PBS w temperaturze pokojowej (około 23 C) przez dwie do pięciu godzin. Na nieadsorbującej płytce (Nunc #269620), miesza się 100 pm lub 26 pm [ 125 I]-antygenu z serią rozcieńczeń badanego Fab (np. zgodnie z oznaczeniem przeciwciała anty VEGF, Fab-12, w pracy Presta et al., (1997) Cancer Res. 57: ). Badany region Fab poddaje się inkubacji przez noc; czas inkubacji może być jednak dłuższy (np. 65 godzin) w celu zapewnienia osiągnięcia stanu równowagi. Następnie mieszaniny przenosi się na płytkę wychwytującą antygen w celu przeprowadzenia trwającej jedną godzinę inkubacji w temperaturze pokojowej. Następnie usuwa się roztwór, a płytką przemywa osiem razy 0,1% roztworem Tween-20 w PBS. Po wyschnięciu płytek dodaje się płyn scyntylacyjny (MicroScint-20; Packard) w ilości 150 µl/dołek, po czym impulsy promieniowania gamma zlicza się przy użyciu licznika Topcount (Packard) przez dziesięć minut. Do badań konkurencyjnego wiązania antygenu wybiera się stężenia każdego Fab zapewniające nie więcej niż 20% maksymalnego stopnia związania antygenu. W innym wariancie wynalazku Kd lub wartość Kd mierzy się techniką rezonansu plazmonów powierzchniowych przy użyciu aparatu BIAcore lub BIAcore (BIAcore, Inc., Piscataway, NJ) w temperaturze 25 C z chipami CM5 z immobilizowanym antygenem na poziomie ~10 jednostek odpowiedzi (RU). W skrócie, chipy biosensorowe z karboksymetylowanego dekstranu (CM5, BIAcore Inc.) aktywuje się chlorowodorkiem N-etylo-N'-(3-dimetyloaminopropylo)-karbodiimidu (EDC) i N-hydroksysukcynoimidem (NHS) zgodnie z instrukcjami dostawcy. Antygen rozcieńcza się 10mM octanem sodu o ph 4,8 do stężenia 5 µg/ml (~0,2 µm) przed wstrzyknięciem z prędkością 5 µl/minutę do momentu uzyskania około 10 jednostek odpowiedzi (RU) sprzężonego białka. Po wstrzyknięciu antygenu do mieszaniny wstrzykuje się 1M roztwór etanoloaminy w celu zablokowania nieprzereagowanych grup. W ramach pomiarów kinetyki, serie dwukrotnych rozcieńczeń Fab (0,78 nm do 500 nm) wstrzykuje się do PBS z zawartością 0,05% Tween 20 (PBST) w temperaturze 25 C, z prędkością około 25 µl/min. Stałe asocjacji (k on ) i dysocjacji (k off ) oblicza się w oparciu o prosty model wiązania jeden na jeden Langmuira (oprogramowanie BIAcore Evaluation Software, wersja 3.2) poprzez równoległe dopasowanie sensorogramów asocjacji i dysocjacji. Równowagową stałą dysocjacji (Kd) oblicza się jako stosunek k off /k on Zob. np. Chen, Y. et al., (1999) J. Mol Biol 293: Jeśli stała asocjacji zmierzona w powyższym oznaczeniu metodą rezonansu plazmonów powierzchniowych przekracza 10 6 M-1 S-1, może zostać zmierzona techniką gaszenia fluorescencji, mierzącą przy użyciu spektrometru, na przykład spektrometru z zatrzymaniem przepływu (Aviv Instruments) lub spektrometrem SLM-Aminco serii 8000 wyposażonym w kuwetę z mieszaniem, wzrost lub spadek intensywności emisji fluorescencji (częstotliwość ekscytacji = 295 nm; częstotliwość emisji = 340 nm, filtr środkowoprzepustowy 16 nm) w temperaturze 25 C dla 20nM przeciwciała antagonistycznego względem antygenu (postać Fab ) w PBS, ph 7,2, w obecności rosnących stężeń antygenu.
16 15 [0065] W jednym z wariantów, stałą asocjacji lub k on według niniejszego wynalazku oznacza się tą samą opisaną powyżej techniką rezonansu plazmonów powierzchniowych przy użyciu aparatu BIAcore lub BIAcore (BIAcore, Inc., Piscataway, NJ) w temperaturze 25 C z chipami CM5 z immobilizowanym antygenem na poziomie ~10 jednostek odpowiedzi (RU). W skrócie, chipy biosensorowe z karboksymetylowanego dekstranu (CM5, BIAcore Inc.) aktywuje się chlorowodorkiem N-etylo-N'-(3-dimetyloaminopropylo)-karbodiimidu (EDC) i N-hydroksysukcynoimidem (NHS) zgodnie z instrukcjami dostawcy. Antygen rozcieńcza się 10mM octanem sodu o ph 4,8 do stężenia 5 µg/ml (~0,2 µm) przed wstrzyknięciem z prędkością 5 µl/minutę do momentu uzyskania około 10 jednostek odpowiedzi (RU) sprzężonego białka. Po wstrzyknięciu antygenu do mieszaniny wstrzykuje się 1M roztwór etanoloaminy w celu zablokowania nieprzereagowanych grup. W ramach pomiarów kinetyki, serie dwukrotnych rozcieńczeń Fab (0,78 nm do 500 nm) wstrzykuje się do PBS z zawartością 0,05% Tween 20 (PBST) w temperaturze 25 C, z prędkością około 25 µl/min. Stałe asocjacji (k on ) i dysocjacji (k off ) oblicza się w oparciu o prosty model wiązania jeden na jeden Langmuira (oprogramowanie BIAcore Evaluation Software, wersja 3.2) poprzez równoległe dopasowanie sensorogramów asocjacji i dysocjacji. Równowagową stałą dysocjacji (Kd) obliczano jako stosunek k off /k on. Zob. np. Chen, Y. et al., (1999) J. Mol Biol 293: Jeżeli jednak stała asocjacji mierzona powyższą techniką rezonansu plazmonów powierzchniowych przekracza 10 6 M -1 S -1 stałą asocjacji korzystnie oznacza się techniką gaszenia fluorescencji, mierzącą przy użyciu spektrometru, na przykład spektrometru z zatrzymaniem przepływu (Aviv Instruments) lub spektrometrem SLM-Aminco serii 8000 wyposażonym w kuwetę z mieszaniem, wzrost lub spadek intensywności emisji fluorescencji (częstotliwość ekscytacji = 295 nm; częstotliwość emisji = 340 nm, filtr środkowoprzepustowy 16 nm) w temperaturze 25 C dla 20nM przeciwciała antagonistycznego względem antygenu (postać Fab ) w PBS, ph 7,2, w obecności rosnących stężeń antygenu. [0066] Sformułowanie zasadniczo mniejszy lub zasadniczo różny w rozumieniu niniejszego opisu oznacza stopień różnicy pomiędzy dwiema wartościami numerycznymi (zazwyczaj jedną związana z przeciwciałem będącym przedmiotem wynalazku, a drugą związaną z przeciwciałem referencyjnym/porównawczym) wystarczająco duży, by osoba biegła w dziedzinie uznała różnicę pomiędzy obiema wartościami za posiadającą znaczenie statystyczne w kontekście cech biologicznych mierzonych rzeczonym i wartościami (np. wartościami Kd, odpowiedziami HAMA). Różnica pomiędzy rzeczonymi dwiema wartościami wynosi korzystnie więcej niż około 10%, korzystnie więcej niż około 20%, korzystnie więcej niż około 30%, korzystnie więcej niż około 40%, korzystnie więcej niż około 50% wartości dla przeciwciała referencyjnego/porównawczego. [0067] Procentową (%) identyczność sekwencji aminokwasów w odniesieniu do sekwencji peptydu lub polipeptydu definiuje się jako odsetek reszt aminokwasowych sekwencji kandydującej identycznych z resztami aminokwasowymi określonej sekwencji peptydowej lub polipeptydowej po dopasowaniu sekwencji i wprowadzeniu odstępów (jeśli są konieczne) w celu osiągnięcia maksymalnej procentowej identyczności sekwencji, bez uznawania jakichkolwiek konserwatywnych substytucji za element identyczności sekwencji. Dopasowanie sekwencji do celów ustalenia procentowej zgodności sekwencji aminokwasów można osiągnąć na różne sposoby znane w dziedzinie wynalazku, na przykład poprzez zastosowanie publicznie dostępnego oprogramowania komputerowego takiego jak BLAST, BLAST-2, ALIGN lub Megalign (DNASTAR) Osoby biegłe w dziedzinie wynalazku są w stanie wyznaczyć odpowiednie parametry pomiaru wyrównania sekwencji, w tym ewentualne algorytmy wymagane do maksymalnego dopasowania sekwencji na pełnej długości porównywanych sekwencji. Do celów niniejszego wynalazku wartości procentowej identyczności sekwencji aminokwasów generowane są jednak przy użyciu programu komputerowego do porównywania sekwencji ALIGN-2, przy czym pełen kod źródłowy programu ALIGN-2 został podany w Tabeli A poniżej. Program komputerowy do porównywania sekwencji ALIGN-2 został opracowany przez firmę Genentech, a kod źródłowy przedstawiony poniżej w Tabeli A został złożony wraz z dokumentacją użytkownika w Urzędzie Praw Autorskich USA, Waszyngton, D.C., 20559, gdzie został zarejestrowany pod nr. TXU Program ALIGN-2 jest oferowany publicznie przez firmę Genentech, Inc., South San Francisco, Kalifornia, USA, lub może zostać skompilowany z kodu źródłowego przedstawionego poniżej na Figurze 8. Program ALIGN-2 powinien zostać skompilowany do pracy pod systemem operacyjnym UNIX, korzystnie pod systemem Digital UNIX V4.0D. Wszystkie parametry porównania sekwencji ustalane są przez program ALIGN-2 i są niezmienne. [0068] W przypadkach, gdy do porównywania sekwencji aminokwasowych wykorzystuje się program ALIGN-2, procentową identyczność sekwencji aminokwasów danej sekwencji aminokwasowej A z, względem lub w porównaniu z sekwencją aminokwasową B (co można inaczej sformułować jako posiadanie przez dane sekwencję aminokwasową A określoną procentową identyczność sekwencji aminokwasowej z, względem lub w porównaniu z sekwencją aminokwasową B) oblicza się w
17 16 następujący sposób: 100 razy ułamek X/Y, gdzie X jest liczbą reszt aminokwasowych zaliczonych podczas dopasowywania sekwencji A i B przez program do dopasowywania sekwencji ALIGN-2 jako identyczne dopasowania, zaś Y jest całkowita liczbą reszt aminokwasowych w B. Rozumie się przez to, że w przypadkach, gdy długość sekwencji aminokwasowej A nie jest równa długości sekwencji aminokwasowej B, procentowa identyczność sekwencji aminokwasowej A względem B nie będzie równa procentowej identyczności sekwencji aminokwasowej B względem A. [0069] O ile zostanie wyraźnie stwierdzone inaczej, wszystkie opisywane tu wartości procentowej identyczności sekwencji aminokwasowych zostały uzyskane w sposób opisany w poprzednim akapicie przy użyciu programu ALIGN-2. Tabela A
18 17
19 18
20 19
21 20
22 21
23 22
24 23
25 24
26 25
27 26
28 27
29 28
30 29
31 30
32 31
33 32
34 33
35 34
36 35
37 36
38 37
39 38
40 39
41 40
42 41
43 42 [0070] Termin wektor w rozumieniu niniejszego opisu oznacza cząsteczkę kwasu nukleinowego zdolną do transportowania innego kwasu nukleinowego, z którym została połączona. Jednym z rodzajów wektorów jest plazmid, będący kolistą pętlą dwułańcuchowego DNA, do której można przyłączyć dodatkowe odcinki DNA. Innym rodzajem wektora jest wektor fagowy. Innym rodzajem wektora jest wektor wirusowy, w którym do genomu wirusa można przyłączyć dodatkowe odcinki DNA. Niektóre wektory posiadają zdolność autonomicznej replikacji w komórkach gospodarza, do których zostały wprowadzone (np. wektory bakteryjne o bakteryjnym miejscu inicjacji replikacji czy episomalne wektory ssacze). Inne wektory (np. nieepisomalne wektory ssacze mogą zostać zintegrowane z genomem komórki gospodarza przy wprowadzeniu do komórki gospodarza i tym samym ulegać replikacji wraz z genomem gospodarza. Ponadto niektóre wektory są w stanie kierować ekspresją genów, do których zostały przyłączone. Wektory takie określa się tu mianem rekombinowanych wektorów ekspresyjnych lub po prostu wektorów rekombinowanych ). Na ogół wektory ekspresyjne wykorzystywane w technikach rekombinacji DNA mają często postać plazmidu. W niniejszym opisie terminy plazmid i wektor mogą być stosowane zamiennie, ponieważ plazmid jest najczęściej stosowaną postacią wektora. [0071] Stosowane tu zamiennie terminy polinukleotyd lub kwas nukleinowy oznaczają polimery nukleotydów o dowolnej długości i obejmują również DNA i RNA. Nukleotydy mogą być
44 43 deoksyrybonukleotydami, rybonukleotydami, modyfikowanymi nukleotydami lub zasadami i/lub ich analogami lub dowolnymi substratami, które mogą zostać włączone do polimeru przy użyciu polimerazy DNA lub RNA lub w reakcji syntezy. Polinukleotyd może zawierać modyfikowane nukleotydy, na przykład metylowane nukleotydy i ich analogi. Modyfikacja struktury nukleotydowej, jeśli ma miejsce, może zostać wprowadzona przed lub po złożeniu polimeru. Sekwencja nukleotydów może być przerwana elementami nienukleotydowymi. Po zsyntetyzowaniu polinukleotyd może być dalej modyfikowany, na przykład w wyniku sprzężenia z etykietą znakującą. Inne rodzaje modyfikacji obejmują np. hydrofobowe zakończenia łańcucha ( cap ), substytucję jednego lub większej liczby naturalnych nukleotydów analogiem, modyfikacje połączeń międzynukleotydowych, na przykład powiązania nienaładowane (np. metylofosfoniany, fosfotriestry, fosfoamidyniany, karbaminiany itp.) oraz naładowane (np. fosforotioestry, fosforoditioestry itp), powiązania z przyłączonymi resztami, na przykład białkowymi (np. nukleazami, toksynami, przeciwciałami, peptydami sygnałowymi, ply-l-lizyną itp.), powiązania z resztami dystansującymi (np. akrydyna, psoralen itp.), powiązania zawierające odczynniki chelatujące (np. metale, metale radioaktywne, bor, metale utleniające itp.), powiązania z czynnikami alkilującymi, powiązania zmodyfikowane (np. z ankomerami alfa kwasów nukleinowych), jak również niezmodyfikowane postaci polinukleotydów. Ponadto każda z grup hydroksylowych obecnych zazwyczaj w cukrach może zostać zastąpiona na przykład grupami fosfonianowymi, fosforanowymi, w cukrach, zablokowana standardowymi grupami ochronnymi, aktywowana w celu wytworzenia dodatkowych powiązań z dodatkowymi nukleotydami lub sprzężona z trwałym lub półtrwałym podłożem. 5'- i 3 -końcowe grupy OH mogą zostać sfosforylowane lub podstawione aminami lub organicznymi resztami hydrofobowymi wielkości od 1 do 20 atomów węgla. Również inne grupy hydroksylowe mogą zostać przekształcone w standardowe grupy ochronne. Polinukleotydy mogą również zawierać znane w dziedzinie analogi cukrów rybozowych lub deoksyrybozowych, w tym na przykład 2'-O-metylo-, 2'-O-allilo, 2'-fluoro- lub 2'-azydorbozę, karbocykliczne analogi cukrów, cukry α-anomeryczne, cukry epimeryczne takie, jak arabinoza, ksylozy lub liksozy, cukry piranozowe, furanozowe, sedoheptulozy, analogi acykliczne i bezzasadowe analogi nukleozydów, na przykład metylorybozyd. Jedna lub większa liczba połączeń fosfodiestrowych może zostać zastąpiona alternatywnymi grupami łącznikowymi. Rzeczone alternatywne grupy łącznikowe obejmują między innymi warianty, w których fosforan zastąpiony jest przez grupę P(O)S ( tioester ), P(S)S ( ditioester ), "(O)NR 2 ( amidynian ), P(O)R, P(O)OR', CO lub CH 2 ( formacetal ), gdzie R i R' są niezależnie atomem H lub podstawionym lub niepodstawionym alkilem (1-20 C), opcjonalnie zawierającymi łącznik eterowy (-O-), arylem, alkilenem, cykloalkilem, cykloalkilenem lub araldylem. Nie jest konieczne, by wszystkie połączenia w polinukleotydzie były identyczne. Powyższy opis dotyczy wszystkich wzmiankowanych tu polinukleotydów, w tym RNA i DNA. [0072] Termin oligonukleotyd w rozumieniu niniejszego opisu dotyczy z reguły krótkich, z reguły jednołańcuchowych, z reguły syntetycznych polinukleotydów, z reguły, lecz nie koniecznie krótszych niż około 200 nukleotydów. Terminy oligonukleotyd i polinukleotyd nie wykluczają się wzajemnie. Przedstawiony powyżej opis dotyczący polinukleotydów ma również pełne zastosowanie w przypadku oligonukleotydów. [0073] Termin czynnik wzrostu hepatocytów lub HGF w rozumieniu niniejszego opisu dotyczy, o ile nie zostanie zaznaczone inaczej lub o ile kontekst nie będzie wskazywał inaczej, dowolnego natywnego lub wariantowego (czy to natywnego, czy też syntetycznego) polipeptydu HGF zdolnego do aktywowania szlaku sygnałowego HGF/c-met w warunkach umożliwiających zachodzenie tego rodzaju procesów. Termin HGF dzikiego typu z reguły dotyczy polipeptydu zawierającego sekwencje aminokwasową właściwą dla naturalnie występującego białka HGF. Termin sekwencja HGF dzikiego typu z reguły sekwencji aminokwasowej właściwej dla naturalnie występującego białka HGF. [0074] Terminy przeciwciało i immunoglobulina stosowane są wymiennie w najszerszym rozumieniu i obejmują przeciwciała monoklonalne (np. przeciwciała monoklonalne pełnej długości lub nienaruszone), przeciwciała poliklonalne, przeciwciała multiwalencyjne, przeciwciała multispecyficzne (np. przeciwciała bispecyficzne, o ile wykazują pożądana aktywność biologiczną), a ponadto mogą obejmować określone fragmenty przeciwciał (zgodnie z zamieszczonym tu bardziej szczegółowym opisem). Przeciwciało może być przeciwciałem ludzkim, humanizowanym lub o dojrzałym powinowactwie. [0075] Fragmenty przeciwciał stanowią jedynie część nienaruszonych przeciwciał, przy czym części za korzystnie zachowują przynajmniej jedną, korzystnie zaś większość lub wszystkie funkcje związane zazwyczaj z daną częścią w sytuacji, gdy jest ona obecna w nienaruszonym przeciwciele. W jednym z wariantów wynalazku fragment przeciwciała zawiera miejsce wiązania antygenu w nienaruszonym przeciwciele i tym samym zachowuje zdolność wiązania antygenu. W innym wariancie wynalazku
45 44 fragment przeciwciała, na przykład fragment zawierający region Fc, zachowuje co najmniej jedną z funkcji biologicznych związanych zazwyczaj z regionem Fc w sytuacji, gdy jest on obecny w nienaruszonym przeciwciele, na przykład funkcję wiązania FcRn, modulowania czasu półtrwania przeciwciała, cytotoksyczności komórkowej zależnej od przeciwciała (ADCC) i wiązania dopełniacza. W jednym z wariantów fragmentem przeciwciała jest przeciwciało monowalencyjne o czasie półtrwania in vivo zasadniczo podobnym do nienaruszonego przeciwciała. Fragment przeciwciała może na przykład zawierać ramię wiązania antygenu połączone z sekwencją Fc zdolną do zapewnienia rzeczonemu fragmentowi trwałości in vivo. W jednym z wariantów przeciwciało będące przedmiotem wynalazku jest jednoramiennym przeciwciałem opisanym w patencie nr WO 2005/ W jednym z wariantów jednoramienne przeciwciało zawiera mutacje Fc tworzące wybrzuszenia ( knobs ) i zagłębienia ( holes ), zgodnie z opisem patentowym nr WO2005/ Mutację odpowiedzialną za zagłębienie może na przykład stanowić jedna lub więcej mutacji polipeptydu Fc z grupy obejmującej T366A, L368A i/lub Y407V, zaś mutacją odpowiedzialną za wybrzuszenie może być T366W. [0076] Termin przeciwciało monoklonalne w rozumieniu niniejszego opisu oznacza przeciwciało uzyskane z populacji zasadniczo jednorodnych przeciwciał, tj. poszczególnych przeciwciał stanowiących populację i identycznych z wyjątkiem ewentualnych naturalnie występujących mutacji, które mogą występować w niewielkich ilościach. Przeciwciała monoklonalne są wysoce specyficzne, kierowane przeciwko pojedynczemu antygenowi. Ponadto, w przeciwieństwie do preparatów przeciwciał poliklonalnych, które zazwyczaj zawierają różne przeciwciała kierowane przeciwko różnym determinantom (epitopom), każde przeciwciało monoklonalne kierowane jest przeciwko pojedynczemu determinantowi w cząsteczce antygenu. [0077] W rozumieniu niniejszego opisu przeciwciała monoklonalne obejmują w szczególności przeciwciała chimeryczne, w których część lekkiego i/lub ciężkiego łańcucha jest identyczna lub homologiczna względem odpowiednich sekwencji w przeciwciałach pochodzących od określonego gatunku lub należących do szczególnej klasy lub podklasy przeciwciał, podczas gdy pozostała część łańcucha(-ów) jest identyczna lub homologiczna względem odpowiednich sekwencji w przeciwciałach pochodzących od innego gatunku lub należących do innej klasy lub podklasy przeciwciał, a także fragmenty takich przeciwciał, o ile wykazują one pożądaną aktywność biologiczną (patent nr US 4,816,567 i Morrison et al., Proc. Natl. Acad. Sci. USA 81: (1984)). [0078] Humanizowanymi formami przeciwciał innych niż ludzkie (np. mysich) są chimeryczne przeciwciała zawierające minimalną sekwencję pochodzącą z immunoglobuliny innej niż ludzka. W większości humanizowane przeciwciała są ludzkimi immunoglobulinami (przeciwciałami biorcy), w których reszty hiperzmiennego regionu biorcy są zastąpione resztami z hiperzmiennych regionów gatunków innych niż człowiek (przeciwciało dawcy), na przykład myszy, szczura, królika lub innego niż człowiek gatunku z rodziny naczelnych, charakteryzujących się pożądaną specyficznością, powinowactwem i pojemnością. W niektórych przypadkach reszty regionu zrębowego (FR) ludzkiej immunoglobuliny zastępowane są odpowiednimi resztami pochodzącymi z innych gatunków. Ponadto, humanizowane przeciwciała mogą zawierać reszty nie występujące ani w przeciwciele biorcy, ani w przeciwciele dawcy. Modyfikacje te maja na celu dalsze udoskonalenie działania przeciwciała. Z reguły przeciwciało humanizowane będzie zawierało zasadniczo wszystkie spośród co najmniej jednej, a zazwyczaj dwóch domen zmiennych, w których wszystkie lub zasadniczo wszystkie pętle hiperzmienne odpowiadają pętlom immunoglobuliny innej niż ludzka, a wszystkie lub zasadniczo wszystkie regiony FR są regionami o sekwencji ludzkiej immunoglobuliny. Humanizowane przeciwciało opcjonalnie będzie również zawierać co najmniej część stałego regionu immunoglobuliny (Fc), zazwyczaj immunoglobuliny ludzkiej. Więcej informacji można znaleźć w pracach Jones et al., Nature 321: (1986); Riechmann et al., Nature 332: (1988); i Presta, Curr. Op. Struct. Biol. 2: (1992). Patrz również następujące artykuły przeglądowe i cytowane tam odnośniki: Vaswani i Hamilton, Ann. Allergy, Asthma & Immunol. 1: (1998); Harris, Biochem. Soc. Transactions 23: (1995); Hurle i Gross, Curr. Op. Biotech. 5: (1994). [0079] Termin antygen oznacza określony z góry antygen, który może być selektywnie wiązany przez przeciwciało. Docelowy antygen może być polipeptydem, węglowodanem, kwasem nukleinowym, lipidem, haptenem lub innym występującym w naturze lub syntetycznym związkiem chemicznym. Korzystnie docelowy antygen jest polipeptydem. Akceptorową ludzką sekwencją zrębową w rozumieniu niniejszego opisu jest sekwencja zrębowa zawierająca sekwencję aminokwasów regionów zrębowych VL lub VH pochodzącą z regionu zrębowego immunoglobuliny ludzkiej lub z ludzkiej konsensusowej sekwencji zrębowej. Akceptorowa ludzka sekwencja zrębowa pochodząca: z sekwencji zrębowej ludzkiego przeciwciała lub ludzkiej konsensusowej sekwencji
46 45 zrębowej może zawierać te same aminokwasy, co rzeczona sekwencja, lub może zawierać określone z góry modyfikacje aminokwasów. W przypadku obecności określonych z góry modyfikacji aminokwasów, w sekwencji obecnych jest nie więcej niż 5 modyfikacji aminokwasów, zaś korzystnie 4 lub mniej albo 3 lub mniej modyfikacji aminokwasów. W przypadku obecności określonych z góry modyfikacji aminokwasów w VH, w korzystnym wariancie modyfikacje te mają miejsce jedynie w trzech pozycjach: 71H, 73H i 78H; na przykład reszty aminokwasowe w tych pozycjach mogą mieć postać 71A, 73T i/lub 78A. W jednym z wariantów akceptorowa ludzka sekwencja zrębowa VL jest identyczna z sekwencją zrębową VL ludzkiej immunoglobuliny lub z ludzką konsensusową sekwencją zrębową. [0080] Ludzką konsensusową sekwencją zrębową jest sekwencja zrębowa zawierająca reszty aminokwasowe najczęściej występujące w wyborze sekwencji zrębowych VL lub VH immunoglobulin ludzkich. Z reguły wybór sekwencji zrębowych VL lub VH immunoglobulin ludzkich zawiera się w podgrupie sekwencji domen zmiennych. Z reguły podgrupą sekwencji jest podgrupa zgodna z opisem w pracy Kabat et al. W jednym z wariantów podgrupą dla VL jest podgrupa kappa I z pracy Kabat et al. W jednym z wariantów podgrupą dla VH jest podgrupa III z pracy Kabat et al. [0081] Konsensusowa sekwencja zrębowa VH z podgrupy III zawiera sekwencję konsensusową uzyskaną z sekwencji aminokwasów tworzących podgrupę III zmiennej domeny łańcucha ciężkiego w klasyfikacji wg pracy Kabat et al. W jednym z wariantów sekwencja zrębowa VH z podgrupy III zawiera co najmniej część lub wszystkie z następujących sekwencji: EVQLVESG-GGLVQPGGSLRLSCAAS (SEQ ID NO: 42)-H1-WVRQAPGKGLEWV (SEQ ID NO: 43)- H2-RFTISRDNSKNT-LYLQMNSLRAEDTAVYYC (SEQ ID NO: 44)-H3-WGQGTLVTVSS (SEQ ID NO: 45). [0082] Konsensusowa sekwencja zrębowa VL z podgrupy I zawiera sekwencję konsensusową uzyskaną z sekwencji aminokwasów tworzących podgrupę I zmiennej domeny łańcucha lekkiego kappa w klasyfikacji wg pracy Kabat et al. W jednym z wariantów sekwencja zrębowa VH z podgrupy I zawiera co najmniej część lub wszystkie z następujących sekwencji: DIQMTQSPSSLSASVGDRVTITC (SEQ ID NO: 46)-L1-WYQQKPGKAPKLLIY (SEQ ID NO: 47)-L2- GVPSRFSGSGSGTDFTLTISSLQPEDFATYYC (SEQ ID NO: 4 8)-L3-FGQGTKVEIK (SEQ ID NO: 49). [0083] Niezmodyfikowaną ludzką sekwencją zrębową jest ludzka sekwencja zrębowa posiadająca tę samą sekwencję aminokwasów co akceptorowa ludzka sekwencja zrębowa, np. bez substytucji w obrębie ludzkiej akceptorowej sekwencji zrębowej aminokwasów ludzkich na inne niż ludzkie. [0084] Zmodyfikowanym regionem hiperzmiennym w rozumieniu niniejszego opisu jest region hiperzmienny zawierający jedną lub większą liczbę (tj. od 1 do ok. 16) substytucji aminokwasów. [0085] Niezmodyfikowanym regionem hiperzmiennym w rozumieniu niniejszego opisu jest region hiperzmienny zawierający tę samą sekwencję aminokwasów, co przeciwciało pochodzące od gatunku innego niż człowiek, z którego region ten pochodzi, tj. region niezawierający jednej lub większej liczby substytucji aminokwasów. [0086] Terminy region hiperzmienny, HVR lub HV w rozumieniu niniejszego opisu oznaczają regiony zmiennej domeny przeciwciała, w których zachodzi hiperzmienność sekwencji i/lub które tworzą strukturalne pętle. Z reguły przeciwciało zawiera sześć regionów hiperzmiennych: trzy w domenie VH (H1, H2, H3) i trzy w domenie VL (L1, L2, L3). W niniejszym opisie wykorzystuje się i uwzględnia wiele definicji regionów hiperzmiennych. Najczęściej stosowane są regiony determinujące komplementarność (CDR) Kabata, oparte na zmienności sekwencji (Kabat et al., Sequences of Proteins of Immunological Interest, wyd. 5, Public Health Service, National Institutes of Health, Bethesda, MD, USA). (1991)). Z kolei Chothia odwołuje się do lokalizacji pętli strukturalnych (Chothia i Lesk J. Mol. Biol. 196: (1987)). Regiony hiperzmienne AbM stanowią kompromis pomiędzy regionami CDR Kabata a pętlami strukturalnymi Chothii; są one wykorzystywane przez program do modelowania przeciwciał AbM firmy Oxford Molecular. Kontaktowe regiony hiperzmienne opierają się na analizie dostępnych struktur krystalograficznych kompleksów. Poniżej przedstawiono reszty każdego z powyższych regionów hiperzmiennych.
47 46 Pętla Kabat AbM Chothia Kontakt L1 L24-L34 L24-L34 L26-L32 L30-L36 L2 L50-L56 L50-L56 L50-L52 L46-L55 L3 L89-L97 L89-L97 L91-L96 L89-L96 H1 H31-H35B H26-H35B H26-H32 H30-H35B (Numeracja wg Kabata) H1 H31-H35 H26-H35 H26-H32 H30-H35 (Numeracja wg Chotii) H2 H50-H65 H50-H58 H53-H55 H47-H58 H3 H95-H102 H95-H102 H96-H101 H93-H101 [0087] Regiony hiperzmienne mogą zawierać rozszerzone regiony hiperzmienne zgodne z poniższym opisem: lub (L1), lub (L2) i (L3) w VL oraz (H1), lub (H2) i , lub (H3) w VH. W każdej z powyższych definicji reszty aminokwasowe w domenie zmiennej ponumerowane są zgodnie z pracą Kabat et al., supra. [0088] Resztami zrębowymi lub FR są reszty domeny zmiennej inne niż reszty regionów hiperzmiennych zgodnych z podaną tu definicją. [0089] Przeciwciałem ludzkim jest przeciwciało posiadające sekwencję aminokwasów odpowiadającą przeciwciału wytwarzanemu przez organizm człowieka i/lub wytworzonemu którąkolwiek technik wytwarzania ludzkich przeciwciał zgodnie z ujawnieniem w niniejszym opisie. Powyższa definicja ludzkiego przeciwciała wyklucza w szczególności humanizowane przeciwciało zawierające reszty wiążące antygen pochodzące od gatunków innych niż człowiek. [0090] Przeciwciałem o dojrzałym powinowactwie jest przeciwciało zawierające jedną lub więcej zmian w jednym lub większej liczbie regionów CDR, prowadzących do poprawy powinowactwa przeciwciała do antygenu w porównaniu z przeciwciałem macierzystym, nie posiadającym ww. zmian. Korzystnie, przeciwciała o dojrzałym powinowactwie będą charakteryzowały się nanomolarnym lub wręcz pikomolarnym powinowactwem do antygenu docelowego. Przeciwciała o dojrzałym powinowactwie wytwarzane s metodami znanymi w dziedzinie wynalazku. W pracy Marks et al. Bio/Technology 10: (1992) opisano dojrzewanie powinowactwa w procesie tasowania domen VH i VL. Losową mutagenezę CDR i/lub reszt zrębowych opisują prace: Barbas et al. Proc. Nat. Acad. Sci, USA 91: (1994); Schier et al. Gene 169: (1995); Yelton et al. J. Immunol. 155: (1995); Jackson et al., J. Immunol. 154(7): (1995); oraz Hawkins et al, J. Mol. Biol. 226: (1992). [0091] Przeciwciałem blokującym lub antagonistycznym jest przeciwciało hamujące lub zmniejszające aktywność biologiczną wiązanego przez siebie antygenu. W korzystnym wariancie przeciwciała blokujące lub antagonistyczne zasadniczo lub całkowicie hamują aktywność biologiczną antygenu. [0092] Przeciwciałem agonistycznym w rozumieniu niniejszego opisu jest przeciwciało naśladujące co najmniej jedną z funkcji przedmiotowego polipeptydu. [0093] Zaburzenie jest stanem, w który odniesionoby korzyść z leczenia substancją/cząsteczką lub metodą będącą przedmiotem wynalazku. Termin ten obejmuje zaburzenia lub choroby przewlekłe i ostre, w tym stany chorobowe predysponujące ssaka do danego zaburzenia. Nieograniczające zakresu przykłady leczonych schorzeń obejmują guzy łagodne i złośliwe; nowotwory niebiałaczkowe i limfoidalne; zaburzenia neuronalne, glejowe, astrocytarne, podwzgórzowe i inne zaburzenia gruczołowe, zaburzenia makrofagalne, nabłonkowe, zrębowe i blastoceliczne oraz zaburzenia zapalne, immunologiczne i inne zaburzenia związane z angiogenezą. [0094] Termin choroba proliferacji komórek i choroba proliferacyjna dotyczą zaburzeń związanych z pewnym stopniem nieprawidłowości w proliferacji komórek. W jednym z wariantów chorobą proliferacyjną jest rak. [0095] Termin nowotwór w rozumieniu niniejszego opisu dotyczy wszystkich przypadków nowotworowego wzrostu i proliferacji komórek, czy to złośliwych, czy też łagodnych oraz wszystkich komórek i tkanek przednowotworowych i nowotworowych. Terminy rak, rakowy, choroba
48 47 proliferacji komórek. choroba proliferacyjna i nowotwór w rozumieniu niniejszego opisu nie wykluczają się wzajemnie. [0096] Terminy rak i rakowy dotyczą lub opisują stan fizjologiczny występujący u ssaków, zazwyczaj charakteryzujący się nieregulowanym wzrostem/proliferacją komórek. Przykłady nowotworów obejmują między innymi raka karcynoma, chłoniaka, raka blastoma, mięsaka i białaczkę. W bardziej szczegółowym ujęciu przykłady nowotworów obejmują raka płaskokomórkowego, drobnokomórkowego raka płuca, niedrobnokomórkowego raka płuca, gruczolakoraka płuca, raka płaskokomórkowego płuca, raka otrzewnej, raka wątrobowokomórkowego, raka przewodu pokarmowego, raka trzustki, glejaka, raka szyjki macicy, raka jajnika, raka wątroby, raka pęcherza, wątrobiaka, raka piersi, raka jelita grubego, raka odbytu i jelita grubego, raka endometrium lub macicy, raka gruczołu ślinowego, raka nerki, raka wątroby, raka stercza, raka sromu, raka tarczycy, raka wątroby i różne rodzaje raka głowy i szyi. [0097] Deregulacja angiogenezy może prowadzić do wielu zaburzeń, dla których zagrożeniem mogą być kompozycje i metody będące przedmiotem wynalazku. Schorzenia te onejmuja zarówno schorzenia nienowtworowe, jak i nowotworowe. Schorzenia nowotworowe obejmują między innymi schorzenia opisane powyżej. Nienowotworowe schorzenia obejmują między innymi: niepożądany lub nieprawidłowy przerost, zapalenie stawów, reumatoidalne zapalenie stawów, łuszczyca, płytka łuszczycowa, sarkoidoza, miażdżyca, płytka miażdżycowa, retinopatia cukrzycowa i inne proliferacyjne retinopatie, w tym retinopatia wcześniaków, pozasoczewkowy rozrost włóknisty, jaskra neowaskularna, zwyrodnienie plamki żółtej związane z wiekiem, cukrzycowy obrzęk plamki, neowaskularyzacja rogówki, neowaskularyzacja przeszczepu rogówki, odrzucenie przeszczepu rogówki, neowaskularyzacja siatkówki/naczyniówki, neowaskularyzacja kąta przesączania (rubeosis), jaskra neowaskularyzacyjna, restenoza naczyń, naczyniaki tętniczo-żylne (AVM), oponiaki, naczyniaki, naczyniakowłókniaki, powiększenie tarczycy w tym choroba Basedowa, przeszczep rogówki i innych tkanek, przewlekły stan zapalny, zapalenie płuc, ostre uszkodzenie płuc/ards, posocznica, pierwotne nadciśnienie płucne, złośliwe wysięki płucne, obrzęk mózgowy (np. związany z ostrym udarem/urazem/uszkodzeniem wewnątrzczaszkowym), zapalenie maziówki, łuszczka w RZS, myositis ossificans, osteopatia przerostowa, zapalenie kości i stawów (ZKS), wodobrzusze oporne na leczenie, wielotorbielowate jajniki, zapalenie endometrium, choroby trzeciego przedziału płynów (zapalenie trzustki, zespół przedziałowy, pasożyty, choroby jelit), włókniaki macicy, przedwczesny poród, przewlekłe stany zapalne takie, jak IBD (choroba Crohna i wrzodziejące zapalenie jelit), odrzucenie allogennego przeszczepu nerki, zapalenie jelit, zespół nerczycowy, niepożądany lub niepożądany przyrost masy tkankowej (nierakowej), stawy krwotoczne, blizny przerostowe, hamowanie porostu włosów, zespół Oslera-Webera, ziarniak ropotwórczy, pozasoczewkowy rozrost włóknisty, skleroderma, jaglica, zrosty naczyniowe, zapalenie maziówki, zapalenie skóry, stan przedrzucawkowy, wodobrzusze, wysięk osierdziowy (związany z zapaleniem osierdzia) oraz wysięk opłucnowy. [0098] W rozumieniu niniejszego opisu termin leczenie dotyczy interwencji klinicznej w ramach prób zmiany naturalnego cyklu leczonego indywiduum lub komórki. Leczenie może być prowadzone bądź w celach profilaktycznych, bądź w przebiegu patologii klinicznej. Pożądane skutki leczenia obejmują zapobieganie wystąpieniu lub nawrotowi choroby, złagodzenie objawów, zmniejszenie ewentualnych bezpośrednich lub pośrednich patologicznych konsekwencji choroby, zapobieganie przerzutom, zmniejszenie odsetków progresji choroby, poprawa lub złagodzenie stanu choroby oraz remisja lub poprawa rokowań. W niektórych wariantach przeciwciała będące przedmiotem wynalazku stosowane są do opóźnienia rozwoju choroby lub zaburzenia. [0099] Skuteczna ilość" dotyczy ilości skutecznej do osiągnięcia, przy wymaganych dawkach i przy wymaganym okresie podawania, pożądanego wyniku terapeutycznego lub profilaktycznego. [0100] Terapeutycznie skuteczna ilość substancji/cząsteczki będącej przedmiotem wynalazku agonistycznej lun antagonistycznej, może być różne w zależności od takich czynników jak stan choroby, wiek, płeć, masa osobnika oraz zdolność substancji/cząsteczki, agonistycznej lub antagonistycznej, do wywierania pożądanej odpowiedzi w organizmie osobnika. Terapeutycznie skuteczna ilość jest również ilością, dla której nad ewentualnym toksycznym lub szkodliwym działaniem substancji/cząsteczki, agonistycznej lub antagonistycznej, przeważa działanie terapeutycznie korzystne. Profilaktycznie skuteczna ilość" dotyczy ilości skutecznej do osiągnięcia, przy wymaganych dawkach i przy wymaganym okresie podawania, pożądanego wyniku profilaktycznego. Zazwyczaj, choć nie koniecznie, ponieważ dawkę profilaktyczną stosuje się u pacjentów przed wystąpieniem choroby lub w jej wczesnym stadium, ilość skuteczna profilaktycznie będzie mniejsza niż ilość skuteczna terapeutycznie.
49 48 [0101] Termin środek cytotoksyczny" w rozumieniu niniejszego opisu dotyczy substancji hamującej lub uniemożliwiającej czynność komórek i/lub powodującej zniszczenie komórek. Termin ten obejmuje izotopy radioaktywne (np. At 211, I 131, I 125, Y 90, Re 186, Re 188, Sm 153, Bi 212, P 32 oraz radioaktywne izotopy Lu), chemioterapeutyki, np. metotreksat, adriamycynę, alkaloidy winka (winkrystyna, winblastyna, etopozyd), doksorubicyna, melfalan, mitomycyna C, chlorambucyl, daunorubicyna lub inne interkalatory, enzymy i ich fragmenty, na przykład enzymy nukleolityczne, antybiotyki i toksyny takie, jak toksyny niskocząsteczkowe lub enzymatycznie aktywne toksyny pochodzenia bakteryjnego, grzybowego, roślinnego lub zwierzęcego, w tym ich fragmenty i/lub warianty, oraz szereg środków przeciwnowotworowych i przeciwrakowych ujawnionych poniżej. Inne środki cytotoksyczne zostały opisane poniżej. Środek rakobójczy powoduje zniszczenie komórek nowotworowych. [0102] Chemioterapeutyk" jest związkiem chemicznym wykorzystywanym w leczeniu raka. Przykłady chemioterapeutyków obejmują środki alkilujące takie, jak tiotepa i CYTOXAN syklofosfamid, sulfoniany alkilowe takie,jak busulfan, improsulfan i piposulfan; azyrydyny takie, jak benzodopa, karbokwon, meturedopa i uredopa; etylenoiminy i metylamelaminy, w tym altretamina, trietylenomelamina, trietylenofosforamid, trietylenotiofosforamid itrimetylolomelamina; acetogeniny (zwłaszcza bullatacyna i bullatacynon); delta-9-tetrahydrokanabinol (dronabinol, MARINOL ); betalapachon; lapachol; kolchicyny; kwas betulinowy; kamptotecyna (oraz jej syntetyczny analog topotekan (HYCAMTIN ), CPT-11 (irinotekan, CAMPTOSAR ), acetylokamptotecyna, skopolektyna i 9-aminokamptotecyna); briostatyka; kallistatyna; CDC-1065 (w tym jego syntetyczne analogi - adozelesyna, karzelesyan i bizelesyna); podofilotoksyna; kwas podofilinowy; tenipozyd; kryptoficyny (w szczególności kryptoficyna 1 i kryptoficyna 8); dolastatyna; duokarmycyna (w tym jej syntetyczne analogi, KW-2189 i CB1-TM1); eleuterobina; pankratystatyna; sarkodiktyina, spongistatyna; iperyty azotowe takie, jak chlorambucyl, chlomafazyna, cholofosfamid, estramustyna, ifosfamid, mechloretamina, chlorowodorek tlenku mechloretaminy, melfalan, nowembichina, fenesteryna, prednimustyna, trofosfamid, iperyt uracylowy, nitrozomoczniki takie, jak karmustyna, chlorozotocyna, fotemustyna, lomustyna, nimustyna i ranimnustyna; antybiotyki, takie, jak antybiotyki enodiynowe (np. kalicheamycyna, zwłaszcza kalicheamycyna gamma 1l i kalicheamycyna omega l1 (zob. np. Angew. Chem. Intl. Ed. Engl., 33: (1994)); dynemycyna, w tym dynamycyna A; esperamycyna; a także chromofor neokarzynostatynowy i pokrewne chromofory enediynowych antybiotyków chromobiałkowych), aklacynomyzyny, aktynomycyna, autramycyna, azaseryna, bleomycyny, kaktynomycyny, karabicyna, karminomycyna, karzynofilina, chromomycyny, daktynomycyna, daunorubicyna, detorubicyna, 6-diazo-5-okso-L- norleucyna, ADRIAMYCIN doksorubicyna (w tym morfolinodoksorubicyna, cyjanomorfolinodoksorubicyna, 2-pirolinodoksorubicyna i deoksysoksorubicyna), epirubicyna, ezorubicyna, idarubicyna, marcellomycyna, mitomycyny, na przykład mitomycyna C, kwas mikofenolowy, nogalamycyna, oliwomycyny, peplomycyna, potfiromycyna, puromycyna, kwelamycyna, rodorubicyna, streptonigryna, streptozocyna, tubercydyna, ubenimeks, zynostatyna, zorubicyna; antymetabolity, takie jak metotreksat i 5- fluorouracyl (5-FU), analogi kwasu foliowego, takie jak denopteryna, metotreksat, pteropteryna, trimetreksat; analogi puryny, takie jak fludarabina, 6-merkaptopuryna, tiamipryna, tioguanina; analogi pirymidyny, takie jak ancytabina, azacytydyna, 6-azaurydyna, karmofur, cytarabina, didezoksyurydyna, doksyflurydyna, enocytabina, floksyurydyna; androgeny, takie jak kalusteron, propionian dromostanolonu, epitiostanol, mepitiostan, testolakton; antagoniści hormonów kory nadnerczy, jak aminoglutetymid, mitotan, trilostan; środki uzupełniające działanie kwasu foliowego, takie jak kwas frolinowy, aceglaton, glikozyd aldofosfamidu, kwas aminolewulinowy, amsakryna, bestrabucil, bisantren, edatraksat, defofamina, demekolcyna, diazykwon, elfomityna, octan eliptynium, epotilon, etoglucyd; azotan galu, hydroksymocznik, lentynian, lonidamina, majtansynoidy takie, jak majtansyna i ansamitocyny, mitoguazon, mitoksantron, mopidamol, nitraeryna, pentostatyna, fenamet, pirarubicyna, losoksantron, 2-etylohydrazyd; prokarbazyn; kompleks polisacharydowy PSK (JHS Natural Products, Eugene, OR), razoksan, rizoksyna, sizofiran; spirogerman, kwas tenuazonowy, triazykwon, 2,2',2 - trichlorotrietyloamina, trikoteceny (zwłaszcza toksyna T-2, werekuryna A, roridyna A i anguidyna); uretan, windezyna, (ELDISINE, FILDESIN ); dakarbazyna, mannomustyna, mitobronitol, mitolaktol, pipobroman, gacytozyna, arabinozyd ( Ara-C ), tiotepa, taksoidy, np. paklitaksel (TAXOL, Bristol- Myers Squibb Oncology, Pronceton, NJ, USA), ABRAXANE - nanocząsteczkowa formulacja paklitakselu związanego z albuminą, niezawierająca cremophoru (American Pharmaceutical Partners, Schaumberg, Illinois) i doksetaksel (TAXOTERE, Rhone-Poulenc Rorer, Antony, Francja), chlorambucyl, gemcytabina (GEMZAR ); 6-tioguanina, merkaptopuryna, metotreksat, analogi platyny, takie jak cisplatyna i karbopoplatyna, winblastyna, (VELBAN ); platyna, etopozyd (VP-16), ifosfamid, mitomycyna C, mitoksantron, winkrystyna (ONCOVIN ); oksaliplatyna; leukowowina; winorelbina (NAVELBINE ); nowantron, edatreksat, daunomycyna, daunomycyna, aminopteryna, ibandronian, inhibitor topoizomerazy RFS 2000, difluorometyloornityna (DMFO), retinoidy takie, jak kwas retinowy,
50 49 kapecytabina (XELODA ) i farmaceutycznie dopuszczalne sole, kwasy lub pochodne dowolnego z powyższych związków, a także skojarzenia dwóch lub większej liczby powyższych związków, na przykład CHOP, skrót skojarzonego leczenia cyklofosfamidem, doksorubicyną, winkrystyną i prednizolonem, lub FOLFOX skrót schematu leczenia zawierającego oksaliplatynę (ELOXA-TIN ) w połączeniu z 5-FU i leukowowiną. [0103] Definicja ta obejmuje również środki przeciwhormonalne o działaniu regulującym, zmniejszającym, blokującym lub hamującym działanie hormonów promujących wzrost raka, często podawane w leczeniu układowym, czyli ogólnoustrojowym. Środki te mogą same być hormonami. Przykłady obejmują antyestrogeny i selektywne modulatory receptorów estrogenowych (SERM), takie jak na przykład tamoksyfen (w tym NOLVADEX tamoksyfen), EVTSTA raloksyfen, droloksyfen, 4- hydroksytamoksyfen, trioksyfen, keoksyfen, LY117018, onapriston i FARESTON toremifen; antyprogesterony; środki zmniejszające ilość receptorów estrogenowych (ERD); środki o działaniu hamującym lub zamykającym jajniki, na przykład agoniści hormonu uwalniającego hormon luteinizujący (LHRH) takie, jak LUPRON i ELIGARD octan leuprolidu, octan gosereliny, octan busereliny i tripterelina; inne antyandrogeny takie,jak flutamid, nilutamid i bikalutamid; oraz inhibitory aromatazy hamujące enzym aromatazę, regulujący produkcję estrogenów w nadnerczach, jak na przykład, 4(5)-imidazole, aminoglutetimid, MEGASE octan megestrolu, AROMASIN eksemestan, formestan, fadrozol, RIVISOR worozol, FEMARA letrozol i ARIMIDEX anastrozol. Ponadto taka definicja chemioterapeutyków obejmuje bisfosfoniany takie, jak klodronian (na przykład, BONEFOS lub OS-TAC ), DIDROCAL etidronian, NE-58095, ZOMETA kwas zoledronowy/zoledronian, FOSAMAX alendronian, AREDIA pamidronian, SKELID tiludronian lub ACTONEL risedronian; a także troksacytabinę (1,3-dioksolanowy nukleozydowy analog cytozyny); oligonukleotydy antysensowne, zwłaszcza hamujące ekspresję genów w szlakach sygnałowych uczestniczących w nieprawidłowej proliferacji komórek, na przykład PKC-alfa, Raf, H-Ras i receptor czynnika wzrostu naskórka (EGF-R); szczepionki takie jak THERATOPE i szczepionki stosowane w terapii genowej, na przykład szczepionka ALLOVECTIN, szczepionka LEUVECTIN i szczepionka VAPID ; inhibitor topoizomerazy 1 LURTOTECAN ; ABARELIX rmrh; ditosylan lapatynibu (niskocząsteczkowy podwójny inhibitor kinaz tyrozynowych ErbB-2 i EGFR, znany również jako GW572016); oraz sole, kwasy lub pochodne dowolnego z powyższych związków. [0104] Środek hamujący wzrost w rozumieniu niniejszego opisu oznacza związek lub kompozycję hamującą wzrost komórki zależny od aktywacji HGF/c-met czy to in vitro, czy in vivo. Tak więc środek hamujący wzrost może być środkiem znacznie zmniejszającym odsetek HGF/c-met-zależnych komórek w fazie S. Przykłady środków hamujących wzrost obejmują środki blokujące progresję w cyklu komórkowym (w fazie innej niż faza S), takie jak środki indukujące zatrzymanie cyklu w fazie G1 lub w fazie M. Klasyczne blokery fazy M obejmują alkaloidy winka (winkrystynę i winblastynę), taksany i inhibitory topoizomerazy II takie jak doksorubicyna, epirubicyna, daunorubicyna, etopozyd i bleomycyna. Środki powodujące zatrzymanie cyklu komórkowego w fazie G1 rozciągają swe działanie również na zatrzymanie w fazie S; są to na przykład środki alkilujące DNA, takie jak tamoksyfen, prednizon, dakarbazyna, mechloretamina, cisplatyna, metotreksat, 5-fluorouracyl i ara-c. Dalsze informacje można znaleźć w pozycji The Molecular Basis of Cancer, Mendelsohn and Israel, (red.), rozdział 1 pt. Cell cycle regulation, oncogenes, and antineoplastic drugs autorstwa Murakami et al. (WB Saunders: Philadelphia, 1995), zwłaszcza na str. 13. Taksany (paklitaksel i docetaksel są lekami przeciwnowotworowymi pochodzącymi z drzewa cisowego. Docetaxel (TAXOTERE, Rhone-Poulenc Rorer), pochodzący z cisu europejskiego, jest półsyntetycznym analogiem paklitakselu (TAXOL, Bristol-Myers Squibb). Paklitaksel i docetaksel promują tworzenie mikrotubuli z dimerów tubulinowych i stabilizują mikrotubule zapobiegając ich depolimeryzacji, co prowadzi do hamowania mitozy w komórkach. [0105] Doksorubicyna jest antybiotykiem antracyklinowym. Pełna nazwa chemiczna doksorubicyny to (8S-cis)-10-[(3-amino-2,3,6-trideoksy-a-L-likso-heksapiroanozyl)oksy]-7,8,9,10-tetrahydro-6,8,11- trihydroksy-8-(hydroksyacetylo)-1-metoksy-5,12-naftacenodion. Generowanie wariantowych przeciwciał charakteryzujących się zmniejszoną odpowiedzią HAMA lub brakiem tej odpowiedzi [0106] Zmniejszenie lub eliminacja odpowiedzi HAMA jest istotnym aspektem rozwoju klinicznego odpowiednich środków terapeutycznych. Zob. np. Khaxzaeli et al., J. Natl. Cancer Inst. (1988), 80:937; Jaffers et al., Transplantation (1986), 41: 572; Shawler et al., J. Immunol. (1985), 135:1530; Sears et al., J. Biol. Response Mod. (1984), 3:138; Miller et al., Blood (1983), 62:988; Hakimi et al., J. Immunol. (1991), 147:1352; Reichmann et al., Nature (1988), 332:323; Junghans et al., Cancer Res. (1990), 50:1495. Zgodnie z przedstawionym opisem, wynalazek dotyczy przeciwciał humanizowanych
51 50 tak, aby zmniejszyć lub wyeliminować odpowiedź HAMA. Następnie przy użyciu metod znanych w dziedzinie wynalazku, z których niektóre opisano poniżej, można uzyskać warianty tych przeciwciał. [0107] Na przykład sekwencja aminokwasów opisywanego tu przeciwciała może posłużyć jako sekwencja wyjściowa (macierzysta) dla procesu dywersyfikacji sekwencji zrębowych i/lub hiperzmiennych. Wybraną sekwencję zrębową, do której przyłączona jest wyjściowa sekwencja hiperzmienna określa się tu jako akceptorową ludzką sekwencję zrębową. Choć akceptorowe ludzkie sekwencje zrębowe mogą być uzyskane z ludzkich immunoglobulin (ich regionów VL i/lub VH) lub być ich pochodnymi, akceptorowe ludzkie sekwencje zrębowe korzystnie są uzyskiwane z ludzkich konsensusowych sekwencji zrębowych lub są ich pochodnymi, ponieważ, jak wykazano, sekwencje te charakteryzują się minimalną lub zerowa immunogennością u pacjentów ludzkich. [0108] W przypadkach, gdy sekwencja akceptorowa pochodzi z immunoglobuliny ludzkiej opcjonalnie możliwy jest wybór ludzkiej sekwencji zrębowej w oparciu o jej homologię z donorową sekwencją zrębową poprzez ułożenie donorowej sekwencji zrębowej obok szeregu ludzkich sekwencji zrębowych i wybór najbardziej homologicznej sekwencji zrębowej na sekwencje akceptorową. [0109] W jednym z wariantów wynalazku ludzkie sekwencje konsensusowe są uzyskiwane z konsensusowych sekwencji zrębowych z podgrupy III dla domeny VH i/lub z podgrupy kappa I dla domeny VL, lub są ich pochodnymi. [0110] Tak więc ludzka akceptorowa sekwencja zrębowa VH może zawierać jedną, dwie, trzy lub wszystkie poniższe sekwencje zrębowe: FR1, zawierająca sekwencję EVQLVESGGGLVQPGGSLRLSCAAS (SEQ ID NO: 42), FR2, zawierająca sekwencję WVRQAPGKGLEWV (SEQ ID NO: 43), FR3, zawierająca sekwencję RFTISX1DX2SKNTX3YLQMNSLRAEDTAVYYC (SEQ ID NO: 50), gdzie X1 = A lub R, X2 = T lub N, a X3 = A lub L, FR4, zawierająca sekwencję WGQGTLVTVSS (SEQ ID NO: 45). [0111] Przykłady konsensusowych sekwencji zrębowych VH obejmują: ludzką konsensusową sekwencję zrębową VH z podgrupy I minus CDR Kabata (SEQ ID NO: 19); ludzką konsensusową sekwencję zrębową VH z podgrupy I minus rozszerzone regiony hiperzmienne (SEQ ID NO: 20-22); ludzką konsensusową sekwencję zrębową VH z podgrupy II minus CDR Kabata (SEQ ID NO: 23); ludzką konsensusową sekwencję zrębową VH z podgrupy II minus rozszerzone regiony hiperzmienne (SEQ ID NO: 24-26); ludzką konsensusową sekwencję zrębową VH z podgrupy III minus CDR Kabata (SEQ ID NO: 27); ludzką konsensusową sekwencję zrębową VH z podgrupy III minus rozszerzone regiony hiperzmienne (SEQ ID NO: 28-30); ludzką akceptorową sekwencję zrębową VH minus CDR Kabata (SEQ ID NO: 31); ludzką akceptorową sekwencję zrębową VH minus rozszerzone regiony hiperzmienne (SEQ ID NO: 32-33); ludzką akceptorową sekwencję zrębową VH nr 2 minus CDR Kabata (SEQ ID NO: 34); lub ludzką akceptorową sekwencję zrębową VH nr 2 minus rozszerzone regiony hiperzmienne (SEQ ID NO: 35-37). [0112] W jednym z wariantów, ludzka akceptorowa sekwencja zrębowa VH zawiera jedną, dwie, trzy lub wszystkie poniższe sekwencje zrębowe: FR1, zawierająca sekwencję EVQLVESGGGLVQPGGSLRLSCAAS (SEQ ID NO: 42), FR2, zawierająca sekwencję WVRQAPGKGLEWV (SEQ ID NO: 43), FR3, zawierająca sekwencję RFTISADTSKNTAYLQMNSLRAEDTAVYYC (SEQ ID NO: 51), RFTISADTSKNTAYLQMNSLRAEDTAVYYCA (SEQ ID NO: 52), RFTISADTSKNTAYLQMNSLRAEDTAVYYCAR (SEQ ID NO: 53),
52 51 RFTISADTSKNTAYLQMNSLRAEDTAVYYCS (SEQ ID NO: 54) lub RFTISADTSKNTAYLQMNSLRAEDTAVYYCSR (SEQ ID NO: 55), FR4, zawierająca sekwencję WGQGTLVTVSS (SEQ ID NO: 45). [0113] W jednym z wariantów, ludzka akceptorowa sekwencja zrębowa VL zawiera jedną, dwie, trzy lub wszystkie poniższe sekwencje zrębowe: FR1, zawierająca sekwencję DIQMTQSPSSLSASVGDRVTITC (SEQ ID NO: 46), FR2, zawierająca sekwencję WYQQKPGKAPKLLIY (SEQ ID NO: 47), FR3, zawierająca sekwencję GVPSRFSGSGSGTDFTLTISSLQPEDFATYYC (SEQ ID NO: 48), FR4, zawierająca sekwencję FGQGTKVEIK (SEQ ID NO: 49). Przykłady konsensusowych sekwencji zrębowych VL obejmują: ludzką konsensusową sekwencję zrębową VL kappa z podgrupy I (SEQ ID NO: 38); ludzką konsensusową sekwencję zrębową VL kappa z podgrupy II (SEQ ID NO: 39); ludzką konsensusową sekwencję zrębową VL kappa z podgrupy III (SEQ ID NO: 40); lub ludzką konsensusową sekwencję zrębową VL kappa z podgrupy IV (SEQ ID NO: 41) [0114] Choć sekwencja akceptorowa może być identyczna z wybraną ludzka sekwencją zrębową, czy to pochodzącą z ludzkiej immunoglobuliny, czy też z ludzkiej sekwencji konsensusowej, w niniejszym wynalazku rozważa się możliwość, że sekwencja akceptorowa może zawierać wcześniej określone substytucje aminokwasów w porównaniu z ludzką konsensusową sekwencją zrębową. Rzeczone określone wcześniej substytucje są korzystnie minimalne, zazwyczaj obejmują jedynie cztery, trzy, dwie lub jedną różnicę w sekwencji aminokwasów w porównaniu z sekwencją immunoglobuliny ludzkiej lub konsensusowej sekwencji zrębowej. [0115] W akceptorowe sekwencje zrębowe VL i/lub VH wprowadzane są reszty regionów hiperzmiennych przeciwciał pochodzących z gatunków innych niż człowiek. Na przykład możliwe jest wprowadzenie reszt odpowiadających resztom regionu CDR Kabata, resztom hiperzmiennych pętli Chothii, resztom AbM i/lub resztom kontaktowym. Opcjonalnie wprowadzane są następujące reszty rozszerzonego regionu hiperzmiennego: (L1), (L2) i (L3), (H1), lub (H2) i , , lub (H3). [0116] Podczas gdy w niniejszym opisie omawia się włączenie reszt regionów hiperzmiennych, rozumie się, że cel ten można osiągnąć na różne sposoby, na przykład kwas nukleinowy kodujący pożądaną sekwencję aminokwasów może zostać wygenerowany na drodze mutacji kwasu nukleinowego kodującego sekwencję zmiennej domeny mysiej, w wyniku czego reszty zrębowe tej domeny zostaną przekształcone w reszty zrębowe ludzkiej akceptorowej sekwencji zrębowej, lub na drodze mutacji kwasu nukleinowego kodującego ludzką sekwencję domeny zmiennej, w wyniku czego reszty domeny hiperzmiennej zostaną przekształcone w reszty sekwencji innej niż ludzka, lub tez na drodze zsyntetyzowania kwasu nukleinowego kodującego pożądaną sekwencję itp. [0117] W podanych przykładach warianty bezpośrednich graftów regionów hiperzmiennych generowano techniką mutagenezy kwasów nukleinowych kodujących ludzkie sekwencje akceptorowe wg Kunkela, stosując oddzielny oligonukleotyd dla każdego regionu hiperzmiennego. Kunkel et al., Methods Enzymol. 154: (1987). Przy użyciu rutynowych technik w regionie zrębowym i/lub w regionie hiperzmiennym można wprowadzić odpowiednie zmiany mające na celu poprawę i ponowne ustanowienie właściwych oddziaływań między regionem hiperzmiennym a antygenem. [0118] Technika ekspresji fagemidowej (w niektórych kontekstach określana tu również jako technika ekspresji fagowej) może zostać wykorzystana w charakterze wygodnej i szybkiej metody generowania i przeszukiwania dużej liczb różnych potencjalnych przeciwciał wariantowych w bibliotece wygenerowanej na drodze randomizacji sekwencji. Osoba biegła w dziedzinie ma jednak dostęp do innych metod wytwarzania i badania zmodyfikowanych przeciwciał. [0119] Technologia ekspresji fagowej (fagemidowej) dostarczyła potężnego narzędzia generowania i selekcji nowych białek wiążących ligand, na przykład antygen. Zastosowanie technik ekspresji fagowej (fagemidowej) umożliwia wygenerowanie dużych bibliotek wariantowych białek, które można szybko sortować pod kątem sekwencji wiążących z dużym powinowactwem cząsteczkę docelową. Kwasy nukleinowe kodujące wariantowe polipeptydy z reguły przyłączone są do sekwencji nukleotydowwej
53 52 kodującej białko płaszcza wirusa, na przykład białko genu III lub białko genu VIII. Opracowano monowalencyjne systemy ekspresji fagemidowej, w których sekwencja nukleotydów kodująca białko lub polipeptyd przyłączona jest do sekwencji nukleotydów kodujących część białka genu III (Bass, S., Proteins, 8:309 (1990); Lowman and Wells, Methods: A Companion to Methods in Enzymology, 3:205 (1991)). W monowalencyjnym fagemidowym systemie ekspresji gen fuzyjny ulega ekspresji na niskim poziomie; ekspresji ulegają również białka genu dzikiego typu III, dzięki czemu zachowana jest zakaźność cząstek. Metody generowania bibliotek peptydów i przeszukiwania tych bibliotek ujawniono w wielu patentach (np. patent nr US 5,723,286, nr US 5,432,018, nr US 5,580,717, nr US 5,427,908 i nr US 5,498,530). [0120] Biblioteki przeciwciał lub polipeptydów wiążących antygen przygotowano różnymi sposobami, w tym zmieniając pojedynczy gen poprzez wprowadzanie losowych sekwencji DNA lub poprzez klonowanie rodziny pokrewnych genów. Metody wyświetlania przeciwciał lub fragmentów wiążących antygen przy użyciu fagów/fagemidów opisano w patentach nr US 5,750,373, US 5,733,743, US 5,837,242, US 5,969,108, US 6,172,197, US 5,580,717 i US 5,658,727. Biblioteki następnie przeszukuje się pod kątem ekspresji antygenów lub białek wiążących antygeny posiadających pożądane właściwości. [0121] Metody substytuowania wybranego aminokwasu w kwas nukleinowy szablonu są dobrze znane w dziedzinie; niektóre z nich zostały tu opisane. Na przykład, substytucji reszt w regionie hiperzmiennym można dokonać metodą Kunkela. Zob. np. Kunkel et al., Methods Enzymol. 154: (1987). [0122] Sekwencja oligonukleotydów zawiera jeden lub większą liczbę zaprojektowanych zestawów kodonów dla zmienianych reszt regionów hiperzmiennych. Zestaw kodonów jest zestawem różnych trypletowych sekwencji nukleotydowych służących do kodowania pożądanych wariantowych aminokwasów. Zestawy kodonów mogą być przedstawiane przy użyciu symboli oznaczających poszczególne nukleotydy lub równomolowe mieszaniny nukleotydów, jak przedstawiono poniżej w przypadku kodu IUB. KODY IUB G Guanina A Adenina T Tymina C Cytozyna R (A lub G) Y (C lub T) M (A lub C) K (G lub T) S (C lub G) W (A lub T) H (A lub C lub T) B (C lub G lub T) V (A lub C lub G) D (A lub G lub T) N (A lub C lub G lub T) [0123] Na przykład w zestawie kodonów DVK, D może być nukleotydem A lub G lub T; V może być nukleotydem A lub G lub C; zaś K może być nukleotydem G lub T. Omawiany zestaw kodonów może zawierać 18 różnych kodonów i kodować aminokwasy Ala, Trp, Tyr, Lys, Thr, Asn, Lys, Ser, Arg, Asp, Glu, Gly i Cys. [0124] Zestawy oligonukleotydów lub starterów można zsyntetyzować przy użyciu standardowych metod. Można na przykład metodą syntezy w fazie stałej zsyntetyzować zestaw oligonukleotydów zawierających sekwencje reprezentujące wszystkie możliwe triplety nukleotydów chodzące w skład
54 53 zestawu kodonów, kodujące pożądaną grupę aminokwasów. W dziedzinie znana jest synteza oligonukleotydów z wybraną degeneracją w określonych pozycjach. Tego rodzaju zestawy nukleotydów posiadające określone zestawy kodonów mogą zostać zsyntezowane przy użyciu dostępnych na rynku syntetyzerów kwasów nukleinowych (oferowanych na przykład przez Applied Biosystems, Foster City, CA, USA), albo mogą zostać nabyte na rynku (na przykład od firmy Life Technologies, Rockville, MD, USA). W związku z powyższym zsyntetyzowany zestaw nukleotydów posiadający dany zestaw kodonów zazwyczaj zawiera wielość oligonukleotydów o różnych sekwencjach, przy czym różnice ustalane są przez zestaw kodonów w ogólnej sekwencji. Oligonukleotydy stosowane według wynalazku posiadają sekwencje pozwalające na ich hybrydyzację do szablonu kwasu nukleinowego kodującego domenę zmienną i mogą zawierać ponadto miejsca restrykcyjne na potrzeby klonowania. [0125] W jednej z metod sekwencje kwasów nukleinowych kodujących wariantowe aminokwasy mogą zostać utworzone na drodze mutagenezy za pośrednictwem oligonukleotydów. Technika ta jest dobrze znana w dziedzinie i została opisana w pracy Zoller et al., Nucleic Acids Res. 10: (1987). W skrócie, sekwencje kwasów nukleinowych kodujące wariantowe aminokwasy tworzy się na drodze hybrydyzacji zestawu oligonukleotydów kodującego pożądane zestawy kodonów do szablonu DNA, gdzie szablon jest jednołańcuchową formą plazmidu zawierającą sekwencję szablonową kwasu nukleinowego regionu zmiennego. Po hybrydyzacji przy użyciu polimerazy DNA syntetyzuje się cały drugi, komplementarny łańcuch szablonu, który w ten sposób będzie zawierał starter oligonukleotydowy i zestawy kodonów dostarczone przez zestaw oligonukleotydów. [0126] Z reguły stosuje się oligonukleotydy o długości co najmniej 25 nukleotydów. Optymalny oligonukleotyd będzie posiadał od 12 do 15 nukleotydów w pełni komplementarnych względem szablonu po którejkolwiek stronie nukleotydu(-ów) kodującego(-ych) mutację(-e). Zapewnia to prawidłową hybrydyzację oligonukleotydu do jednołańcuchowego szablonu DNA. Oligotydy łatwo syntetyzuje się metodami znanymi w dziedzinie, takimi, jak na przykład opisana w pracy Crea et al., Pro. Nat'l. Acad. Sci. USA, 75:5765 (1978). [0127] Szablon DNA generowany jest przez wektory, które pochodzą albo z wektorów bakteriofaga M13 (odpowiednie są handlowo dostępne wektory M13mp18 i M13mp19 ), albo z wektorów zawierających miejsce inicjacji replikacji faga w pojedynczym łańcuchu, zgodnie z opisem w pracy Viera et al., Meth. Enzymol., 153:3 (1987). Tak więc DNA, które ma zostać zmutowane, może zostać wprowadzone do jednego z tych wektorów w celu wygenerowania jednołańcuchowego szablonu. Wytwarzanie jednołańcuchowego szablonu opisano w sekcjach pracy Sambrook et al., supra. [0128] W celu zmiany natywnej sekwencji DNA, oligonukleotyd hybrydyzuje się do jednołańcuchowego szablonu w odpowiednich warunkach hybrydyzacji. Następnie dodaje się enzym polimeryzujący DNA, zazwyczaj polimeraza DNA T7 lub fragment Klenow polimerazy DNA I w celu zsyntetyzowania komplementarnego łańcucha szablonu z wykorzystaniem oligonukleotydu jako startera w procesie syntezy. W ten sposób tworzy się cząsteczkę heterodupleksową, w której jeden łańcuch koduje zmutowana postać genu 1, zaś drugi (oryginalny szablon koduje natywną, niezmodyfikowaną sekwencję genu 1. Otrzymana cząsteczka heterodipleksowa transformowana jest następnie do odpowiedniej komórki gospodarza, zazwyczaj prokariotycznej, takiej, jak E. coli JM101. Po wyhodowaniu, komórki nanosi się na płytki agarozowe i poddaje badaniu przesiewowemu przy użyciu oligonukleotydowego startera znakowanego radioizotopowo fosforanem-32 w celu zidentyfikowania klonów bakteryjnych zawierających zmutowane DNA. [0129] Powyższą metodę można modyfikować, wytwarzając cząsteczkę homodupleksową, w której oba łańcuchy plazmidu zawierają mutację(-e). Modyfikacje przebiegają w następujący sposób: Jednołańcuchowy oligonukleotyd przyłącza się do jednołańcuchowego szablonu w sposób opisany powyżej. Mieszaninę trzech deoksyrybonukleozydów: deoksyryboadenozyny (datp), deoksyryboguanozyny (dgtp) i deoksyrybotymidyny (dtt), miesza się z modyfikowaną tiodeoksyrybocytozyną o symbolu dctp-(as) (dostępną w handlu dzięki firmie Amersham). Mieszaninę tę dodaje się do kompleksu szablon-oligonukleotyd. W momencie dodania do tej mieszaniny polimerazy DNA generowany jest łańcuch DNA identyczny z szablonem z wyjątkiem zmutowanych zasad. Ponadto, nowy łańcuch DNA będzie zawierał dctp-(as) zamiast dctp, co służy jego ochronie przed trawieniem restrykcyjną endonukleazą. Po posegmentowaniu łańcucha szablonu dwułańcuchowego heterodupleksu przy użyciu odpowiedniego enzymu restrykcyjnego, łańcuch szablonu może zostać poddany trawieniu nukleazą ExoIII lub inną odpowiednią nukleazą w odcinku następującym za regionem zawierającym miejsce(-a) będące przedmiotem mutagenezy. Reakcję następnie przerywa się, otrzymując cząsteczkę jedynie częściowo jednołańcuchową. Następnie przy
55 54 użyciu polimerazy DNA w obecności wszystkich czterech trifosforanów deoksyrybonukleotydowych, ATP i ligazy DNA tworzy się pełny dwułańcuchowy homodupleks DNA. Tak otrzymana cząsteczka homodupleksu może następnie być transformowana do odpowiedniej komórki gospodarza. [0130] Jak wzmiankowano powyżej, sekwencja zestawu oligonukleotydów ma długość wystarczającą do hybrydyzacji do szablonowego kwasu nukleinowego i może ponadto, choć nie jest to konieczne, zawierać miejsca restrykcji. Szablon DNA może być generowany przez wektory, które pochodzą albo z wektorów bakteriofaga M13, albo z wektorów zawierających miejsce inicjacji replikacji faga w pojedynczym łańcuchu, zgodnie z opisem w pracy Viera et al., Meth. Enzymol., 153:3 (1987). Tak więc DNA, które ma zostać zmutowane, musi zostać wprowadzone do jednego z tych wektorów w celu wygenerowania jednołańcuchowego szablonu. Wytwarzanie jednołańcuchowego szablonu opisano w sekcjach pracy Sambrooket al., supra. [0131] Zgodnie z inną metodą, bibliotekę można przygotować zapewniając zestawy oligonukleotydów dla górnego i dolnego odcinka, gdzie każdy zestaw zawiera wielość oligonukleotydów o różnych sekwencjach, różne sekwencje ustalane są przez zestawy kodonów umieszczonych w sekwencji oligonukleotydów. Zestawy oligonukleotydów górnych i dolnych odcinków wraz z sekwencją nukleotydów kodujących domenę zmienną mogą być wykorzystane w reakcji PCR w celu wygenerowania biblioteki produktów PCR. Produkty PCR można określać jako kasety kwasu nukleinowego, ponieważ można je łączyć z innymi pokrewnymi lub niepokrewnymi sekwencjami nukleotydów, na przykład odpowiadającymi białkom płaszcza wirusowego i domenom dimeryzacyjnym z wykorzystaniem znanych technik biologii molekularnej. [0132] Sekwencja starterów PCR zawiera jeden lub większą liczbę zaprojektowanych zestawów kodonów kodujących powierzchnie dostępne dla rozpuszczalnika i wysoce zróżnicowane pozycje w regionie hiperzmiennym. Jak opisano powyżej, zestaw kodonów jest zestawem różnych trypletowych sekwencji nukleotydowych służących do kodowania pożądanych wariantowych aminokwasów. [0133] Czynniki selekcjonujące przeciwciała spełniające pożądane kryteria, dobrane na drodze odpowiednich etapów przesiewania/selekcji, mogą zostać wyizolowane i poddane klonowaniu z zastosowaniem standardowych technik rekombinacyjnych. Wektory, komórki gospodarzy i metody rekombinacji [0134] W celu rekombinacyjnego wytwarzania przeciwciała będącego przedmiotem wynalazku, kodujący je kwas nukleinowy jest wyodrębniany i wprowadzany do replikowalnego wektora w celu dalszego klonowania (amplifikacji DNA) lub ekspresji. DNA kodujące przeciwciało jest łatwo wyodrębniane i sekwencjonowane z zastosowaniem konwencjonalnych procedur (np. dzięki zastosowaniu sond oligonukleotydowych zdolnych do specyficznego wiązania genów kodujących ciężki i lekki łańcuch przeciwciała). Dostępnych jest wiele wektorów. Wybór wektora zależy częściowo od komórki gospodarza, która ma zostać zastosowana. Z reguły korzystne komórki gospodarza są pochodzenia prokariotycznego lub eukariotycznego (z reguły ssaczego). Generowanie przeciwciał z wykorzystaniem prokariotycznych komórek gospodarza: Konstruowanie wektora [0135] Sekwencje polinukleotydowe kodujące polipeptydowe komponenty przeciwciała będącego przedmiotem wynalazku można uzyskać w oparciu o standardowe techniki rekombinacyjne. Pożądane sekwencje polinukleotydowe mogą być wyodrębnione z komórek wytwarzających przeciwciała, na przykład komórek hybrydowych, a następnie poddane sekwencjonowaniu. Ewentualnie, polinukleotydy można syntetyzować przy użyciu syntetyzera nukleotydów lub z zastosowaniem technik PCR. Po otrzymaniu, sekwencje kodujące polipeptydy wprowadza się do rekombinowanego wektora zdolnego do replikacji i ekspresji heterologicznych polinukleotydów w prokariotycznych komórkach gospodarza. Do celów niniejszego wynalazku może zostać wykorzystanych wiele dostępnych i znanych w dziedzinie wektorów. Wybór odpowiedniego wektora będzie zależał głównie od rozmiaru kwasu nukleinowego, który ma zostać wprowadzony do wektora oraz od komórki gospodarza, która ma zostać poddana transformacji przy użyciu wektora. Każdy wektor zawiera różne elementy, zależnie od funkcji (amplifikacja lub ekspresja heterologicznego polinukleotydu lub obie te funkcje) i zgodności z komórką gospodarza, w której ma rezydować. Elementy wektora z reguły obejmują, choć nie ograniczają się do poniższych: miejsce inicjacji replikacji, marker selekcji, promotor, miejsce wiązania rybosomu (RBS), sekwencja sygnałowa, insert heterologicznego kwasu nukleinowego oraz sekwencja terminacji transkrypcji.
56 55 [0136] Z reguły wektory plazmidowe zawierające replikon i sekwencje sterujące pochodzące z gatunków zgodnych z komórkami gospodarza stosowane są z tymi właśnie komórkami. Wektor zazwyczaj zawiera miejsce replikacji a także sekwencje markerowe, mogące zapewniać selekcję fenotypów transformowanych komórek. Na przykład komórki E. coli zazwyczaj transformuje się przy użyciu pbr322, plazmidu pochodzącego z gatunku E. coli. pbr322 zawiera geny kodujące oporność na ampicylinę (Amp) i tetracyklinę (Tet), tym samym zapewniając dogodny sposób identyfikacji transformowanych komórek. pbr322, jego pochodne lub inne mikrobiotyczne plazmidy lub bakteriofagi mogą również zawierać, lub zostać poddane modyfikacji tak, by zawierały, promotory, które mogą być wykorzystywane przez drobnoustroje do ekspresji endogennych białek. Przykłady pochodnych pbr322 wykorzystywanych do ekspresji określonych przeciwciał opisano szczegółowo w pracy Carter et al., patent nr US 5,648,237. [0137] Ponadto, wektory fagowe zawierające replikon i sekwencje sterujące, zgodne z mikroorganizmem gospodarza, mogą być wykorzystane jako wektory transformujące w odniesieniu do rzeczonych komórek gospodarza. Na przykład bakteriofag taki, jak λgem.tm.-11, może być wykorzystany do wygenerowania rekombinowanego wektora, który może zostać użyty so transformacji podatnych komórek gospodarza takich, jak E. coli LE392. [0138] Wektor ekspresyjny będący przedmiotem wynalazku może zawierać dwie lub większą liczbę par promotor-cistron, kodujących każdy z komponentów polipeptydowych. Promotor jest nie ulegającą translacji sekwencją regulacyjną umieszczoną powyżej (5') cistronu modulującego jego ekspresję. Promotory prokariotyczne zazwyczaj należą do jednej dwóch klas - promotorów indukowalnych i konstytutywnych. Promotor indukowalny jest promotorem inicjującym podwyższony poziom transkrypcji kontrolowanego przez siebie cistronu w odpowiedzi na zmiany w warunkach hodowli, np. obecności lub braku substancji odżywczej lub zmiany temperatury. [0139] Znanych jest wiele promotorów rozpoznawanych przez wiele potencjalnych komórek gospodarzy. Wybrany promotor może być operacyjnie połączony z DNA cistronu kodującego łańcuch lekki lub ciężki poprzez usunięcie promotora ze źródłowego DNA metodą trawienia enzymami restrykcyjnymi i wprowadzenie wyizolowanej sekwencji promotora do wektora będącego przedmiotem wynalazku. DO bezpośredniej amplifikacji i/lub ekspresji docelowych genów może zostać użyta zarówno natywna sekwencja promotora, jak i wiele heterologicznych promotorów. W niektórych wariantach wykorzystuje się heterologiczne promotory, ponieważ z reguły pozwalają one na uzyskanie wyższej transkrypcji i wyższe wydajności ekspresji genu docelowego w porównaniu z natywnym promotorem ekspresji docelowego polipeptydu. [0140] Promotory nadające się do wykorzystania w prokariotycznych komórkach gospodarza obejmują promotor PhoA, systemy promotorów β-galaktamazy i laktozy, system promotora tryptofanu (trp) oraz promotory hybrydowe, takie jak promotor tac lub trc. Do zastosowania nadają się również jednak inne promotory działające w komórkach bakterii (na przykład inne znane promotory bakteryjne lub fagowe). Sekwencje nukleotydowe tych promotorów zostały opublikowane, co pozwala przeszkolonemu pracownikowi połączenie ich z cistronami kodującymi docelowe łańcuchy lekki i ciężkie (Siebenlist et al. (1980) Cell 20: 269) przy użyciu sekwencji łącznikowych lub adaptorowych w celu wprowadzenia wymaganych miejsc restrykcji. [0141] W jednym z aspektów wynalazku każdy cistron w rekombinowanym wektorze zawiera komponent sekwencji sygnałowej sekrecji, sterującej translokacją ulegających ekspresji polipeptydów przez błonę komórkową. Z reguły sekwencja sygnałowa może być komponentem wektora lub też może stanowić część DNA docelowego polipeptydu wprowadzanego do wektora. Wybrana do celów niniejszego wynalazku sekwencja sygnałowa powinna być sekwencją rozpoznawaną i przetwarzaną (np. ciętą przez peptydazę sygnałową) w komórce gospodarza. W przypadku prokariotycznych komórek gospodarza nierozpoznających i nieprzetwarzających sekwencji sygnałowych natywnych dla heterologicznych polipeptydów, sekwencję sygnałową zastępuje się prokariotyczną sekwencją sygnałową wybraną na przykład z grupy obejmującej fosfatazę zasadową, penicylinazę, Ipp lub trwałe termicznie sekwencje liderowe enterotoksyny II (STII), LamB, PhoE, PelB, OmpA i MBP. W jednym z wariantów wynalazku sekwencje sygnałowe stosowane w obu cistronach systemu ekspresji są sekwencjami sygnałowymi STII lub jej wariantami. [0142] W innym aspekcie wynalazku wytwarzanie immunoglobulin według wynalazku może zachodzić w cytoplazmie komórki gospodarza, a tym samym nie wymagać obecności sekwencji sygnałowych sekrecji w każdym z cistronów. W takim przypadku lekkie i ciężkie łańcuchy immunoglobuliny ulegają ekspresji, zwijaniu i łączeniu w funkcjonalne immunoglobuliny wewnątrz cytoplazmy. Niektóre szczepy gospodarza (np. szczepy E. coli trxb - ) zapewniają warunki cytoplazmatyczne sprzyjające tworzeniu
57 56 wiązań disiarczkowych, tym samym umożliwiając właściwe zwijanie i łączenie ulegających ekspresji podjednostek białkowych. Proba i Pluckthun, Gene, 159:203 (1995). [0143] Niniejszy wynalazek ujawnia system ekspresji, w którym ilościowe stosunki podlegających ekspresji komponentów polipeptydowych mogą być modulowane w celu zmaksymalizowania wydajności wydzielanych i prawidłowo skonstruowanych przeciwciał będących przedmiotem wynalazku. Modulacje taką osiąga się przynajmniej częściowo poprzez równoległą modulację sił translacyjnych komponentów polipeptydowych. [0144] Jedną z technik modulowania siły translacji jest technika ujawniona w pracy Simmons et al., patent nr US 5,840,523. Wykorzystuje ona region inicjalizacji translacji (TIR) w cistronie. Dla danego regionu TIR istnieje możliwość stworzenia serii wariantowych sekwencji aminokwasowych lub nukleotydowych o różnych siłach translacyjnych, co zapewnia dogodny sposób dopasowywania tego czynnika do pożądanego poziomu ekspresji określonego łańcucha. Warianty TIR mogą być generowane zwykłymi technikami mutagenetycznymi prowadzącymi do zmian kodonów mogących zmieniać sekwencje aminokwasów, choć preferowane są ciche zmiany sekwencji nukleotydowej. Modyfikacje TIR mogą obejmować na przykład modyfikacje liczby sekwencji Shine-Dalgarno lub odstępów pomiędzy nimi, wraz z modyfikacjami sekwencji sygnałowej. Inną metodą generowania zmutowanych sekwencji sygnałowych jest wygenerowanie banku kodonów na początku sekwencji kodującej, nie zmieniającego sekwencji aminokwasów w sekwencji sygnałowej (tj. wygenerowanie cichych zmian). Cel ten może zostać osiągnięty poprzez zmianę trzeciego nukleotydu w każdym kodonie. Ponadto niektóre aminokwasy takie, jak leucyna, seryna i arginina posiadają wielokrotne pierwsze i drugie pozycje kodonu, co może zwiększyć złożoność generowanego banku. Omawiana metoda mutagenezy została dokładnie opisana w pracy Yansura et al. (1992) METHODS: A Companion to Methods in Enzymol. 4: [0145] Korzystnie generuje się zestaw wektorów z różna siłą TIR w każdym cistronie. Tak ograniczony zestaw zapewnia porównanie poziomów ekspresji każdego łańcucha, a także wydajności pożądanych produktów-przeciwciał w różnych kombinacjach sił TIR. Siły TIR można zmierzyć poprzez ilościowe oznaczenie poziomu ekspresji genu reporterowego, jak to zostało szczegółowo opisane w pracy Simmons et al., patent nr US 5,840,523. W oparciu o porównanie siły translacji wybierane są pożądane regiony TIR mające podlegać łączeniu z konstruktami wektorów ekspresyjnych będących przedmiotem wynalazku. [0146] Prokariotyczne komórki gospodarza nadające się do ekspresji przeciwciał będących przedmiotem wynalazku obejmują bakterie podkrólestw Archaebacteria i Eubacteria, takie jak organizmy Gram-ujemne lub Gram-dodatnie. Przykłady użytecznych bakterii obejmują rodzaj Escherichia (np. E. coli), Bacilli (np. B. subtilis), Enterobacteria, gatunki Pseudomonas (np. P. aeruginosa), Salmonella typhimurium, Serratia marcescans, Klebsiella, Proteus, Shigella, Rhizobia, Vitreoscilla lub Paracoccus. W jednym z wariantów stosuje się komórki gram-ujemne. W jednym z wariantów jako komórki gospodarza dla wynalazku stosuje się komórki E. coli. Przykłady szczepów E. coli obejmują szczep W3110 (Bachmann, Cellular and Molecular Biology, t. 2 (Washington, D.C.: American Society for Microbiology, 1987), str ; nr depozytu ATCC 27,325) oraz jego pochodne, w tym szczep 33D3 o genotypie W3110 ΔfhuA (ΔtonA) ptr3 lac Iq lacl8 ΔompTΔ(nmpcfepE) degp41 kan R (patent nr US 5,639,635). Odpowiednie są również inne szczepy oraz ich pochodne, na przykład E. coli 294 (ATCC 31,446), E. coli B, E. coli λ 1776 (ATCC 31,537) i E. coli RV308(ATCC 31,608). Przykłady te mają charakter ilustracyjny i nie ograniczający. Metody konstruowania pochodnych którychkolwiek z wyżej wymienionych bakterii o określonych genotypach są znane w dziedzinie wynalazku i opisane na przykład w pracy Bass et al., Proteins, 8: (1990). Zazwyczaj konieczne jest dokonanie wyboru odpowiednich bakterii, biorąc pod uwagę możliwość replikacji replikonu w komórkach bakteryjnych. Na przykład gdy replikon dostarczany jest przez dobrze poznane plazmidy takie, jak pbr322, pbr325, pacyc177 czy pkn410, jako odpowiednie komórki gospodarza mogą zostać użyte gatunki E. coli, Serratia lub Salmonella. Zazwyczaj komórki gospodarza powinny wydzielać minimalne ilości enzymów proteolitycznych, zas do hodowli komórkowej mogą być dodatkowo wprowadzane dodatkowe inhibitory proteazy. Wytwarzanie przeciwciał [0147] Komórki gospodarza są transformowane opisanymi powyżej wektorami ekspresyjnymi i hodowane na konwencjonalnych pożywkach zmodyfikowanych w zależności od potrzeb w kierunku indukcji promotorów, wyboru transformowanych komórek lub amplifikacji genów kodujących pożądane sekwencje.
58 57 [0148] Transformacja oznacza wprowadzenie DNA do prokariotycznej komórki gospodarza w taki sposób, aby DNA było replikowalne, czy to jako element pozachromosomalny, czy też jako element zintegrowany w chromosomie. W zależności od użytej komórki gospodarza transformacji dokonuje się standardowymi technikami właściwymi dla danego rodzaju komórek. W przypadku komórek bakteryjnych zawierającymi znaczne bariery w ścianie komórkowej zazwyczaj stosuje się traktowanie wapniem z wykorzystaniem chlorku wapnia. Inna metoda transformacji wykorzystuje glikol polietylenowy/dmso. Jeszcze inną stosowaną techniką jest elektroporacja. [0149] Komórki prokariotyczne wykorzystywane do otrzymywania polipeptydów będących przedmiotem wynalazku hodowane są na podłożach znanych w dziedzinie i odpowiednich do hodowania wybranych komórek gospodarza. Przykłady odpowiednich podłoży obejmują bulion Lurii (LB) uzupełniony koniecznymi substancjami odżywczymi. W niektórych wariantach podłoża zawierają również środek selekcyjny, wybrany w oparciu o konstrukcję wektora ekspresyjnego, mający na celu selektywne umożliwianie wzrostu komórek prokariotycznych zawierających wektor ekspresyjny. Na przykład w celu wyhodowania komórek, w których zachodzi ekspresja genu opornego na ampicylinę, do podłoża dodaje się ampicylinę. [0150] Możliwe jest uwzględnienie w odpowiednich stężeniach wszelkich wymaganych dodatków oprócz źródeł węgla, azotu i nieorganicznych fosforanów, wprowadzanych oddzielnie lub w charakterze mieszaniny z innym dodatkiem lub podłożem, na przykład w postaci złożonego podłoża azotowego. Opcjonalnie, podłoże hodowlane może zawierać jeden lub większą liczbę reduktorów wybranych z grupy obejmującej glutation, cysteinę, cystaminę, tioglikolan, ditioerytrytol i ditiotreitol. [0151] Prokariotyczne komórki gospodarzy hodowane są w odpowiednich temperaturach. Na przykład w przypadku hodowli E. coli, temperatura zawiera się korzystnie w zakresie od około 20 C do około 39 C, korzystniej od około 25 C do około 37 C, zaś jeszcze korzystniej około 30 C. ph podłoża może być dowolnym ph z zakresu od około 5 do około 9, w zależności głównie od organizmu gospodarza. W przypadku E. coli, ph wynosi korzystnie od około 6,8 do około 7,4, zaś korzystniej około 7,0. [0152] Jeśli w wektorze ekspresyjnym będącym przedmiotem wynalazku stosuje się indukowalny promotor, ekspresja białka indukowana jest w warunkach odpowiednich do aktywacji promotora. W jednym z aspektów wynalazku do kontroli transkrypcji polipeptydów wykorzystuje się promotory PhoA. Zgodnie z powyższym, transformowane komórki gospodarza hoduje się w celu indukcji na podłożu z ograniczoną ilością fosforanu. Korzystnie podłożem z ograniczoną ilością fosforanu jest podłoże C.R.A.P (zob. np. Simmons et al., J. Immunol. Methods (2002), 263: ). Możliwe jest wykorzystanie szeregu innych induktorów w zależności od konstruktu wektora, zgodnie ze stanem wiedzy w dziedzinie. [0153] W jednym z wariantów wynalazku podlegające ekspresji polipeptydy będące przedmiotem niniejszego wynalazku są wydzielane i odzyskiwane z periplazmy komórek gospodarza. Odzyskanie białka obejmuje zazwyczaj przerwanie komórki mikroorganizmu, zazwyczaj metodami takimi, jak szok osmotyczny, sonikacja lub liza. Po rozerwaniu komórki, szczątki komórkowe lub całe komórki usuwane są poprzez odwirowanie lub filtrację. Białka mogą być dalej oczyszczane, na przykład techniką chromatografii powinowactwa na żywicach. Ewentualnie białka mogą zostać przeniesione na podłoże hodowlane i tam wyodrębnione. Komórki mogą być usunięte z hodowli, a nadsącz hodowlany może być przefiltrowany i zatężony w celu dalszego oczyszczania otrzymanych białek. Eksponowane polipeptydy mogą być w dalszej kolejności oczyszczone i zidentyfikowane przy użyciu powszechnie znanych metod takich, jak elektroforeza na żelu poliakryloamidowym (PAGE) i test Western Blot. [0154] W jednym z aspektów wynalazku produkcja przeciwciał prowadzona jest w dużych ilościach w procesie fermentacji. Do produkcji rekombinowanych białek na wielka skalę dostępnych jest wiele procedur fermentacji półciągłej. Fermentacje prowadzone w dużej skali charakteryzują się pojemnością co najmniej 1000 litrów, korzystnie od około 1000 litrów do litrów. W reaktorach fermentacyjnych do rozprowadzania tlenu i substancji odżywczych, zwłaszcza glukozy (preferowanego źródła węgla/energii) wykorzystuje się mieszadła wirnikowe. Fermentacja w małej skali dotyczy z reguły fermentacji w reaktorze fermentacyjnym o pojemności nie większej niż około 100 litrów, mieszczącej się w zakresie od około 1 litra do około 100 litrów. [0155] W procesie fermentacji indukcja ekspresji białka zazwyczaj inicjowana jest po okresie hodowania komórek w odpowiednich warunkach do pożądanej gęstości, np. OD 550 około , na którym to etapie komórki znajdują się we wczesnej fazie stacjonarnej. Możliwe jest wykorzystanie szeregu induktorów w zależności od konstruktu wektora, zgodnie ze stanem wiedzy w dziedzinie i opisem przedstawionym powyżej. Przed indukcją komórki mogą być hodowane przez krótszy czas.
59 58 Komórki zazwyczaj indukuje się przez około lat, choć można zastosować również krótsze lub dłuższe czasy indukcji. [0156] W celu poprawy wydajności i jakości wytwarzania polipeptydów będących przedmiotem wynalazku istnieje możliwość modyfikowania warunków fermentacji. Na przykład w celu poprawy prawidłowego konstruowania i zwijania wydzielanych polipeptydów przeciwciała, istnieje możliwość zastosowania dodatkowych wektorów, warunkujących ekspresję białek opiekuńczych takich, jak białka Dsb (DsbA, DsbB, DsbC, DsbD i/lub DsbG) lub FkpA (cis,trans-izomeraza peptydyloprolilowa o aktywności białka opiekuńczego) w celu kotransformacji prokariotycznych komórek gospodarza. Wykazano, że białka opiekuńcze ułatwiają zwijanie białek i rozpuszczalność heterologicznych białek wytwarzanych w bakteryjnych komórkach gospodarza. Chen et al. (1999) J Bio Chem. 274: ; Georgiou et al., patent nr US 6,083,715; Georgiou et al., patent nr US 6,027,888; Bothmann i Pluckthun (2000) J. Biol. Chem. 275: ; Ramm i Pluckthun (2000) J. Biol. Chem. 275: ; Arie et al. (2001) Mol. Microbiol. 39: [0157] W celu zminimalizowania proteolizy ulegających ekspresji heterologicznych białek (zwłaszcza białek wrażliwych na proteolizę), możliwe jest wykorzystanie określonych szczepów gospodarzy pozbawionych enzymów proteolitycznych. Na przykład komórki gospodarza mogą zostać zmodyfikowane poprzez mutacje genetyczne w genach kodujących znane proteazy bakteryjne takie, jak proteaza III, OmpT, DegP, Tsp, proteaza I, proteaza Mi, proteaza V, proteaza VI i ich kombinacje. Dostępne są szczepy E. coli pozbawione proteaz; zostały one opisane np. w pracy Joly et al. (1998), supra; Georgiou et al., patent nr US 5,264,365; Georgiou et al., patent nr US 5,508,192; Hara et al., Microbial Drug Resistance, 2:63-72 (1996). [0158] W jednym z wariantów wynalazku jako komórki gospodarza w systemie ekspresji będącym przedmiotem wynalazku stosuje się szczepy E. coli pozbawione enzymów proteolitycznych i transformowane plazmidami warunkującymi ekspresję jednego lub większej liczby białek pomocniczych. Oczyszczanie przeciwciał [0159] W jednym z wariantów otrzymywane białko przeciwciała poddawane jest procesowi dalszego oczyszczania w celu uzyskania do dalszych oznaczeń i zastosowań preparatów zasadniczo homogennych. Zastosowanie mogą znaleźć standardowe metody oczyszczania białek znane w dziedzinie wynalazku. Przykładami odpowiednich procedur oczyszczania są następujące procedury: frakcjonowanie na kolumnach immunopowinowactwa lub kolumnach jonowymiennicy, wytrącanie z etanolu, chromatografia HPLC z odwróconą fazą, chromatografia na silikażelu lub żywicy jonowymiennej takiej, jak DEAE, ogniskowanie chromatograficzne, SDS-PAGE, wytrącanie z siarczanu amonu i filtracja na żelu, na przykład Sephadex G-75. [0160] W jednym z aspektów do oczyszczania na bazie immunopowinowactwa pełnej długości przeciwciał jako produktami będącymi przedmiotem wynalazku stosuje się białko A immobilizowane na fazie stałej. Białko A jest białkiem ściany komórkowej pochodzącym z bakteriistaphylococcus aureus, o wielkości 41 kd, wiążącym z wysokim powinowactwem region Fc przeciwciał. Lindmark et al (1983) J. Immunol. Meth. 62:1-13. Faza stała, na której immobilizowane jest białko A jest korzystnie kolumną zawierającą powierzchnię szklaną lub krzemionkową, korzystniej kolumnę szklaną o kontrolowanej porowatości lub kolumnę z kwasem krzemowym. W niektórych zastosowaniach kolumnę powleka się odczynnikiem takim, jak glicerol, w celu zapobieżenia niespecyficznemu wiązaniu zanieczyszczeń. [0161] Jako pierwszy etap oczyszczania preparat pochodzący z hodowli komórkowej opisanej powyżej nanosi się na fazę stałą z immobilizowanym na niej białkiem A w celu umożliwienia specyficznego wiązania przedmiotowego przeciwciała przez białko A. Następnie faza stała jest przemywana w celu usunięcia zanieczyszczeń związanych w sposób niespecyficzny z faza stałą. interest to Protein A. Następnie wreszcie pożądane przeciwciało jest odzyskiwane z fazy stałej drogą wymywania. Generowanie przeciwciał z wykorzystaniem eukariotycznych komórek gospodarza: [0162] Elementy wektora z reguły obejmują, choć nie ograniczają się do poniższych: sekwencja sygnałowa, miejsce inicjacji replikacji, jeden lub większa liczba markerów selekcji, sekwencja wzmacniająca, promotor oraz sekwencja terminacji transkrypcji. (i) Komponent sekwencji sygnałowej
60 59 [0163] Wektor przeznaczony do stosowania w eukariotycznych komórkach gospodarza może również zawierać sekwencję sygnałową lub inny polipeptyd o swoistym miejscu cięcia przy N-końcu dojrzałego białka lub polipeptydu będącego przedmiotem zainteresowania. Wybrana heterologiczna sekwencja sygnałowa jest korzystnie sekwencją rozpoznawaną i przetwarzaną (np. ciętą przez peptydazę sygnałową) w komórce gospodarza. W przypadku ekspresji w komórkach ssaczych dostępne są ssacza sekwencje sygnałowe, a także wirusowe sekwencje liderowe sekrecji, na przykład sygnał gd wirusa herpes simplex. [0164] DNA takiego regionu prekursorowego łączy się w ramce odczytu z DNA kodującym przeciwciało. (ii) Miejsce inicjacji replikacji [0165] Z reguły element miejsca inicjacji replikacji nie jest wymagany w ssaczych wektorach ekspresyjnych. Na przykład miejsce inicjacji SV40 może być zazwyczaj wykorzystywane jedynie z uwagi na fakt, że zawiera sekwencję promotorową wczesnego regionu kodującego. (iii) Gen selekcji [0166] Wektory ekspresyjne i klonujące mogą zawierać gen selekcji, zwany również markerem selekcji. Typowe geny selekcji kodują białka, które (a) zapewniają oporność na antybiotyki lub inne toksyny, np. ampicylinę, neomycynę, metotreksat lub tetracyklinę (b) stanowią uzupełnienie niedoborów auksotropowych w przypadkach, gdy ma to zastosowanie lub (c) dostarczają istotne składniki odżywcze niedostępne z podłoża. [0167] Jeden z przykładowych schematów selekcji wykorzystuje zastosowanie leku do zatrzymania wzrostu komórek gospodarza. Komórki transformowane heterologicznym genem wytwarzają białko zapewniające oporność na lek, a przez to przeżywają reżim selekcji. Przykłady tego rodzaju selekcji dominant obejmują zastosowanie leków takich, jak neomycyna, kwas mykofenolowy czy higromycyna. [0168] Innym przykładem odpowiednich markerów selekcji komórek ssaczych są markery umożliwiające identyfikację komórek zdolnych do wychwytywania kwasów nukleinowych przeciwciał takich, jak geny DHFR, kinazy tymidynowej, metalotioneiny-i i -II, korzystnie geny metalotioneiny naczelnych, deaminazy adenozynowej, dekarboksylazy ornitynowej itp. [0169] Na przykład komórki transformowane genem selekcji DHFR są identyfikowane w pierwszej kolejności przez hodowlę transformowanych komórek na podłożu zawierającym metotreksat (Mtx), kompetytywnego antagonistę DHFR. W przypadku stosowania DHFR dzikiego typu odpowiednią komórką gospodarza jest linia komórek jajowych chomika chińskiego (CHO) pozbawiona aktywności DHFR (np. ATCC CRL-9096). [0170] Ewentualnie, komórki gospodarza (zwłaszcza komórki dzikiego typu zawierające endogenny DHFR) transformowane lub kotransformowane sekwencjami DNA zawierającymi przeciwciało, DHFR dzikiego typu i inny marker selekcji, na przykład 3'-fosfotransferazę aminoglikozydową (APH) mogą zostać wyselekcjonowane poprzez hodowlę w podłożu zawierającym środek selekcjonujący marker selekcji, na przykład antybiotyk aminoglikozydowy, np. kanamycynę, neomycynę lub G418. Patrz patent nr US 4,965,199. (iv) Promotor [0171] Wektory ekspresyjne i klonujące zazwyczaj zawierają promotor rozpoznawany przez organizm gospodarza i operacyjnie połączony z kwasem nukleinowym kodującym polipeptyd przeciwciała. Sekwencje promotorowe występują u eukariotów. Praktycznie wszystkie geny eukariotyczne posiadają region bogaty w AT od około 25 do około 30 zasad przed miejscem, w którym rozpoczynana jest transkrypcja. Inną sekwencją, występującą od 70 do 80 zasad przed przed miejscem, w którym rozpoczynana jest transkrypcja, jest region CNCAAT, gdzie N jest dowolnym nukleotydem. Przy 3'-końcu większości genów eukariotów znajduje się sekwencja AATAAA, mogąca być sygnałem do dodania ogona poliadenozynowego do 3'-końca sekwencji kodującej. Wszystkie powyższe sekwencje stosowanie wprowadza się do eukariotycznych wektorów ekspresyjnych. [0172] Transkrypcja polipeptydu przeciwciała z wektora z wektorów w komórkach ssaków jest sterowana na przykład przez promotory uzyskane z genomów wirusów takich, jak wirus polyoma, wirus ospy ptasiej, adenowirus (na przykład Adenowirus 2), wirus brodawczaka bydlęcego, wirus mięsaka ptaków, cytomegalowirus, retrowirus, wirus zapalenia wątroby typu B i wirus Simian 40 (SV40), z heterologicznych promotorów ssaczych, np. promotora aktynowego lub
61 60 immunoglobulinowego, z promotorów białka szoku cieplnego, pod warunkiem, że promotory te są zgodne z komórkami gospodarza. [0173] Promoitory wczesnego i późnego regionu transkrypcji wirusa SV40 dogodnie uzyskuje się w postaci fragmentu restrykcyjnego SV40, zawierającego również miejsce inicjacji replikacji wirusa SV40. Bezpośredni promotor wczesnego regionu kodującego ludzkiego cytomegalowirusa dogodnie uzyskuje się z wykorzystaniem fragmentu restrykcyjnego HindIII E. System ekspresji DNA w ssaczych komórkach gospodarza przy użyciu wirusa brodawczaka bydlęcego (BPV) w charakterze wektora ujawniono w patencie nr US 4,419,446. Modyfikację tego systemu opisano w patencie nr US 4,601,978. Zob. też Reyes et al., Nature 297: (1982) odnośnie ekspresji ludzkiego cdna interferonu β w komórkach myszy pod kontrolą promotora kinazy tymidynowej z wirusa herpes simplex. Ewentualnie, jako promotor może zostać wykorzystane długie terminalne powtórzenie genomu wirusa mięsaka Rousa. (v) Sekwencja wzmacniająca [0174] Transkrypcja DNA kodującego polipeptyd przeciwciała będącego przedmiotem niniejszego wynalazku przez wyższe eukarioty jest często intensyfikowana poprzez wprowadzenie do wektora sekwencji wzmacniającej. Aktualnie znanych jest wiele sekwencji wzmacniających z genów ssaczych (globiny, elastazy, albuminy, α-fetoproteiny i insuliny). Zazwyczaj jednak stosuje się sekwencje wzmacniające z wirusów pasożytujących w komórkach eukariotycznych. Przykłady obejmują sekwencję wzmacniającą wirusa SV40 w po późnej stronie miejsca inicjacji replikacji (pz ), sekwencję wzmacniającą promotora wczesnego regionu transkrypcji cytomegalowirusa, sekwencję wzmacniającą wirusa polyoma po późnej stronie miejsca inicjacji replikacji oraz sekwencje wzmacniające adenowirusów. W celu uzyskania dalszych informacji na temat sekwencji wzmacniających aktywacje promotorów eukariotycznych zob. też Yaniv, Nature 297:17-18 (1982). Sekwencja wzmacniająca może zostać dołączona do wektora w pozycji 5' lub 3' względem sekwencji kodującej polipeptyd przeciwciała, jednak korzystnie dołączana jest w miejscu 5' względem promotora. (vi) Składniki terminacji transkrypcji [0175] Wektory ekspresji stosowane w eukariotycznych komórkach gospodarzy zazwyczaj zawierają również sekwencje wymagane do terminacji transkrypcji i stabilizacji mrna. Sekwencje takie dostępne są często w regionach 5'-nieulegających translacji, niekiedy 3'-nieulegających translacji, eukariotycznych lub wirusowych łańcuchów DNA lub cdna. Regiony te zawierają segmenty nukleotydowe transkrybowane w postaci poliadenylowanych fragmentów w nieulegającej translacji części mrna kodującego przeciwciało. Innym użytecznym elementem terminacji transkrypcji jest poliadenylowany region bydlęcego hormonu wzrostu. Zob. opis patentowy WO94/11026 i ujawniony tamże wektor ekspresyjny. (vii) Wybór i transformacja komórek gospodarza [0176] Odpowiednie komórki gospodarzy do klonowania lub ekspresji DNA w wektorach będących przedmiotem niniejszego wynalazku obejmują opisane tu komórki wyższych eukariotów, w tym komórki kręgowców. Propagacja komórek kręgowców w hodowli (hodowla tkankowa) stała się procedurą rutynową. Przykładami użytecznych komórek ssaczych są: linia małpich komórek nerkowych CV1 transformowana wektorem SV40 (COS-7, ATCC CRL 1651); linia ludzkich płodowych komórek nerkowych (293 lub komórki 293 subklonowane do wzrostu w hodowli zawiesinowej, Graham et al., J. Gen Virol. 36:59 (1977)); noworodkowe komórki nerkowe chomika (BHK, ATCC CCL 10); komórki jajowe chomika chińskiego/- DHFR (CHO, Urlaub et al., Proc. Natl. Acad. Sci. USA 77:4216 (1980)); mysie komórki Sertoliego (TM4, Mather, Biol. Reprod. 23: (1980)); małpie komórki nerkowe (CVl ATCC CCL 70); komórki nerkowe koczkodana saba (VERO-76, ATCC CRL-1587); ludzkie komórki raka szyjki macicy (HELA, ATCC CCL 2); psie komórki nerkowe (MDCK, ATCC CCL 34); komórki wątrobowe szczura bawolego (BRL 3A, ATCC CRL 1442); ludzkie komórki płucne (W138, ATCC CCL 75); ludzkie komórki wątrobowe (Hep G2, HB 8065); mysie komórki guza sutka (MMT , ATCC CCL51); komórki TRI (Mather et al., Annals N.Y. Acad. Sci. 383: (1982)); komórki MRC 5; komórki FS4; i ludzka linia komórek hepatoma (Hep G2). [0177] Komórki gospodarza transformowane są opisanymi powyżej wektorami ekspresyjnymi lub klonującymi w celu wytwarzania przeciwciała i hodowane na konwencjonalnych pożywkach modyfikowanych w zależności od potrzeb w kierunku indukcji promotorów, wyboru transformowanych komórek lub amplifikacji genów kodujących pożądane sekwencje. (viii) Hodowla komórek gospodarza
62 61 [0178] Komórki gospodarza wykorzystywane do wytwarzania przeciwciał będących przedmiotem wynalazku mogą być hodowane na wielu podłożach. Do hodowli komórek gospodarzy nadają się dostępne handlowo podłoża takie, jak podłoże Ham F10 (Sigma), podłoże MEM (Sigma), RPMI-1640 (Sigma) i podłoże Eagle a w modyfikacji Dulbecco ((DMEM), Sigma). Ponadto w charakterze podłoża hodowlanego dla komórek gospodarza można wykorzystać dowolne podłoża opisane w pracy Ham et al., Meth. Enz. 58:44 (1979), Barnes et al., Anal. Biochem.102:255 (1980), patentach nr US 4,767,704; US 4,657,866; US 4,927,762; US 4,560,655; lub US 5,122,469; WO 90/03430; WO 87/00195; lub US Re. 30,985. Każde z tych podłoży może zostać w razie potrzeby uzupełnione hormonami i/lub innymi czynnikami wzrostu (na przykład insuliną, transferyną lub czynnikiem wzrostu naskórka), solami (na przykład chlorkiem sodu, solami wapnia, magnezu i fosforanami), buforami (na przykład HEPES), nukleotydami (na przykład adenozyna i tymidyną), antybiotykami (na przykład gentamycyną), pierwiastkami śladowymi (definiowanymi jako związki nieorganiczne zazwyczaj obecne w końcowych stężeniach rzędu mikromolowego) i glukozą lub równoważnym źródłem energii. Możliwe jest również uwzględnienie wszelkich innych dodatków w odpowiednich stężeniach, znanych osobie biegłej w dziedzinie wynalazku. Warunki hodowli, takie jak temperatura, ph itp., są takie, jak stosowane uprzednio dla komórek gospodarza selekcjonowanych w celu ekspresji i będą znane osobie posiadającej zwykłe wyszkolenie w dziedzinie. (ix) Oczyszczanie przeciwciała [0179] W przypadku wykorzystywania technik rekombinacyjnych przeciwciało może być wytworzone we wnętrzu komórki lub bezpośrednio wydzielone do podłoża. Jeśli przeciwciało wytwarzane jest we wnętrzu komórki, w pierwszym etapie usuwa się szczątki stałe - czy to komórki gospodarza, czy też ich zlizowane fragmenty - na przykład poprzez odwirowanie lub ultrafiltrację. W przypadku, gdy przeciwciało jest wydzielane do podłoża, nadsącze z tego rodzaju systemów ekspresji są zazwyczaj w pierwszej kolejności zatężane przy użyciu komercyjnie dostępnych filtrów do zatężania białek, na przykład jednostek ultrafiltracyjnych Amicon lub Millipore Pellicon W którymkolwiek z poprzedzających etapów można zastosować inhibitor proteazy taki, jak PMSF, w celu hamowania proteolizy przez antybiotyki w celu zapobieżenia wzrostowi przypadkowych zanieczyszczeń. [0180] Przygotowana w komórkach kompozycja przeciwciała może zostać poddana oczyszczaniu, na przykład techniką chromatografii na hydroksyloapatycie, elektroforezy żelowej, dializy oraz chromatografii powinowactwa, przy czym preferowaną techniką oczyszczania jest chromatografia powinowactwa. Przydatność białka A jako liganda powinowactwa zależy od gatunku i izotypu domeny Fc immunoglobuliny obecnej w przeciwciele. Białko A może być wykorzystane do oczyszczania przeciwciał opartych na ludzkich łańcuchach ciężkich izotypów γ1, γ2, lub γ4 (Lindmark et al., J. Immunol. Meth.62:1-13 (1983)).Białko G zaleca się dla wszystkich izotypów mysich oraz ludzkiego γ3 (Guss et al., EMBO J. 5: (1986)). Złożem, do którego przyłącza się ligand powinowactwa, jest najczęściej agaroza, ale dostępne są inne złoża. Złoża stabilne mechanicznie, takie jak szkło lub poli(styrenodiwinylo)benzen o kontrolowanej wielkości porów, pozwalają na wyższe szybkości przepływu i krótsze czasy obróbki niż możliwe do uzyskania w przypadku agarozy. Gdy przeciwciało zawiera domenę C H 3, do oczyszczania stosuje się żywicę Bakerbond ABX (J. T. Baker, Phillipsburg, NJ). Dostępne są również inne techniki oczyszczania białek, takie jak frakcjonowanie na kolumnie jonowymiennej, wytrącanie etanolem, HPL.C z odwróco- nymi fazami, chromatografia na krzemionce, chromatografia na heparyna-sepharose, chromatografia na żywicy aniono- lub kationowymiennej, takiej jak kolumna z kwasem poliaspartylowym), ogniskowanie chromatograficzne, SDS-PAGE i wytrącanie siarczanem amonu, zależnie od odzyskiwanego przeciwciała. [0181] Po jakimkolwiek wstępnym etapie(-ach) oczyszczania, mieszaninę zawierającą przeciwciało będące przedmiotem zainteresowania i zanieczyszczenia można poddać chromatografii oddziaływań hydrofobowych w niskim ph z zastosowaniem buforu do elucji o ph pomiędzy 2,5 a 4,5, korzystnie prowadzonej przy niskim stężeniu soli (np. od około 0 do 0,25 M). Oznaczenia aktywności [0182] Przeciwciała będące przedmiotem niniejszego wynalazku mogą zostać scharakteryzowane pod względem właściwości fizykochemicznych i funkcji biologicznych przy użyciu różnych oznaczeń znanych w dziedzinie wynalazku. [0183] Oczyszczone immunoglobuliny mogą być dalej charakteryzowane w serii oznaczeń obejmującej między innymi, sekwencjonowanie N-terminalne, analiza aminokwasów, wysokosprawna chromatografia cieczowa (HPLC) z wykluczaniem cząsteczek na podstawie rozmiaru w warunkach niedenaturujących, spektrometria mas, chromatografia jonowymienna i trawienie papainą.
63 62 [0184] W niektórych wariantach wynalazku wytwarzane immunoglobuliny analizowane są pod kątem ich aktywności biologicznej. W niektórych wariantach immunoglobuliny będące przedmiotem niniejszego wynalazku badane są pod kątem ich powinowactwa wiązania antygenu. Testy wiązania antygenu znane w dziedzinie i mogące znaleźć zastosowanie w niniejszym wynalazku obejmują bez ograniczeń wszelkie bezpośrednie lub konkurencyjne testy wiązania przy użyciu technik takich, jak Western Blot, test radioimmunologiczny, test immunoenzymatyczny (ELISA), sandwiczowe testy immunologiczne, testy immunoprecypitacyjne, testy immunofluorescencyjne i testy immunologiczne z zastosowaniem białka A. Ilustracyjny test wiązania antygenu przedstawiono poniżej w sekcji Przykłady. [0185] W jednym z wariantów niniejszego wynalazku rozważa się zmodyfikowane przeciwciało posiadające niektóre, ale nie wszystkie funkcje efektorowe, co czyni to przeciwciało pożądanym kandydatem do wielu zastosowań, w których ważny jest czas półtrwania przeciwciała in vivo, jednak niektóre funkcje efektorowe (takie jak funkcja dopełniacza lub ADCC) są zbędne lub szkodliwe. W niektórych wariantach mierzy się aktywności Fc wytworzonej immunoglobuliny w celu upewnienia się, że zachowane są jedynie pożądane własności. W celu potwierdzenia redukcji/zubożenia aktywności CDC i/lub or ADCC można wykonać oznaczenia cytotoksyczności in vitro i/lub in vivo. Można na przykład wykonać testy wiązania receptora Fc (FcR) w celu upewnienia się, że przeciwciało nie posiada zdolności wiązania FcγR (a tym samym prawdopodobnie nie posiada aktywności ADCC), lecz zachowuje zdolność wiązania FcRn. W podstawowych komórkach stosowanych w przenoszeniu. ADCC, tj. komórkach NK, ekspresji ulega jedynie FcγRIII, podczas gdy w monocytach ekspresji ulegają FcγRI, FcγRII i FcγRIII. Ekspresję FcR w komórkach krwiotwórczych podsumowano w Tabeli 3 na str. 464 pracy Ravetch i Kinet, Annu. Rev. Immunol 9: (1991). Przykład metody oznaczenia in vitro aktywności ADCC badanej cząsteczki opisano w patentach nr US 5,500,362 lub US 5,821,337. Zazwyczaj komórki efektorowe w tego rodzaju oznaczeniach obejmują jednojądrzaste komórki krwi obwodowej (PBMC) i naturalnych zabójców (NK). Ewentualnie lub dodatkowo aktywność ADCC cząsteczki będącej przedmiotem zainteresowania może być oceniana in vivo, np. w modelu zwierzęcym takim, jak ujawniony w pracy Clynes et al. PNAS (USA) 95: (1998). Można również przeprowadzić testy wiązania C1q w celu potwierdzenia, że przeciwciało nie jest w stanie wiązać C1q, a tym samym nie posiada aktywności CDC. W celu oceny aktywacji dopełniacza wykonuje się test CDC, np. zgodnie z opisem w pracy Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996). Oznaczenia wiązania FcRn oraz klirensu/czasu półtrwania in vivo można również wykonać metodami znanymi w dziedzinie, np. metodami opisanymi w sekcji Przykłady. Przeciwciała humanizowane [0186] Niniejszy wynalazek obejmuje przeciwciała humanizowane. W dziedzinie wynalazku znanych jest wiele sposobów humanizowania przeciwciał innych niż przeciwciała ludzkie. Na przykład, do humanizowanego przeciwciała może być wprowadzona jedna lub większa liczba reszt aminokwasowych ze źródła innego lub ludzkie. Rzeczone reszty aminokwasowe ze źródła innego niż ludzkie często nazywane są resztami importowanymi, zazwyczaj pochodzącymi z importowanej domeny zmiennej. Humanizację można zasadniczo wykonać według metody opisanej przez Wintera i współpracowników (Jones et al. (1986) Nature 321: ; Riechmann et al. (1988) Nature 332: ; Verhoeyen et al. (1988) Science 239: ), podstawiając sekwencje regionu hiperzmiennego w miejsce odpowiednich sekwencji przeciwciała ludzkiego. Zgodnie z powyższym, tego rodzaju humanizowanymi przeciwciałami są przeciwciała chimeryczne (patent nr US 4,816,567) gdzie znacznie mniejsza cześć niż nienaruszona ludzka domena zmienna została podstawiona odpowiednią sekwencją pochodzącą od gatunku innego niż człowiek. W praktyce, humanizowane przeciwciała są zazwyczaj ludzkimi przeciwciałami, w których niektóre reszty regionów hiperzmiennych i ewentualnie niektóre reszty FR podstawione są resztami pochodzącymi z analogicznych miejsc w przeciwciałach gryzoni. [0187] W celu zmniejszenia antygenności bardzo ważny jest wybór ludzkich domen zmiennych, zarówno łańcucha lekkiego, jak i ciężkiego, które mają znaleźć zastosowanie w wytwarzaniu humanizowanych przeciwciał. Według metody tzw. najlepszego dopasowania, sekwencja zmiennej domeny przeciwciała gryzonia porównywana jest z cała biblioteką znanych ludzkich domen zmiennych. Jako sekwencję ludzką do użycia w humanizowanym przeciwciele przyjmuje się sekwencję ludzką najbardziej zbliżoną do sekwencji gryzonia (Sims et al. (1993) J. Immunol. 151:2296; Chothia et al. (1987) J. Mol. Biol. 196:901. Inna metoda wykorzystuje szczególną sekwencję zrębową pochodzącą z konsensusowej sekwencji wszystkich ludzkich przeciwciał lub szczególnej podgrupy łańcuchów lekkich lub ciężkich. Ta sama sekwencja zrębowa może być
64 63 wykorzystana w kilku różnych przeciwciałach humanizowanych (Carter et al. (1992) Proc. Natl. Acad. Sci. USA, 89:4285; Presta et al. (1993) J. Immunol., 151:2623. [0188] Ponadto istotne jest, aby przeciwciała były humanizowane z zachowaniem wysokiego powinowactwa do antygenu oraz innych korzystnych właściwości biologicznych. Aby osiągnąć ten cel, w jednej z metod humanizowane przeciwciała przygotowuje się w procesie analizy macierzystych sekwencji i rozmaitych konceptualnych produktów humanizowanych z zastosowaniem trójwymiarowych modeli sekwencji macierzystych i humanizowanych. Trójwymiarowe modele immunoglobulin są powszechnie dostępne i znane osobom biegłym w dziedzinie. Dostępne są programy komputerowe ilustrujące i wyświetlające prawdopodobne trójwymiarowe struktury konformacyjne wybranych kandydujących sekwencji immunoglobulinowych. Zbadanie wyświetlanych struktur umożliwia analizę prawdopodobnej roli reszt w funkcjonowaniu kandydujących sekwencji immunoglobulinowych, tj. analizy reszt mających wpływ na zdolność wiązania antygenu przez kandydującą immunoglobulinę. W ten sposób można wybrać i połączyć sekwencje FR biorcy i zaimportować sekwencje w taki sposób, by osiągnąć pożądane właściwości przeciwciała, na przykład zwiększone powinowactwo w kierunku docelowego antygenu(-ów). Z reguły reszty regionu hiperzmiennego są bezpośrednio i najbardziej zasadniczo związane z wpływem na wiązanie antygenu. Warianty przeciwciał [0189] W jednym z aspektów wynalazek ujawnia fragment przeciwciała zawierający modyfikacje na styku polipeptydów Fc tworzących region Fc, gdzie rzeczone modyfikacje ułatwiają i/lub promują heterodimeryzację. Modyfikacje te polegają na wprowadzeniu wybrzuszenia w pierwszym polipeptydzie Fc i zagłębienia w drugim polipeptydzie Fc, tak, aby wybrzuszenie mieściło się w zagłębieniu, co ma promować tworzenie kompleksów pierwszego i drugiego polipeptydu Fc. Metody generowania przeciwciał z omawianymi modyfikacjami są znane w dziedzinie i opisane np. w patencie nr US 5,731,168. [0190] W niektórych wariantach wynalazku rozważa się zastosowanie opisanych modyfikacji sekwencji aminokwasowych. Na przykład pożądane może być poprawienie powinowactwa wiązania i/lub innych właściwości biologicznych przeciwciała. Wariantowe sekwencje aminokwasowe przeciwciała przygotowuje się wprowadzając odpowiednie modyfikacje nukleotydowe do kwasu nukleinowego kodującego przeciwciało lub na drodze syntezy peptydów. Modyfikacje takie obejmują na przykład delecje i/lub insercje i/lub substytucje reszt w sekwencjach aminokwasów w cząsteczce przeciwciała. W celu wytworzenia końcowego konstruktu stosuje się dowolne kombinacje delecji, insercji i substytucji, pod warunkiem, że końcowy konstrukt posiada pożądane cechy. Modyfikacje aminokwasów w przedmiotowej sekwencji przeciwciała mogą być wprowadzone w momencie wykonywania tej sekwencji. [0191] Przydatną metodą identyfikacji określonych reszt lub regionów przeciwciała będących preferowanymi miejscami mutagenezy jest metoda nazywana skanującą mutagenezą alaninową ( alanine scanning mutagenesis ), opisana w pracy Cunningham i Wells (1989) Science, 244: W metodzie tej wskazuje się resztę lub grupę docelowych reszt (np. reszt naładowanych takich, jak arg, asp, his, lys i glu), a następnie zastępuje aminokwasem obojętnym lub ujemnie naładowanym (najkorzystniej alanina lub polialaniną) w celu wywołania oddziaływania aminokwasów z antygenem. Następnie pozycje aminokwasowe wykazujące funkcjonalną czułość na substytucje udoskonala się wprowadzając dodatkowe lub inne warianty w miejscach substytucji. Tak więc, podczas, gdy miejsce wprowadzenia wariacji sekwencji aminokwasów jest określone z góry, nie ma konieczności określania z góry charakteru mutacji per se. Na przykład w celu zanalizowania skutku wprowadzenia mutacji w danym miejscu w docelowym kodonie lub regionie prowadzi się skanującą mutaganezę alaninową lub mutagenezę losową, a ulegające ekspresji immunoglobuliny bada się pod kątem pożądanej aktywności. [0192] Insercje w sekwencjach aminokwasów obejmują fuzje terminalne przy końcu aminowym i/lub karboksylowym, charakteryzujące się długością od pojedynczej reszty do polipeptydów zawierających sto lub większą liczbę reszt, a także międzysekwencyjne insercje pojedynczych lub wielu reszt aminokwasowych. Przykłady insercji terminalnych obejmują przeciwciała z N-terminalną reszta metionylową lub przeciwciała połączone z polipeptydami cytotoksycznymi. Inne insercyjne warianty przeciwciała obejmują przyłączenie do N- lub C-końca przeciwciała enzymu (np. ADEPT) lub polipeptydu zwiększającego czas półtrwania przeciwciała w osoczu. [0193] Innym rodzajem wariantu jest wariant z substytucją aminokwasu. Warianty te posiadają co najmniej jedną resztę aminokwasową w cząsteczce przeciwciała zastąpioną inną resztą
65 64 aminokwasową. Miejsca będące przedmiotem największego zainteresowania pod kątem mutagenezy substytucyjnej obejmują regiony hiperzmienne, jednak rozważane są również modyfikacje FR. Substytucje konserwatywne przedstawiono w Tabeli 2 pod nagłówkiem Korzystne substytucje. Jeśli tego rodzaju substytucje prowadzą do zmiany aktywności biologicznej, możliwe jest wprowadzenie bardziej zasadniczych zmian, oznaczonych w Tabeli 2 nagłówkiem Przykładowe substytucje), lub zgodnie z przedstawionym poniżej opisem klas aminokwasów, a następnie zbadanie produktów. TABELA 2 Reszta wyjściowa Przykładowe substytucje Korzystne substytucje Ala (A) Val; Leu; Ile Val Arg (R) Lys; Gln; Asn Lys Asn (N) Gln; His; Asp, Lys; Arg Gln Asp (D) Glu; Asn Glu Cys (C) Ser; Ala Ser Gln (Q) Asn; Glu Asn Glu (E) Asp; Gln Asp Gly (G) Ala Ala His (H) Asn; Gln; Lys; Arg Arg Ile (I) Leu; Val; Met; Ala; Phe; Norleucyna Leu Leu (L) Norleucyna; Ile; Val; Met; Ala; Phe Ile Lys (K) Arg; Gln; Asn Arg Met (M) Leu; Phe; Ile Leu Phe (F) Trip; Leu; Val; Ile; Ala; Tyr Tyr Pro (P) Ala Ala Ser (S) Thr Thr Thr (T) Val; Ser Ser Trp (W) Tyr; Phe Tyr Tyr (Y) Trp; Phe; Thr; Ser Phe Val (V) Ile; Leu; Met; Phe; Ala; Norleucyna Leu [0194] Znaczące modyfikacje właściwości biologicznych przeciwciała osiąga się poprzez wybór substytucji różniących się znacznie wpływem na (a) strukturę szkieletu polipeptydowego w obszarze substytucji, np. konformacja spirali lub kartki, (b) zmianę hydrofobowości cząsteczki w miejscu docelowym lub (c) zmianę objętości łańcucha bocznego. Aminokwasy można grupować na podstawie podobieństw właściwości ich łańcuchów bocznych (za A. L. Lehninger, w: Biochemistry, wyd. 2, str , Worth Publishers, Nowy Jork (1975)): (1) niepolarne: Ala (A), Val (V), Leu (L), Ile (I), Pro (P), Phe (F), Trp (W), Met (M) (2) polarne nienaładowane: Gly (G), Ser (S), Thr (T), Cys (C), Tyr (Y), Asn (N), Gln (Q) (3) kwaśne: Asp (D), Glu (E) (4) zasadowe: Lys (K), Arg (R), His(H) [0195] Ewentualnie, występujące w naturze reszty można podzielić na grupy w oparciu o wspólne właściwości łańcuchów bocznych: (1) hydrofobowe: Norleucyna, Met, Ala, Val, Leu, Ile; (2) obojętne hydrofilowe: Cys, Ser, Thr, Asn, Gln; (3) kwaśne: Asp, Glu; (4) zasadowe: His, Lys, Arg; (5) reszty wpływające na orientację łańcucha: Gly, Pro; (6) aromatyczne: Trp, Tyr, Phe.
66 65 [0196] Substytucje niekonserwatywne obejmują wymianę członka jednej z tych klas na członka innej klasy. Tego rodzaju podstawione reszty mogą również być wprowadzane w miejsca substytucji konserwatywnej lub, co bardziej korzystne, w pozostałe miejsca (niekonserwatywne). [0197] Jeden z wariantów substytucyjnych obejmuje substytucje jednej lub większej liczby reszt w regionie hiperzmiennym przeciwciała macierzystego (np. przeciwciała humanizowanego lub ludzkiego). Z reguły uzyskany(-e) wariant(y) wybrane do dalszego rozwoju będą charakteryzowały sią lepszymi właściwościami biologicznymi niż przeciwciało macierzyste, z którego zostały wygenerowane. Dogodny sposób generowania tego rodzaju wariantów substytucyjnych obejmuje dojrzewanie powinowactwa przy użyciu ekspresji w wektorach fagowych. W skrócie, dokonuje się mutacji kilku miejsc (np. 6-7 miejsc) w hiperzmiennych regionach w celu wygenerowania wszystkich możliwych substytucji aminokwasów w każdym z miejsc. Wytworzone w ten sposób przeciwciała ulegają ekspresji z nitkowatych cząsteczek faga jako fuzje z produktem genu III M 13 umieszczonego w każdej cząsteczce. Warianty ulegające ekspresji fagowej są następnie badane pod kątem aktywności biologicznej (np. powinowactwa wiązania) w sposób ujawniony w niniejszym opisie. W celu zidentyfikowania miejsc regionu hiperzmiennego stanowiących dobrych kandydatów do modyfikacji można przeprowadzić skanująca mutagenezę alaninową w celu zidentyfikowania reszt w regionach hiperzmiennych mających znaczny wkład w wiązanie antygenu. Ewentualnie lub dodatkowo korzystne może być zanalizowanie struktury krystalicznej kompleksu antygen-przeciwciało w celu wskazania punktów kontaktu między cząsteczka przeciwciała a antygenem. Tego rodzaju reszty kontaktowe i reszty sąsiadujące z nimi są kandydatami do substytucji z zastosowaniem omawianych tu technik. Po wygenerowaniu takich wariantów panel wariantów poddaje się badaniu zgodnie z przedstawionym opisem. Do dalszego rozwoju można wybrać przeciwciała charakteryzujące się lepszymi właściwościami w jednym lub większej liczbie odpowiednich oznaczeń. [0198] Cząsteczki kwasów nukleinowych kodujących wariantowe sekwencje aminokwasowe przeciwciała przygotowuje się przy użyciu szeregu metod znanych w dziedzinie wynalazku. Metody te obejmują między innymi wyodrębnienie ze źródła naturalnego (w przypadku wariantów sekwencji aminokwasowej występujących w naturze) lub przygotowanie na drodze mutagenezy prowadzonej za pośrednictwem oligonukleotydów (czyli ukierunkowanej, mutagenezy PCR i mutagenezy kasetowej wcześniej przygotowanej wariantowej lub niewariantowej wersji przeciwciała. [0199] Pożądane może być wprowadzenie jednej lub większej liczby modyfikacji aminokwasowych w regionie Fc polipeptyfów immunoglobulinowych będących przedmiotem wynalazku, prowadzące do wygenerowania wariantu regionu Fc. Region Fc może zawierać ludzką sekwencję regionu Fc region (np. ludzki region IgG1, IgG2, IgG3 lub IgG4 Fc), zawierającą modyfikację aminokwasów (np. substytucję) w jednym lub większej liczbie pozycji aminokwasowych, w tym cysteiny w regionie zawiasowym. [0200] Zgodnie z niniejszym opisem i wiedzą panującą w dziedzinie rozważa się, że w niektórych wariantach przeciwciało wykorzystywane w metodach będących przedmiotem wynalazku może zawierać jedną lub większą liczbę modyfikacji w porównaniu z odpowiednim przeciwciałem dzikiego typu, np. w regionie Fc. Przeciwciała te zachowywałyby jednak zasadniczo te same cechy wymagane do ich użyteczności terapeutycznej, co odpowiednie przeciwciała dzikiego typu. Na przykład uważa się, że możliwe jest wprowadzenie określonych modyfikacji w regionie Fc, prowadzących do zmiany (tj. poprawy lub osłabienia) wiązania C1q i/lub cytotoksyczności zależnej od dopełniacza (CDC), np. zgodnie z opisem patentowym nr WO99/ Zob. też Duncan i Winter, Nature 322: (1988); patent nr US 5,648,260; US 5,624,821 i WO94/29351 pod kątem innych przykładów wariantów regionu Fc. Immunokoniugaty [0201] Wynalazek dotyczy również immunokoniugatów, czyli koniugatów typu przeciwciało-lek (ADC), zawierających przeciwciało sprzężone ze środkiem cytotoksycznym, na przykład chermioterapeutykiem, lekiem, środkiem hamującym wzrost, toksyną (np. enzymatycznie aktywną toksyną pochodzenia bakteryjnego, grzybowego, roślinnego lub zwierzęcego) lub isotopu radioaktywnego (tj. radiokoniugatów). [0202] Zastosowanie koniugatów typu przeciwciało-lek do miejscowego dostarczania środków cytotoksycznych lub cytostatycznych, tj. laków zabijających lub hamujących wzrost komórek nowotworowych w leczeniu raka (Syrigos i Epenetos (1999) Anticancer Research 19: ; Niculescu-Duvaz i Springer (1997) Adv. Drg Del. Rev. 26: ; patent nr US 4,975,278) teoretycznie umożliwia na kierowane dostarczanie reszty lekowej do guza i wewnątrzkomórkową akumulacje leku w guzie, podczas gdy układowe podanie niesprzężonych środków leczniczych
67 66 prowadziłoby do niedopuszczalnego poziomu toksyczności dla zwykłych komórek oprócz komórek nowotworowych, które miałyby zostać wyeliminowane (Baldwin et al., (1986) Lancet pp. (15 Mar. 1986):603-05; Thorpe, (1985) Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review, w: Monoclonal Antibodies '84: Biological And Clinical Applications, A. Pinchera et al. (red)., str ). W ten sposób próbuje się osiągnąć maksymalną skuteczność przy minimalnej toksyczności Jako przydatne w omawianych strategiach opisywano przeciwciała zarówno poliklonalne, jak i monoklonalne (Rowland et al., (1986) Cancer Immunol. Immunother., 21: ). Leki stosowane w tych metodach obejmują daunomycynę, doksorubicynę, metotreksat i windezynę (Rowland et al., (1986) supra). Do toksyn wykorzystywanych w koniugatach typu przeciwciało-toksyna należą toksyny bakteryjne takie, jak toksyny dyfterii, toksyny roślinne takie, jak rycyna, toksyny niskocząsteczkowe takie, jak geldanamycyna (Mandler et al (2000) Jour. of the Nat. Cancer Inst. 92(19): ; Mandler et al. (2000) Bioorganic & Med. Chem. Letters 10: ; Mandler et al. (2002) Bioconjugate Chem. 13: ), majtansynoidy (EP ; Liu et al., (1996) Proc. Natl. Acad. Sci. USA 93: ) i kalicheamycyna (Lode et al. (1998) Cancer Res. 58:2928; Hinman et al (1993) Cancer Res. 53: ). Toksyny mogą wywierać wpływ cytotoksyczny i cytostatyczny poprzez mechanizmy obejmujące wiązanie tubuliny, wiązanie DNA lub hamowanie topoizomerazy. Niektóre leki cytotoksyczne wykazują tendencję do braku aktywności lub mniejszej aktywności po sprzężeniu do dużych przeciwciał lub białkowych ligandów receptorowych. [0203] ZEVALIN (tiuksetan ibritumomabu, Biogen/Idec) jest koniugatem przeciwciała z radioizotopem złożonym z monoklonalnego mysiego IgG1 kappa, skierowanego przeciwko antygenowi CD20 występującego na powierzchni normalnych i złośliwych limfocytów B i radioizotopu 111 In lub 90 Y, przyłączonego tiomocznikowym łącznikiem-odczynnikiem chelatującym (Wiseman et al (2000) Eur. Jour. Nucl. Med. 27(7):766-77; Wiseman et al. (2002) Blood 99(12): ; Witzig et al. (2002) J. Clin. Oncol. 20(10): ; Witzig et al (2002) J. Clin. Oncol. 20(15): ). Choć preparat ZEVALIN wykazuje aktywność przeciwko chłoniakowi nieziarniczemu (NHL) z rozrostem limfocytów B, jego podanie u większości pacjentów prowadzi do ciężkich, długotrwałych cytopenii. MYLOTARG (gemtuzumab-ozogamycyna, Wyeth Pharmaceuticals), lecznicze przeciwciało sprzężone składające się z przeciwciała hucd33 połączonego z kalicheamycyną, zostało w 2000 r. zatwierdzone do stosowania w postaci zastrzyków w leczeniu ostrej białaczki szpikowej (Drugs of the Future (2000) 25(7): 686; patenty nr US 4,970,198; US 5,079,233; US 5,585,089; US 5,606,040; US 5,693,762; US 5,739,116; US 5,767,285; US 5,773,001). Kantuzumab-mertanzyna (Immunogen, Inc.), lecznicze przeciwciało sprzężone składające się z przeciwciała huc242 połączonego disiarczkowym łącznikiem SPP z lekową resztą majtansynoidu, DM1, przeszedł do drugiej fazy badań klinicznych w leczeni nowotworów, w których zachodzi ekspresja CanAg, takich, jak rak jelita grubego, trzustki, żołądka i inne. MLN-2704 (Millennium Pharm., BZL Biologics, Immunogen Inc.), lecznicze przeciwciało sprzężone składające się z monoklonalnego przeciwciała skierowanego przeciwko specyficznemu antygenowi błonowemu stercza (PSMA) połączonego z lekową resztą majtansynoidu, DM1, jest w fazie rozwoju w związku z potencjalnym zastosowaniem w leczeni u nowotworów prostaty. Peptydy aurystatynowe - aurystatyna E (AE) i monometyloaurystatyna(mmae) - syntetyczne analogi dolastatyny, zostały sprzężone z chimerycznymi monoklonalnymi przeciwciałami cbr96 (swoistymi dla antygenu Lewis Y na komórkach guzów) i cac10 (swoistymi dla CD30 w komórkach nowotworów hematologicznych) (Doronina et al (2003) Nature Biotechnology 21(7): ) i aktualnie znajdują się w fazie rozwoju. [0204] Chemioterapeutyki przydatne w generowaniu immunokoniugatów zostały opisane powyżej. Do aktywnych enzymatycznie toksyn i ich fragmentów, które mogą znaleźć zastosowanie należą łańcuch A toksyny dyfterii, niewiążące aktywne fragmenty toksyny dyfterii, łańcuch A egzotoksyny (z Pseudomonas aeruginosa), łańcuch A rycyny, łańcuch A abryny, łańcuch A modekcyny, alfa-sarcyna, białkaaleurites fordii białka diantyny, białka Phytolaca americana (PAPI, PAPII i PAPS) inhibitor Momordica charantia, kurcyna, krotyna, inhibitor Sapaonaria officinalis, gelonina, mitogellina, restyktocyna, fenomycyna, enomycyna oraz trikoteceny. Do produkcji przeciwciał sprzężonych z izotopami radioaktywnymi dostępny jest szereg radionuklidów. Przykłady obejmują 212 Bi, 131 1, 131 In, 90 Y i 186 Re. Koniugaty przeciwciała i środka cytotoksycznego wytwarza się z zastosowaniem wielu dwufunkcyjnych środków sprzęgających białka, takich jak propionian N-sukcynoimidylo-3-(2- pirydyloditiolu) (SPDP), iminotiolan (IT), dwufunkcyjne pochodne imidoestrów; (takie jak chlorowodorek adypoimidanu dimetylu), aktywne estry (takie jak suberynian disukcynoimidylu), aldehydy (takie jak aldehyd glutarowy), związki bis-azydowe (takie jak bis(pazydobenzoilo)heksanodiamina), pochodne bis-diazoniowe (takie jak bis-(pdiazoniobenzoilo)etylenodiamina), diizocyjaniany (takie jak toluileno-2,6-diizocyjanian) oraz związki z dwoma aktywnymi atomami fluoru (takie jak 1,5-difluoro-2,4-dinitrobenzen). Na przykład
68 67 immunotoksyna rycynowa może zostać przygotowana w sposób opisany w pracy Vitetta et al., Science, 238: 1098 (1987). Przykładowym odczynnikiem chelatującym służącym do sprzęgania radionuklidu z przeciwciałem jest znakowany izotopem węgla 14 C 1-isotiocyjanatobenzylo-3- metylodietylenotriaminopentaoctowy (MX-DTPA). Zob. patent nr WO 94/ [0205] W niniejszym dokumencie rozważa się również koniugaty przeciwciała i jednej lub większej liczby toksyn niskocząsteczkowych, na przykład kalicheamycyny, majtansynoidów, trikotecenu i CC1065 oraz pochodnych tych toksyn posiadających działanie toksyczne. Majtansyna i majtansynoidy [0206] W jednym z wariantów wynalazku przeciwciało (w pełnej długości lub we fragmencie) będące przedmiotem wynalazku sprzężone jest z jednym lub większą liczbę cząsteczek majtansynoidu. [0207] Majtansynoidy są inhibitorami przemian mitotycznych, których działanie polega na hamowaniu polimeryzacji tubulin. Majtansyna została po raz pierwszy wyodrębniona z wschodnioafrykańskiego krzewu Maytenus serrata (patent nr US 3,896,111). Następnie odkryto, że majtansynoidy, na przykład majtansynol i C-3 estry majtansynolu, są również wytwarzane przez niektóre drobnoustroje (patent nr US 4,151,042). Syntetyczny majtansynol, jego pochodne i analogi ujawniono na przykład w patentach nr US 4,137,230; US 4,248,870; US 4,256,746; US 4,260,608; US 4,265,814; US 4,294,757; US 4,307,016; US 4,308,268; US 4,308,269; US 4,309,428; US 4,313,946; US 4,315,929; US 4,317,821; US 4,322,348; US 4,331,598; US 4,361,650; US 4,364,866; US 4,424,219; US 4,450,254; US 4,362,663 i US 4,371,533. Koniugaty majtansynoid-przeciwciało [0208] W ramach prób podniesienia wskaźnika terapeutycznego do przeciwciał specyficznie wiążących antygeny komórek nowotworowych dołączono majtansynę i majtansynoidy. Immunokoniugaty zawierające majtansynoidy oraz ich zastosowania terapeutyczne ujawniono na przykład w patentach nr US 5,208,020, US 5,416,064 i EP B1. W pracy Liu et al., Proc. Natl. Acad. Sci. USA 93: (1996) opisano immunokoniugaty zawierające majtansynoid oznaczony jako DM1, połączony z monoklonalnym przeciwciałem C242 skierowanym przeciwko ludzkiemu rakowi odbytu i jelita grubego. Stwierdzono wysoką toksyczność koniugatu względem hodowlanych komórek raka jelita grubego, a także aktywność przeciwnowotworową w teście wzrostu guza in vivo. W pracy Chari et al., Cancer Research 52: (1992) opisano immunokoniugaty, w których majtansynoid wiązano poprzez łącznik disiarczkowy z mysim przeciwciałem A7, wiążącym antygen występujący w liniach komórkowych ludzkiego raka jelita grubego lub z innym mysim przeciwciałem monoklonalnym TA.1, wiążącym onkogen HER-2/neu. Cytotoksycznośc koniugatu TA.1-majtansynoid zbadano in vitro w linii komórek ludzkiego raka piersi SK-BR-3, w których zachodzi ekspresja 3 x 10 5 antygenów powierzchniowych HER-2 na jedną komórkę. Koniugat przeciwciała i leku charakteryzował się stopniem cytotoksyczności podobnym do wolnego majtansynoidu, który można było podnieść zwiększając liczbę cząsteczek majtansynoidu na cząsteczkę przeciwciała. Koniugat A7-majtansynoid charakteryzował się niską cytotoksycznością układową w organizmie myszy. Koniugaty majtansynoid-przeciwciało (immunokoniugaty) [0209] Koniugaty przeciwciało-majtansynoid przygotowuje się łącząc przeciwciało z cząsteczką majtansynoidu na drodze reakcji chemicznej bez zmniejszania aktywności biologicznej czy to przeciwciała, czy też cząsteczki majtansynoidu. Średnia liczba 3-4 cząsteczek majtansynoidu skoniugowanego z cząsteczką przeciwciała wykazywała skuteczność w zwiększaniu cytotokcyczności względem komórek docelowych bez negatywnego wpływu na działanie lub rozpuszczalność przeciwciała, choć wzrostu cytotoksyczności względem niesprzężonego przeciwciała należałoby oczekiwać nawet przy jednej cząsteczce toksyny na jedna cząsteczkę przeciwciała. Majtansynoidy są dobrze znane w dziedzinie wynalazku i mogą być zsyntetyzowane znanymi technikami lub wyodrębnione z naturalnych źródeł. Odpowiednie majtansynoidy ujawniono na przykład w patencie nr US 5,208,020 oraz w innych wzmiankowanych powyżej patentach i publikacjach o charakterze innym niż patentowy. Korzystnymi majtansynoidami są majtansynol i analogi majtansynolu modyfikowane w pierścieniu aromatycznym lub w innych pozycjach cząsteczki majtansynoidu, na przykład różne estry majtansynolu. [0210] W dziedzinie znanych jest wiele grup łączących pozwalających na przygotowanie koniugatów przeciwciało-majtansynoid, w tym na przykład grupy opisane w patentach nr US 5,208,020 lub EP B1, oraz w pracy Chari et al., Cancer Research 52: (1992). Grupy łączące obejmują grupy disiarczkowe, grupy tioeterowe, grupy kwasolabilne grupy fotolabilne, grupy peptydazolabilne
69 68 lub esterazolabilne, jak to ujawniono w wyżej wymienionych patentach, przy czym preferowane są grupy disiarczkowe i tioeterowe. [0211] Koniugaty przeciwciała i majtansynoidu można wytworzyć z zastosowaniem wielu dwufunkcyjnych środków sprzęgających białka, takich jak propionian N-sukcynoimidylo-3-(2- pirydyloditiolu) (SPDP), cykloheksano-1-karboksylan sukcynoimidylo-4-n-maleimidometylu), iminotiolan (IT), dwufunkcyjne pochodne imidoestrów; (takie jak chlorowodorek adypoimidanu dimetylu), aktywne estry (takie jak suberynian disukcynoimidylu), aldehydy (takie jak aldehyd glutarowy), związki bis-azydowe (takie jak bis(p-azydobenzoilo)heksanodiamina), pochodne bisdiazoniowe (takie jak bis-(p-diazoniobenzoilo)etylenodiamina), diizocyjaniany (takie jak toluileno-2,6- diizocyjanian) oraz związki z dwoma aktywnymi atomami fluoru (takie jak 1,5-difluoro-2,4- dinitrobenzen). Szczególnie korzystne środki sprzęgające obejmują propionian N-sukcynimidylo-3-(2- pirydyloditiolu) (SPDP) (Carlsson et al., Bio-chem. J. 173: [1978]) i pentanoan N- sukcynimidylo-4-(2-pirydylotiolu) (SPP), wytwarzające połączenia disiarczkowe. [0212] Łącznik może być przyłączony do cząsteczki majtansynoidu w różnych pozycjach, w zależności od rodzaju połączenia. Na przykład połączenie estrowe może być utworzone w reakcji z grupą hydroksylową z zastosowaniem konwencjonalnych technik sprzęgania. Reakcja może zajść w pozycji C-3, w której znajduje się grupa hydroksylowa, pozycji C-14 modyfikowanej grupą hydroksymetylową, pozycji C-15 modyfikowanej grupą hydroksylową i pozycji C-20, w której znajduje się grupa hydroksylowa. W preferowanym wariancie połączenie tworzy się w pozycji C-3 majtansynolu lub jego analogu. Kalicheamycyna [0213] Inny przedmiotowy immunokoniugat składa się z przeciwciała sprzężonego z jedną lub dwiema cząsteczkami kalicheamycyny. Antybiotykowa rodzina kalicheamycyn jest w stanie dokonywać przerw w podwójnych łańcuchach DNA w stężeniach subpikomolarnych. Sposób przygotowywania koniugatów związków z rodziny kalicheamycyny można znaleźć w patentach nr US 5,712,374, US 5,714,586, US 5,739,116, US 5,767,285, US 5,770,701, US 5,770,710, US 5,773,001, US 5,877,296 (wszystkie w posiadaniu American Cyanamid Company). Nadające się do zastosowania strukturalne analogi kalicheamycyny obejmują między innymi γ 1, α I 2, α I 3, N-acetylo-γ I 1, PSAG i θ I 1 (Hinman et al., Cancer Research 53: (1993), Lode et al., Cancer Research 58: (1998) oraz wyżej wymienione patenty w posiadaniu American Cyanamid). Innym lekiem przeciwnowotworowym, do którego można przyłączyć przeciwciało, jest QFA, będący związkiem przeciwfolianowym. Zarówno kalicheamycyna, jak i QFA posiadają wewnątrzkomórkowe miejsca oddziaływania i nie przechodzą łatwo przez błonę osocza. W związku z tym komórkowy wychwyt ww. środków na drodze internalizacji za pośrednictwem przeciwciała znacznie zwiększa ich działanie cytotoksyczne. Inne środki cytotoksyczne [0214] Innymi środkami przeciwnowotworowymi, które mogą być sprzężone z przeciwciałami będącymi przedmiotem wynalazku są na przykład BCNU, streptozoicyna, winkrystyna i 5-fluorouracyl, rodzina środków znana pod grupową nazwą kompleksu LL-E33288 opisana w patentach nr US 5,053,394 i US 5,770,710, a także esperamycyny (patent nr US 5,877,296). [0215] Do aktywnych enzymatycznie toksyn i ich fragmentów, które mogą znaleźć zastosowanie należą łańcuch A toksyny dyfterii, niewiążące aktywne fragmenty toksyny dyfterii, łańcuch A egzotoksyny (z Pseudomonas aeruginosa), łańcuch A rycyny, łańcuch A abryny, łańcuch A modekcyny, alfa-sarcyna, białkaaleurites fordii białka diantyny, białka Phytolaca americana (PAPI, PAPII i PAP-S), inhibitor Momordica charantia, kurcyna, krotyna, inhibitor Sapaonaria officinalis, gelonina, mitogellina, restyktocyna, fenomycyna, enomycyna oraz trikoteceny. Zob np. patent WO 93/21232 opublikowany 28 października [0216] W niniejszym wynalazku ponadto rozważa się immunokoniugat wytworzony pomiędzy przeciwciałem a związkiem o aktywności nukleolitycznej (np. rybonukleazą, endonukleazą DNA taką jak deoksyrybonukleaza, DNaza). [0217] W celu selektywnego zniszczenia guza przeciwciało może zawierać atom o wysokiej radioaktywności. Do produkcji przeciwciał sprzężonych z izotopami radioaktywnymi dostępny jest szereg radionuklidów. Przykłady obejmują At 211, I 131, I 125, Y 90, Re 186, Re 188, Sm 153, Bi 212, P 32, Pb 212 oraz radioaktywne izotopy Lu. W przypadkach, gdy koniugat wykorzystywany jest do detekcji, może zawierać atom właściwy dla badań scyntygraficznych, na przykład Tc 99m lub I 123, albo etykietę spinową do badań obrazowych metoda jądrowego rezonansu magnetycznego (NMR) (znanego również jako
70 69 rezonans magnetyczny, MRI), na przykład również jod-123, jod-131, ind-111, fluor-19, węgiel-13, azot-15, tlen-17, gadolin, mangan lub żelazo. [0218] Etykiety radioaktywne lub inne etykiety mogą być wprowadzane do koniugatu znanymi sposobami. Na przykład peptyd może zostać wytworzony na drodze biosyntezy lub chemicznej syntezy aminokwasów z wykorzystaniem odpowiednich prekursorów aminokwasów zawierających na przykład fluor 19 zamiast wodoru. Znaczniki radioaktywne takie, jak Tc 99m lub I 123, Re 186, Re 188 czy In 111 mogą zostać przyłączone poprzez wchodzącą w skład peptydu resztę cysteinową. Itr-90 może zostać przyłączony przez resztę lizynową. Przyłączenie jodu-123 może zostać osiągnięte metodą IODOGEN (Fraker et al (1978) Biochem. Biophys. Res. Commun. 80: Inne metody zostały szczegółowo opisane w pracy Monoclonal Antibodies in Immunoscintigraphy (Chatal, CRC Press 1989). [0219] Koniugaty przeciwciała i środka cytotoksycznego można wytworzyć z zastosowaniem wielu dwufunkcyjnych środków sprzęgających białka, takich jak propionian N-sukcynoimidylo-3-(2- pirydyloditiolu) (SPDP), cykloheksano-1-karboksylan sukcynoimidylo-4-n-maleimidometylu), iminotiolan (IT), dwufunkcyjne pochodne imidoestrów; (takie jak chlorowodorek adypoimidanu dimetylu), aktywne estry (takie jak suberynian disukcynoimidylu), aldehydy (takie jak aldehyd glutarowy), związki bis-azydowe (takie jak bis(p-azydobenzoilo)heksanodiamina), pochodne bisdiazoniowe (takie jak bis-(p-diazoniobenzoilo)etylenodiamina), diizocyjaniany (takie jak toluileno-2,6- diizocyjanian) oraz związki z dwoma aktywnymi atomami fluoru (takie jak 1,5-difluoro-2,4- dinitrobenzen). Na przykład immunotoksyna rycynowa może zostać przygotowana w sposób opisany w pracy Vitetta et al., Science 238:1098 (1987). Przykładowym odczynnikiem chelatującym służącym do sprzęgania radionuklidu z przeciwciałem jest znakowany izotopem węgla 14 C kwas 1- isotiocyjanatobenzylo-3-metylodietylenotriaminopentaoctowy (MX-DTPA). Zob. patent nr WO 94/ Łącznik może być łącznikiem łamliwym, ułatwiającym uwalnianie leku cytotoksycznego w komórce. Na przykład można zastosować łącznik kwasolabilny, peptydazolabilny, fotolabilny, dimetylowy lub łącznik zawierający grupę disiarczkową (Chari et al., Cancer Research 52: (1992); U.S. nr US 5,208,020). [0220] Jako związki będące przedmiotem badania rozważa się, jednak bez ograniczania się do tych związków, ADC przygotowywane z zastosowaniem odczynników sieciujących: BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-emcs, sulfo-gmbs, sulfo-kmus, sulfo-mbs, sulfo-siab, sulfo-smcc i sulfo-smpb oraz SVSB (4- winylosulfonobenzoesan sukcynoimidylu), które są odczynnikami dostępnymi handlowo (np. oferowanymi przez Pierce Biotechnology, Inc., Rockford, IL., USA). Zob. str , Applications Handbook and Catalog. Przygotowanie koniugatów przeciwciało-lek [0221] W koniugatach typu przeciwciało-lek (ADC) będących przedmiotem wynalazku przeciwciało (Ab) skoniugowane jest z jedną lub większą liczbę reszt lekowych (D), np. z około 1 do około 20 resztami lekowymi na cząsteczkę przeciwciała poprzez łącznik (L). Koniugaty ADC w wzorze I mogą być przygotowane na kilka sposobów, z wykorzystaniem reakcji chemii organicznej, warunków i odczynników znanych osobom biegłym w dziedzinie tym z wykorzystaniem: (1) reakcji nukleofilowej grupy przeciwciała z biwalencyjnym odczynnikiem łącznikowym z wytworzeniem kowalencyjnego wiązania Ab-L i następczej reakcji z resztą kwasową D; oraz (2) reakcji nukleofilowej grupy reszty lekowej z biwalencyjnym odczynnikiem łącznikowym z wytworzeniem kowalencyjnego wiązania D-L i następczej reakcji grupą nukleofilową przeciwciała. Ab-(L-D) p I [0222] Grupy nukleofilowe w cząsteczkach przeciwciał obejmują między innymi: (i) N-końcowe grupy aminowe, (ii) grupy aminowe w łańcuchach bocznych, np. w lizynie, (iii) grupy tiolowe w łańcuchach bocznych, np. w cysteinie oraz (iv) cukrowych grup hydroksylowych lub aminowych w przypadku przeciwciała glikozylowanego. Grupy aminowe, tiolowe i hydroksylowe są nukleofilowe i mogą reagować z wytworzeniem wiązań kowalencyjnych z grupami elektrofilowymi w resztach łącznikowych i odczynnikach łącznikowych, w tym z: (i) estrami aktywnymi takimi jak estry NHS, estry HOBt, halomrówczany i halogenki kwasowe; (ii) halogenki alkilowe i benzylowe takie, jak haloacetamidy; (iii) aldehydy,ketony, grupy karboksylowe i maleimidowe. W niektórych przeciwciałach występują międzyłańcuchowe ugrupowania disiarczkowe, np. mostki cysteinowe. Przeciwciała można ureaktywnić w kierunku koniugacji z odczynnikami łącznikowymi poprzez traktowanie reduktorem, takim, jak DTT (ditiotreitol). Każdy mostek cysteinowy może więc teoretycznie utworzyć dwa reaktywne nukleofile tiolowe. Dodatkowe grupy nukleofilowe możne wprowadzić w cząsteczkę
71 70 przeciwciała w reakcji reszt lizynowych z 2-iminotiolanem (odczynnikiem Trauta), co prowadzi do konwersji grup aminowych do grup tiolowych. [0223] Koniugaty ADC będące przedmiotem wynalazku mogą być również wytworzone na drodze modyfikacji przeciwciała polegającej na wprowadzeniu reszt elektrofilowych, które mogłyby wchodzić w reakcje z podstawnikami nukleofilowymi w cząsteczce odczynnika łączącego lub leku. Reszty cukrowe glikozylowanych przeciwciał mogą być utleniane, na przykład odczynnikami nadjodanowymi, z wytworzeniem grup aldehydowych lub ketonowych, które mogą wchodzić w reakcje z grupą aminową w cząsteczce odczynnika łączącego lub leku. Powstałe w ten sposób iminowe zasady Schiffa mogą stanowić trwałe połączenia lub mogą być poddane redukcji, na przykład odczynnikami borowodorkowymi, z wytworzeniem trwałych połączeń aminowych. W jednym z wariantów reakcja węglowodanowej części glikozylowanego przeciwciała z galaktooksydazą lub metanadjodanem sodu może dostarczyć grupy karbonylowe (aldehydowe i ketonowe), które mogą zostać poddane reakcji z odpowiednimi grupami w cząsteczce leku (Hermanson, Bioconjugate Techniques). W innym wariancie białka zawierające N-terminalne reszty seryny lub treoniny mogą być poddane reakcji z metanadjodanem sodu z wytworzeniem aldehydu w miejsce pierwszego aminokwasu (Geoghegan & Stroh, (1992) Bioconjugate Chem. 3: ; US 5,362,852). Aldehyd taki może być użyty w reakcji z nukleofilem w reszcie leku lub łącznika. [0224] analogicznie, grupy nukleofilowe w reszcie leku mogą obejmować między innymi grupy aminowe, tiolowe, hydroksylowe, hydrazydowe, oksymowe, hydrazynowe, tiosemikarbazonowe, karboksylohydrazynowe oraz arylohydrazydowe mogące reagować z wytworzeniem wiązań kowalencyjnych z grupami elektrofilowymi w resztach łącznikowych i odczynnikach łącznikowych, w tym z: (i) estrami aktywnymi takimi jak estry NHS, estry HOBt, halomrówczany i halogenki kwasowe; (ii) halogenki alkilowe i benzylowe takie, jak haloacetamidy; (iii) aldehydy,ketony, grupy karboksylowe i maleimidowe. [0225] Ewentualnie białko fuzyjne zawierające przeciwciało i środek cytotoksyczny może zostać wytworzone np. przy użyciu technik rekombinacji lub syntezy peptydów. Długość łańcucha DNA może obejmować odpowiednie regiony kodujące dwie części koniugatu, czy to w pozycjach sąsiadujących, czy też rozdzielonych regionem kodującym peptyd łącznikowy bez zniszczenia pożądanych właściwości koniugatu. [0226] W jeszcze innym wariancie przeciwciało może być skoniugowane z receptorem (na przykład streptawidyną) w celu wykorzystania do wstępnego nakierowywania na guz nowotworowy, przy czym koniugat przeciwciało-receptor jest podawane pacjentowi, następnie niezwiązane przeciwciało usuwane jest z krążenia środkiem oczyszczającym, a następnie następuje podanie liganda (np. awidyny) sprzężonego ze środkiem cytotoksycznym (np. radionuklidem). Pochodne przeciwciał [0227] Przeciwciała będące przedmiotem niniejszego wynalazku mogą być poddane dalszym modyfikacjom w celu uwzględnienia w nich dalszych niebiałkowych reszt, znanych w dziedzinie wynalazku i łatwo dostępnych. Korzystnie reszty nadające się do tworzenia pochodnych przeciwciała są rozpuszczalnymi w wodzie polimerami. Nieograniczające zakresu wynalazku przykłady polimerów rozpuszczalnych w wodzie obejmują między innymi glikol polietylenowy (PEG), kopolimery glikolu etylenowego/glikolu propylenowego, karboksymetylocelulozę, dekstran, alkohol poliwinylowy, poliwinylopirolidon, poli-1,3-dioksolan, poli-1,3,6-trioksan, kopolimer etylen/bezwodnik kwasu maleinowego, poliaminokwasy (komopolimery lub losowe polimery) oraz dekstran i poli(nwinylopirolidon), homopolimery glikolu polietylenowego i polipropylenowego, kopolimery tlenku polipropylenu/etylenu, polietoksylowane poliole (np. glicerol), alkohol poliwinylowy i mieszaniny powyższych związków. Glikol polietylenowy/aldehyd propionowy może być korzystny w procesie wytwarzania ze względu na swoją trwałość w wodzie. Polimer może mieć dowolną masę cząsteczkową, może też być rozgałęziony lub nierozgałęziony. Liczba polimerów przyłączonych do przeciwciała może być różna, przy czym w przypadku przyłączenia więcej niż jednej cząsteczki polimeru, cząsteczki te mogą być takie same lub różne. Z reguły liczba i/lub rodzaj polimerów wykorzystywanych do tworzenia pochodnych może być ustalona w oparciu o rozważania obejmujące między innymi szczególne właściwości lub funkcje przeciwciała, których ma dotyczyć poprawa; fakt, czy pochodna przeciwciała będzie stosowana w leczeniu w określonych warunkach itp. Formulacje farmaceutyczne [0228] Formulacje terapeutyczne zawierające przeciwciało będące przedmiotem wynalazku przygotowuje się do przechowywania poprzez zmieszanie polipeptydu o pożądanym stopniu czystości
72 71 z opcjonalnymi dopuszczalnymi fizjologicznie substancjami nośnymi, pomocniczymi lub stabilizującymi (Remington's Pharmaceutical Sciences, wyd. 16, Osol, A. (red.), (1980)), w postaci roztworów wodnych, w postaci liofilizowanej lub w postaci innych formulacji suchych. Dopuszczalne substancje nośne, pomocnicze lub stabilizujące są nietoksyczne dla biorców w zastosowanych dawkach i stężeniach i obejmują bufory takie, jak bufor fosforanowy, cytrynianowy, histydynę i inne kwasy organiczne; przeciwutleniacze, w tym kwas askorbinowy i metioninę, środki konserwujące (takie jak chlorek oktadecylodimetylobenzyloamoniowy, chlorek heksametoniowy, chlorek benzalkoniowy, chlorek benzetoniowy, fenol, alkohol butylowy lub benzylowy; alkiloparabeny takie, jak metylo- lub propyloparaben, katechol, rezorcynol,. cykloheksanol, 3-pentanol i m-krezol); niskocząsteczkowe (zawierające mniej niż 10 reszt) polipeptydy; białka, na przykład albumina surowicy, żelatyna lub immunoglobuliny, hydrofilowe polimery takie, jakpoliwinylopirolidon, aminokwasy takie, jak glicyna, glutamina, asparagina, histydyna, arginina lub lizyna, monosacharydy, disacharydy i inne węglowodany, w tym glukoza, mannoza lub dekstryny; środki chelatujące takie, jak EDTA; cukry takie jak sacharoza, mannitol, trehaloza lub sorbitol; przeciwjony tworzące sole takie, jak sód; kompleksy metali (np. kompleksy Zn-białko) i/lub niejonowe środki powierzchniowo czynne takie, jak TWEEN, PLURONICS czy glikol polietylenowy (PEG). [0229] Formulacja według wynalazku może również zawierać jeden lub większą liczbę związków aktywnych zgodnie z potrzebami wyznaczanymi przez leczone wskazanie, korzystnie związków o komplementarnej aktywności, niewchodzących w niepożądane oddziaływania. Cząsteczki takie korzystnie obecne są w skojarzeniu w ilościach skutecznych dla zamierzonych celów. [0230] Składniki aktywne mogą również być umieszczone w mikrokapsułkach, wytworzonych na przykład techniką koacerwacji lub polimeryzacji międzyfazowej, na przykład w odpowiednio w mikrokapsułkach hydroksymetylocelulozowych lub żelatynowych oraz z polimetakrylanu metylu, w koloidalnych układach dostarczania leków (np. w liposomach, mikrosferach albuminowych, nanocząstkach i nanokapsułkach) lub w makroemulsjach. Techniki te zostały opisane w pozycji Remington's Pharmaceutical Sciences, wyd. 16., red. Osol, A. (1980). [0231] Formulacje przeznaczone do zastosowań in vivo muszą być jałowe. Jałowość formulacji można z łatwością uzyskać dzięki filtracji przez jałowe błony filtracyjne. [0232] Możliwe jest przygotowywanie preparatów o przedłużonym uwalnianiu. Odpowiednie przykłady preparatów o przedłużonym uwalnianiu obejmują półprzepuszczalne matryce stałych polimerów hydrofobowych zawierające immunoglobulinę będącą przedmiotem wynalazku, przy czym rzeczone matryce mają postać wyrobów formowanych, np. błon lub mikrokapsułek. Przykładowe matryce do przedłużonego uwalniania obejmują poliestry, hydrożele (np. polimetakrylan 2-hydroksyetylu lub alkohol poliwinylowy), polilaktydy (Pat. nr 3,773,919), kopolimery kwasu L-glutaminowego iγ-lglutaminianu etylu, niedegradowalnego octanu etylenowo-winylowego, degradowalnych kopolimerów kwasu mlekowego-kwasu glikolowego jak na przykład LUPRON DEPOT (mikrosfery z kopolimeru kwasu mlekowego-kwasu glikolowego i octanu leuprolidu do iniekcji) oraz kwas poli-d-(-)-3- hydroksymasłowy. Podczas, gdy polimery takie, jak octan etylenowo-winylowy czy kopolimer kwasu mlekowego-kwasu glikolowego umożliwiają uwalnianie cząsteczek przez ponad 100 dni, niektóre hydrożele uwalniają białka w krótszych okresach. Kiedy zamknięte w matrycy immunoglobuliny pozostają w organizmie przez dłuższy czas, istnieje możliwość ich denaturacji lub agregacji w wyniku narażenia na wilgoć w temperaturze 37 C, prowadząc do utraty aktywności biologicznej i możliwych zmian immunogenności. Istnieje możliwość opracowania racjonalnych strategii stabilizacji przeciwciał w zależności od mechanizmu. Na przykład w przypadku odkrycia, że mechanizm agregacji polega na tworzeniu międzycząsteczkowych wiązań S-S na drodze wymiany tiol-disiarczek, stabilizację można osiągnąć poprzez modyfikację reszt sulfhydrylowych, liofilizację z kwaśnych roztworów, kontrolę zawartości wody, stosowanie odpowiednich dodatków i opracowywanie właściwych kompozycji matrycy polimerowej. Zastosowania [0233] Przeciwciało będące przedmiotem niniejszego wynalazku może być na przykład wykorzystywane w metodach terapeutycznych in vitro, ex vivo i in vivo. Przeciwciała będące przedmiotem wynalazku mogą być wykorzystywane jako antagoniści do częściowego lub pełnego blokowania aktywności określonego antygenu in vitro, ex vivo i/lub in vivo. Ponadto przynajmniej niektóre przeciwciała będące przedmiotem wynalazku mogą neutralizować aktywność antygenów pochodzących z innych gatunków. Zgodnie z powyższym, przeciwciała będące przedmiotem wynalazku mogą być wykorzystywane do hamowania aktywności określonego antygenu, na przykład w hodowli komórkowej zawierającej antygen, u pacjentów będących ludźmi lub u innych pacjentów z
73 72 gromady ssaków, z którym przeciwciało będące przedmiotem wynalazku wchodzi w reakcję krzyżową (np. u szympansów, pawianów, marmozet, makaków i rezusów, świń oraz myszy). W jednym z wariantów przeciwciało będące przedmiotem wynalazku może być wykorzystywane do hamowania aktywności antygenu poprzez zetknięcia przeciwciała z antygenem w taki sposób, że nastąpi zahamowanie aktywności antygenu. Korzystnie antygen jest cząsteczką białka ludzkiego. [0234] W jednym z wariantów przeciwciało będące przedmiotem wynalazku może być wykorzystywane w metodzie hamowania aktywności antygenu u pacjenta cierpiącego na schorzenie, w którym aktywność antygenu jest szkodliwa, polegającej na podawaniu pacjentowi przeciwciała będącego przedmiotem wynalazku w taki sposób, że nastąpi zahamowanie aktywności antygenu. Korzystnie antygen jest cząsteczką białka ludzkiego, a pacjent jest człowiekiem. Ewentualnie pacjent może być ssakiem, u którego zachodzi ekspresja antygenu wiązanego przez przeciwciało. Ponadto, pacjent może być ssakiem, do którego organizmu wprowadzono antygen (np. poprzez podanie antygenu lub poprzez ekspresję transgenu kodującego antygen). Przeciwciało będące przedmiotem wynalazku może być podane pacjentowi ludzkiemu w celach terapeutycznych. Ponadto przeciwciało będące przedmiotem wynalazku może być podane ssakowi innemu niż człowiek, u którego zachodzi ekspresja antygenu z którym immunoglobulina wchodzi w reakcję krzyżową (np. ssaka z gromady naczelnych, świni lub myszy) w celach weterynaryjnych w zwierzęcym modelu choroby ludzkiej. W odniesieniu do ostatniej możliwości, rzeczone modele zwierzęce mogą być użyteczne w ocenie skuteczności terapeutycznej przeciwciał będących przedmiotem wynalazku (np. badania wielkości dawek i czasowych cykli podawania przeciwciała). Do użytecznych terapeutycznie przeciwciał blokujących będących przedmiotem wynalazku należą przykładowo, między innymi, przeciwciała anty HER2, anty VEGF, anty IgE, anty CD11, przeciwciała przeciwinterferonowe i przeciwko czynnikowi tkankowemu. Przeciwciała będące przedmiotem wynalazku mogą być wykorzystywane w leczeniu, hamowaniu, opóźnianiu progresji, zapobieganiu/opóźnianiu nawrotów, polepszaniu lub zapobieganiu chorób, zaburzeń lub schorzeń związanych z nieprawidłowa ekspresją i/lub aktywnością jednej lub większej liczby cząsteczek antygenowych, w tym między innymi guzów złośliwych i łagodnych; nowotworów niebiałaczkowych i limfoidalnych; zaburzeń neuronalnych, glejowych, astrocytarnych, podwzgórzowych i innych zaburzeń gruczołowych, zaburzeń makrofagalnych, nabłonkowych, zrębowych i blastocelicznych; oraz zaburzeń zapalnych, angiogennych i immunologicznych. [0235] W jednym z aspektów blokujące przeciwciało będące przedmiotem wynalazku jest specyficzne względem ligandu antygenu i hamuje aktywność antygenu blokując lub zakłócając oddziaływania ligand-receptor z udziałem ligandu antygenu i w ten sposób hamując odpowiedni szlak sygnałowy i inne zdarzenia cząsteczkowe lub komórkowe. Wynalazek obejmuje również przeciwciała specyficzne względem receptorów, niekoniecznie zapobiegające wiązaniu ligandu, ale zakłócające aktywację receptora, tym samym hamując wszelkie odpowiedzi wywoływane zazwyczaj związaniem ligandu. Wynalazek obejmuje również przeciwciała które albo korzystnie, albo wyłącznie wiążą kompleksy ligand-receptor. Przeciwciało będące przedmiotem wynalazku może również odgrywać rolę agonisty określonego antygenu, tym samym wzmacniając, nasilając lub aktywując wszelką aktywność lub cząstkowe aktywności związane z aktywacją receptora, w której pośredniczy wiązanie ligandu. [0236] W niektórych wariantach wynalazku immunokoniugat złożony z przeciwciała sprzężonego ze środkiem cytotoksycznym podawany jest pacjentowi. W niektórych wariantach immunokoniugat i/lub antygen, z którym jest związany jest/są internalizowany(-e) w komórce, prowadząc do zwiększenia skuteczności terapeutycznej immunokoniugatu w zabijaniu docelowych komórek, z którymi jest on związany. W jednym z wariantów działanie środka cytotoksycznego kierowane jest lub zakłóca funkcje kwasu nukleinowego w komórce docelowej. Przykłady tego rodzaju środków cytotoksycznych obejmują dowolne z wymienionych tu chemioterapeutyków (na przykład majtansynoid lub kalicheamycynę), izotop radioaktywny, rybonukleazę lub endonukleazę DNA. [0237] Przeciwciała będące przedmiotem wynalazku mogą być wykorzystywane same lub w skojarzeniu z innymi kompozycjami w ramach terapii. Na przykład przeciwciało będące przemiotem wynalazku może być podane równocześnie z innym przeciwciałem, chemiopterapeutykiem(-ami) (w tym koktajlami chemioterapeutyków), innym(i) środkiem(-ami) cytotoksycznym(i), środkiem(-ami), przeciwangiogennym(i), cytokinami i/lub środkiem(-ami) hamującym(i) wzrost. W przypadku, gdy przeciwciało będące przedmiotem wynalazku hamuje wzrost guza, szczególnie pożądane może być połączenie go z jednym lub większą liczba środków terapeutycznych, które również hamują wzrost guza. Na przykład przeciwciało będące przedmiotem wynalazku może być skojarzone z przeciwciałem anty VEGF (np. AVASTIN) i/lub przeciwciałami anty ErbB (np. HER-CEPTIN, przeciwciało anty HER2) w schemacie leczenia, tj. w leczeniu dowolnej z opisanych tu chorób, w tym raka odbytu i jelita grubego, przerzutowego raka piersi i raka nerki. Ewentualnie lub dodatkowo pacjent może
74 73 otrzymywać skojarzone leczenie radiacyjne (np. napromieniowanie zewnętrzną wiązką lub leczenie środkiem, na przykład przeciwciałem, znakowanym radioaktywnie). Tego rodzaju omówione powyżej terapie skojarzone obejmują podawanie łączne (gdy dwa lub więcej środków zawartych jest w tej samej formulacji lub w oddzielnych formulacjach) i oddzielne; w takim przypadku, podanie przeciwciała będącego przedmiotem wynalazku może zajść przed i/lub po podaniu terapii wspomagającej lub terapii wspomagających. [0238] Przeciwciało będące przedmiotem wynalazku (oraz wspomagający środek terapeutyczny) jest/są podawane na dowolne właściwe sposoby, w tym pozajelitowo, podskórnie, dootrzewnowo, dopłucnie i donosowo oraz, w przypadku, gdy wymagane jest leczenie miejscowe, podanie na zmianę. Wlewy pozajelitowe obejmują podawanie domięśniowe, dożylne, dotętnicze, dootrzewnowe lub podskórne. Ponadto odpowiednim sposobem podawania przeciwciała jest wlew pulsacyjny, zwłaszcza z zastosowaniem malejących dawek przeciwciała. Dawkowanie może odbywać się dowolną odpowiednią drogą, np. poprzez iniekcje takie, jak iniekcje dożylne lub podskórne, częściowo zależnie od tego, czy podawanie ma charakter krótkotrwały, czy przewlekły. [0239] Kompozycja zawierająca przeciwciało będąca przedmiotem wynalazku będzie formułowana, dawkowana i podawana zgodnie z dobra praktyką medyczną. Czynniki, które należy uwzględnić w tym kontekście obejmują leczone schorzenie, leczony gatunek ssaka, warunki kliniczne danego pacjenta, przyczynę schorzenia, miejsce podania środka, sposób podania, harmonogram podawania i inne czynniki znane praktykującym lekarzom. Przeciwciało nie musi być, ale opcjonalnie jest formułowane z dodatkiem jednego lub większej liczby środków aktualnie stosowanych w zapobieganiu lub leczeniu danego schorzenia. Skuteczna ilość tego rodzaju innych środków zależy od ilości przeciwciał będących przedmiotem wynalazku obecnych w formulacji, rodzaju zaburzenia lub leczenia oraz innych omawianych powyżej czynników. Leki te z reguły stosuje się w tych samych dawkach i przy użyciu tych samych dróg podania jak opisano powyżej albo od około 1 do około 99% dotychczas stosowanych dawek. [0240] W zapobieganiu lub leczeniu choroby odpowiednia dawka przeciwciała będącego przedmiotem wynalazku (samego lub w skojarzeniu z innymi środkami takimi, jak chemioterapeutyki) będzie zależała od rodzaju leczonej choroby, rodzaju przeciwciała, stopnia ciężkości i przebiegu choroby, faktu podawania przeciwciała w celach zapobiegawczych lub leczniczych, wcześniejszych terapii oraz wywiadu klinicznego pacjenta i jego odpowiedzi na przeciwciało, według decyzji lekarza prowadzącego leczenie. Odpowiednim sposobem podawania przeciwciała pacjentowi jest podanie jednorazowe seryjne. W zależności od rodzaju i stopnia ciężkości choroby, wstępną dawką rozważaną do podania pacjentowi jest dawka od około 1 µg/kg do około 15 mg/kg (np. 0,1 mg/kg - 10 mg/kg), czy to, na przykład w jednym lub większej liczbie oddzielnych podań, czy też we wlewie ciągłym. Jedna z typowych dobowych dawek może zawierać się w zakresie od około 1 µg/kg do 100 mg/kg lub więcej, w zależności od czynników wymienionych powyżej. W przypadku wielokrotnego podawania w okresie kilku dni lub dłuższym, w zależności od schorzenia, leczenie podtrzymuje się do wystąpienia pożądanego ustąpienia objawów choroby. Jedna z przykładowych dobowych dawek przeciwciała może zawierać się w zakresie od około 0,05 mg/kg do około 10 mg/kg. Tak więc, pacjentowi może zostać podana jedna lub większa liczba dawek obejmujących około 0,5 mg/kg, 2,0 mg/kg, 4,0mg/kg lub 10 mg/kg (lub dowolne kombinacje tych dawek). Dawki te mogą być podawane okresowo, na przykład raz w tygodniu lub raz na trzy tygodnie (np. tak, że pacjent otrzymuje od około dwóch do około dwudziestu, na przykład około sześciu dawek przeciwciała). Istnieje możliwość podania początkowej, wyższej dawki wysycającej i następczych niższych dawek. Przykładowy schemat dawkowania obejmuje podanie wstępnej dawki wysycającej wynoszącej około 4 mg/kg i następczych cotygodniowych dawek podtrzymujących w wysokości około 2 mg przeciwciała/kg. Użyteczne mogą być jednak również inne schematy dawkowania. Postęp omawianej terapii daje się łatwo monitorować przy użyciu konwencjonalnych technik i oznaczeń. Wyroby przemysłowe [0241] Opisywany jest wyrób przemysłowy zawierający materiały użyteczne w leczeniu, zapobieganiu i/lub rozpoznawaniu opisanych powyżej chorób. Wyrób przemysłowy obejmuje pojemnik i etykietę bądź ulotkę do opakowania umieszczoną na pojemniku lub związaną z pojemnikiem. Odpowiednie pojemniki obejmują na przykład butelki, fiolki, strzykawki itp. Pojemniki mogą być wytwarzane z szeregu materiałów takich, jak szkło czy tworzywa sztuczne. Pojemnik zawiera kompozycję jako taką lub w skojarzeniu z inną kompozycją skuteczną w leczeniu, zapobieganiu i/lub rozpoznawaniu schorzenia i może posiadać jałowy punkt dostępu (na przykład pojemnik może być workiem z roztworem do wlewów dożylnych lub fiolką z korkiem przebijanym przez igłę do iniekcji podskórnych). Co najmniej jednak składnikiem aktywnym kompozycji jest przeciwciało będące przedmiotem
75 74 wynalazku. Etykieta lub ulotka do opakowania informuje, że kompozycja stosowana jest w leczeniu wybranego schorzenia, na przykład raka. Ponadto, wyrób przemysłowy może zawierać (a) pierwszy pojemnik zawierający kompozycję, przy czym kompozycja zawiera przeciwciało będące przedmiotem wynalazku; oraz (b) drugi pojemnik zawierający kompozycję, przy czym kompozycja zawiera dodatkowy środek cytotoksyczny. Wyrób przemysłowy może ponadto zawierać ulotkę do opakowania zawierającą informację, że pierwsza i druga kompozycja przeciwciała mogą być zastosowane w leczeniu wybranego schorzenia, na przykład raka. Ewentualnie, lub dodatkowo, wyrób przemysłowy może ponadto zawierać drugi (lub trzeci) pojemnik zawierający dopuszczalny farmaceutycznie bufor, na przykład wodę bakteriostatyczną do iniekcji (BWFI), roztwór soli fizjologicznej buforowany fosforanem, roztwór Ringera i roztwór dekstrozy. Wyrób może ponadto zawierać inne materiały pożądane z komercyjnego punktu widzenia lub z punktu widzenia użytkownika, w tym inne bufory, rozcieńczalniki, filtry, igły i strzykawki. [0242] Poniżej przedstawiono przykłady metod i kompozycji będących przedmiotem wynalazku. Rozumie się, że realizowane mogą być inne warianty wynalazku, zgodne z ogólnym opisem powyżej. PRZYKŁADY Materiały i metody [0243] Numery reszt są zgodne z podanymi w pracy Kabata (Kabat et al., Sequences of Proteins of Immunological Interest, wyd. 5, Public Health Service, National Institutes of Health, Bethesda, MD, USA (1991)). Stosuje się jednoliterowe skróty aminokwasów. Degenerację sekwencji DNA oznacza się przy użyciu kodu TUB (N = A/C/G/T, D = A/G/T, V = A/C/G, B= C/G/T, H= A/C/T, K = G/T, M = A/C, R = A/G, S = G/C, W= A/T, Y = G/T). [0244] Bezpośrednie wszczepienie regionów hiperzmiennych w ludzką akceptorową konsensusową sekwencję zrębową -Wykorzystywanym w tym celu fagemidem jest monowalencyjny wektor ekspresyjny Fab-g3 (pv0350-2b) posiadający 2 otwarte ramki odczytu kontrolowane przez promotor phoa, zasadniczo zgodnie z opisem przedstawionym w pracy Lee et al., J. Mol. Biol. (2004), 340(5): Pierwsza otwarta ramka odczytu składa się z sekwencji sygnałowej stii przyłączonej do domen VH i CH1 akceptorowego łańcucha ciężkiego, po której następuje skrócone mniejsze białko płaszcza fagowego P3. Zob. Lee et al., supra. [0245] Domeny VL i VH mysiego przeciwciała 5D5 (patrz hybrydoma 5D5.11.6, nr depozytu ATCC HB-11895, data złożenia 23 maja 1995) ułożono obok domen ludzkiej konsensusowej sekwencji kappa I (huki) i ludzkiej sekwencji konsensusowej VH z podgrupy III (huiii). Do wszczepienia regionów HVR użyto akceptorowej sekwencji zrębowej VH, różniącej się od ludzkiej konsensusowej sekwencji domeny VH z podgrupy III w trzech pozycjach: R71A, N73T i L78A (Carter et al., Proc. Natl. Acad. Sci. USA 89:4285 (1992)). W celu wykonania bezpośredniego graftu HVR 5D5 (graftu 5D5), regiony hiperzmienne mysiego przeciwciała 5D5 (mu5d5) wprowadzono pomiędzy ludzkie akceptorowe konsensusowe sekwencje zrębowe. W domenie VL w ludzkiej konsensusowej sekwencji akceptorowej wszczepiono następujące regiony: pozycje (L1), (L2) i (L3). W domenie VH wszczepiono pozycje (H1), (H2) i (H3) (Rycina 1). [0246] Warianty bezpośrednich graftów generowano techniką mutagenezy Kunkela, stosując oddzielny oligonukleotyd dla każdego regionu hiperzmiennego. Właściwe klony oceniano metodą sekwencjonowania DNA. [0247] Miękka randomizacja regionów hiperzmiennych - Sekwencje każdego regionu hiperzmiennego różnicowano w oparciu o strategię miękkiej randomizacji, z tendencją w kierunku zachowania sekwencji mysich regionów hiperzmiennych. Cel ten został osiągnięty w oparciu o strategię zastosowania zatrutego oligonukleotydu, opisana w pracy Gallop et al., J. Med. Chem. 37: (1994). Dla dane pozycji w mutowanym regionie hiperzmiennym kodon kodujący aminokwas dzikiego typu zatruwany jest mieszaniną nukleotydów w stosunku , co daje średnio 50-procentowy odsetek mutacji w każdej z pozycji. [0248] Zrandomizowane oligonukleotydy były wzorowane na sekwencjach mysich regionów hiperzmiennych i obejmowały te same regiony, co regiony definiowane przez bezpośrednie wszczepienie regionów hiperzmiennych. Pozycja aminokwasu na początku sekwencji H2 (pozycja 49) w domenie VH została ograniczona do możliwych opcji A, G, S lub T poprzez zastosowanie kodonu RGC. [0249] Generowanie bibliotek fagów- Pule randomizowanych oligonukleotydów skonstruowanych dla każdego z regionów hiperzmiennych fosforylowano oddzielnie w sześciu reakcjach w objętości 20
76 75 µl, zawierającej 660 ng oligonukleotydu, 50 mm Tris ph 7,5, 10 mm MgCl 2, 1 mm ATP, 20 mm DTT i 5 U kinazy polinukleotydowej, prowadzonych przez 1 h w temperaturze 37 C. Sześć pul sfosforylowanych oligonukleotydów połączono z 20 µl szablonu Kunkela w 50 mm Tris, ph 7,5, 10 mm MgCl 2 w końcowej objętości 500 µl, uzyskując stosunek ilościowy oligonukleotydów do szablonu wynoszący 3. Mieszaninę poddano hybrydyzacji w 90 C przez 4 min., 50 C przez 5 min., a następnie schładzano w lodzie. Nadmiarowe, niezhybrydyzowane oligonukleotydy usunięto przy użyciu zestawy do oczyszczania QIAQUICK PCR (zestaw Qiagen nr 28106) ze zmodyfikowanym protokołem w celu zapobieżenia nadmiernej denaturacji zhybrydyzowanego DNA. Do 500 µl zhybrydyzowanej mieszaniny dodano 150 µl PB, a następnie mieszaninę rozdzielono na dwie kolumny z silikażelem. Po przemyciu każdej kolumny 750 µl PE i dodatkowym obrocie w celu wysuszenia, każdą kolumnę przemywano 110 µl 10 mm Tris, 1 mm EDTA, ph 8. Zhybrydyzowany i oczyszczony szablon (220 µl) uzupełniono dodając 1 µl 100mM ATP, 10 µl 25mM dntp (po 25mM datp, dctp, dgtp and dttp), 15 µl 100mM DTT, 25 µl buforu 10X TM (0,5 M Tris, ph 7,5, 0,1 M MgCl 2 ), 2400 U ligazy T4 i 30 U polimerazy T7 na 3 godziny w temperaturze pokojowej. [0250] Uzupełniony produkt analizowano na żelu Tris-octan-EDTA/agaroza (Sidhu et al., Methods in Enzymology 328: (2000)). Zazwyczaj widoczne były trzy pasma: dolne pasmo odpowiadające poprawnie uzupełnionemu i związanemu produktowi środkowe pasmo odpowiadające uzupełnionemu, ale niezwiązanemu produktowi i górne pasmo odpowiadające produktowi niezawierającemu łańcucha. Górne pasmo jest wynikiem samoistnej aktywności ubocznej polimerazy T7 i jest trudne do uniknięcia (Lechner et al., J. Biol. Chem. 258: (1983)); jednak pasmo to przekształcane jest z 30- krotnie mniejszą wydajnością w porównaniu z pasmem dolnym i zazwyczaj jego udział w bibliotece jest niewielki. Środkowe pasmo jest wynikiem braku 5'-fosforanu w końcowej reakcji wiązania; pasmo to jest przekształcane z dużą wydajnością i daje głównie sekwencję dzikiego typu. [0251] Uzupełniony produkt oczyszczono i elektroporowano do komórek SS320, po czym propagowano w obecności faga pomocniczego M13/KO7 zgodnie z opisem w pracy Sidhu et al., Methods in Enzymology 328: (2000). Rozmiary bibliotek zawierały się w zakresie 1-2 x 10 9 niezależnych klonów. W celu oceny jakości biblioteki sekwencjonowano losowe klony z początkowych bibliotek. [0252] Wybór fagu - Użyto wygenerowanego ludzkiego receptora HGF w fuzji z Fc (HGFR-Fc) (Mark et al., J. Biol. Chem. (1992), 267: ). Otrzymanym białkiem HGFR-Fc powleczono płytki mikrotitracyjne MaxiSorp (Nunc) w stężeniu 5 µg/ml w PBS. W pierwszej rundzie wyboru użyto 8 dołków białka docelowego; w następnych cyklach wyboru używano pojedynczego dołka białka docelowego. Dołki blokowano przez 1 godzinę odczynnikiem Casein Blocker (Pierce). Fag zbierano z nadsączu hodowlanego i wprowadzano do zawiesiny w PBS zawierającym 1 % BSA i 0,05% preparatu TWEEN 20 (PBST). Po wiązaniu zawartości dołków przez 2 godziny niezwiązany fag usunięto poprzez obfite przemycie PBS zawierającym 1 % BSA i 0,05% preparatu TWEEN 20 (PBST). Związany fag wymywano inkubując dołki przy użyciu 50 mm HCl, 0,5 M KCl przez 30 minut. Fag amplifikowano w komórkach Top10 przy użyciu faga pomocniczego M13/KO7 i hodowano przez noc w temperaturze 37 C na podłożu 2YT z dodatkiem karbanecyliny w ilości 50 µg/ml. W celu dokonania oceny stopnia wzbogacenia, miano faga wymytego z dołka powleczonych białkiem docelowym porównano z mianem faga odzyskanego z dołka niepowleczonego białkiem docelowym. [0253] Dla potrzeb dojrzewania powinowactwa biblioteki fagów posortowano przy użyciu metody sortowania roztworów. HFGR-Fc biotynylowano mieszając 500 µl roztworu HGFR-Fc w PBS o stężeniu 3,6 mg/ml i 10 µl 1 M fosforanu potasu, ph 8 z 20 µl 4 mm Sulfo-NHS-LC-biotyny (Pierce). Biotynylowane HGFR-Fc (b-hgfr-fc) oczyszczono na kolumnie NAP5 (Amersham Biosciences) w PBS. Dołki płytki mikrotitracyjnej powleczono przez noc neutrawidyną w stężeniu 10 µg/ml w PBS w temperaturze 4 C, a następnie blokowano przez 1 godzinę odczynnikiem Casein Blocker (Pierce). W pierwszej rundzie panoramowania 200 części zawiesiny faga w PBS zawierającej 0,05% Tween 20 (PBST) i 1% BSA mieszano przez 1 godzinę z 10 nm b-hgfr-fc. Fag związany z b-hgfr-fc wychwytywano w dołkach powleczonych neutrawidyną przez 10 minut, a niezwiązany fag odmyto PBST. Fagi wymywano przez 45 min przy użyciu 20 mm HCl, 500 mm KCl, zobojętniano i propagowano w komórkach XL1 Blue (Stratagene) w obecności faga pomocniczego KO7 (New England Biolabs). Następne rundy sortowania wykonywano w podobny sposób, z następującymi wyjątkami: w rundzie 2., końcowe stężenie b-hgfr-fc wynosiło 5,6 nm, w rundzie 3. końcowe stężenie b-hgfr-fc wynosiło 0,1 nm, w rundzie 4. końcowe stężenie b-hgfr-fc wynosiło 0,5 nm, a 1 godzinę przed wychwytem na neutrawidynie do mieszaniny dodano 780 nm niebiotynylowanego HGFR-Fc.
77 76 [0254] Badanie fagów w teście ELISA - Płytki mikrotitracyjne MaxiSorp powleczono ludzkim human HGFR-Fc w stężeniu 5 µg/ml w PBS, a następnie blokowano odczynnikiem Casein Blocker. Fagi z nadsączy hodowlanych inkubowano przez 1 h w seryjnych rozcieńczeniach HGFR-Fc w PBST zawierającym 1% BSA na płytce mikrotitracyjnej do hodowli tkankowych, a następnie 80 µl mieszaniny przenoszono do dołków powleczonych białkiem docelowym w celu wychwycenia niezwiązanego faga. Płytkę przemyto PBST, po czym dodano przeciwciało anty M13 sprzężone z HRP (Amersham Pharmacia Biotech) (1:5000 w PBST zawierającym 1% BSA) na 40 min. Płytkę przemyto PBST i rozwijano dodając substrat - tetrametylobenzydynę (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA). W celu wyznaczenia IC 50 wykonano wykres absorbancji przy długości fali 405 nm w funkcji docelowego stężenia w roztworze. Wartość tę wykorzystywano jako estymatorową wartość powinowactwa klonu Fab ulegającego ekspresji na powierzchni faga. [0255] Otrzymywanie Fab i oznaczanie powinowactwa - W celu uzyskania ekspresji białka Fab do pomiarów powinowactwa pomiędzy łańcuchem ciężkim a g3 w fagowym wektorze ekspresyjnym wprowadzono kodon stop. Klony transformowano w komórkach E. coli 34B8 i hodowano w podłożu AP5 w temperaturze 30 C (Presta et al. Cancer Res. 57: (1997)). Komórki zebrano poprzez odwirowanie, zawieszono w 10 mm Tris, 1 mm EDTA ph 8 i przerwano przy użyciu mikrofluidyzatora. Fab oczyszczono metodą chromatografii powinowactwa z zastosowaniem białka G. [0256] Oznaczenia powinowactwa wykonywano techniką rezonansu plazmonów powierzchniowych przy użyciu aparatu BIAcore HGFR-Fc immobilizowano (-1000 jednostek odpowiedzi (RU)) na chipie CM5, po czym nastrzykiwano różne stężenia Fab (od 4 do 500 nm) w PBST. Po każdym nastrzyknięciu chip regenerowano 100 mm HCl. W wyznaczonej odpowiedzi w postaci wiązania uwzględniano poprawkę, odejmując wartość RU dla pustej komórki. Do analiz kinetyki użyto modelu 1:1 Langmuira równoczesnego dopasowywania k on i k off. Elektrochemiluminescencyjny test blokowania wiązania HGF / cmet przez OA5D5 [0257] Oczyszczone białko cmet-ig z firmy Genentech (South San Francisco, CA) biotynylowano poprzez inkubację z 20-krotnym nadmiarem molowym NHS-X-biotyny w 0,1 M NaHCO3, ph 8,5 przy użyciu biotynylo-x-nhs (Research Organics, Cleveland, OH). Oczyszczony ludzki dwułańcuchowy HGF wyprodukowany przez Genentech oznaczono BV-TAG (nr kat ) na drodze syntezy z estrem aktywnym NHS, zgodnie ze wskazówkami wytwórcy (BioVeris International, Gaithersburg, MD, USA). Kompleks cmet-ig-biotyna (500 ng/ml), HGF-ruten (250 ng/ml) i serie rozcieńczeń przeciwciała OA5D5 w zakresie ,21 nm inkubowano razem w 100 µl rozcieńczalnika do oznaczenia: PBS + 0,5% BSA / 0,5% Tween 20 /0,033% Proclin (Supelco Inc. Bellefonte PA). Mieszaniny inkubowano przez 2-4 godziny w temperaturze pokojowej, z wytrząsaniem, na zamkniętych polipropylenowych płytkach z 96 okrągło-dennymi dołkami (Coming). Dodano streptawidynowe perełki magnetyczne (Dynabeads, BioVeris). Po końcowej, trwającej 45 min. inkubacji z energicznym wytrząsaniem płytki poddano odczytowi w aparacie BioVeris serii M (BioVeris International, Gaithersburg, MD, USA). KIRA (HGF-zależna fosfofylacja c-met) [0258] Komórki A549 (ATCC, Manassas, VA, USA) trzymano w podłożu hodowlanym (Ham's F12/DMEM 50:50 [Gibco, Grand Island, NY, USA] zawierającym 10% cielęcej surowicy płodowej (FBS, Sigma, St. Louis, MO, USA). W celu przygotowania komórek na potrzeby oznaczenia, komórki z konfluentnych hodowli wyodrębniono przy użyciu odczynnika Accutase (ICN, Aurora, OH, USA) i zaszczepiono na 96-dołkowych płytkach w ilości komórek/dołek. Po prowadzonej przez noc inkubacji w temperaturze 37 C, podłoże hodowlane usunięto a komórki inkubowano przez minut w niedoborze surowicy w podłożu zawierającym 0,1% FBS. W celu oznaczenia zdolności hamowania fosforylacji c-met przez OA-5D5, cząsteczkę poddano serii rozcieńczeń w zakresie od 200 do 0,19 nm w podłożu + 0,1% FBS i naniesiono na płytki do oznaczeń. Po 15 min inkubacji w 37 C dodano HGF (50 ng/ml). Następnie płytki inkubowano w temperaturze 37 C przez następne 10 minut, podłoże usunięto i dodano bufor do lizy komórek, (Cell Signaling Technologies, Cat # 9803, Beverly, MA, USA; uzupełniony koktajlem inhibitorów proteaz z firmy Calbiochem, nr kat, , San Diego, CA, USA). Lizaty analizowano pod kątem obecności forforylowanego c-met w teście elektrochemoluminescencyjnym przy użyciu aparatu BioVeris serii M (BioVeris International, Gaithersburg, MD, USA). Antyfosfotyrozynowe przeciwciało monoklonalne (klon4g10, Upstate, Lake Placid, MY, USA) oznakowano BV-TAG na drodze syntezy z estrem aktywnym NHS zgodnie z wskazówkami wytwórcy (BioVeris) Przeciwciała skierowane przeciw zewnątrzkomórkowej domenie c- met biotynylowano przy użyciu biotinylo-x-nhs (Research Organics, Cleveland, OH). Przeciwciało 4G10 znakowane BV-TAG i biotynylowane mab anty c-met rozcieńczono w buforze do oznaczeń
78 77 (PBS / 0,5% Tween-20 / 0,5% BSA) i dodano w postaci koktajlu do lizatów komórkowych. Po trwającej 1,5-3 h inkubacji w temperaturze pokojowej z energicznym wytrząsaniem dodano streptawidynowe perełki magnetyczne (Dynabeads, BioVeris). Po końcowej, trwającej 45 min. inkubacji z energicznym wytrząsaniem płytki poddano odczytowi w aparacie BioVeris. Hodowla komórek i oznaczenie proliferacji [0259] BaF3 jest mysią komórką limfatyczną zależną od IL-3, w której zazwyczaj nie zachodzi ekspresja c-met i która nie wykazuje odpowiedzi na HGF. Natomiast w komórkach BaF3-hMet otrzymanych w wyniku transfekcji normalnego, pełnej długości cdna właściwego dla ludzkiego c-met (Schwall et al., J. Cell Biol. (1996), 133: ), HGF stymuluje proliferację i przeżycie komórek pod nieobecność IL-3. Komórki BaF3-hMet i BaF3-neo rutynowo pasażowano w RPMI 1640, 5% FBS, 4 µl/l β-merkaptoetanolu, 100 U/ml penicyliny, 100 µg/ml siarczanu streptomycyny, 2 mm L-glutaminy i podłożu kondycjonowanym 5% WEHI jako źródle IL-3. W celu zmierzenia HGF-zależnej proliferacji komórek liczbę komórek po 3 dniach traktowania oznaczono ilościowo dodając 25 µl odczynnika Alarma Blue (Trek Diagnostic Systems; Cleveland, OH, USA) i mierząc natężenie fluorescencji po upływie 6 godzin. Eksperymentami kontrolnymi były badania proliferacji tych samych komórek pod nieobecność HGF. Komórki H358-PSF2 and HGF-PSF8 pasażowano w RPMI 1640, 10% FBS, 100 U/ml penicyliny, 100 µg/ml siarczanu streptomycyny, 2 mm L-glutaminy. Podłoże do oznaczeń stanowiło RPMI 1640 z dodatkiem odpowiednio 0,1%, 0,5% BSA, lub 10% FBS. Oznaczenie wykonano zgodnie z wcześniejszym opisem. Immunoprecypitacja i Western Blot [0260] Komórki H358 stanowią linię komórkową pochodzącą z ludzkiego niedrobnokomórkowego raka płuca (NSCLC). Komórki H358-PSF2 and HGF-PSF8 są trwałymi komórkami transfekowanymi ludzkim HGF (Tsao et al., Neoplasia, Vol.2, No.3, 2000). Komórki te hodowano w RPMI 1640, 10% FBS, 100 U/ml penicyliny, 2000 µg/ml siarczanu streptomycyny i 2 mm L-glutaminy. Wykrywanie reszt tyrozynowych w c-met prowadzono zasadniczo zgodnie z opublikowanym wcześniej opisem (Zioncheck, J Bio Chem, 270(28): , 1995). W skrócie, komórki naniesiono na płytki 60 mm i inkubowano przez noc, a następnie zmieniono podłoże na RPMI 1640, zawierające 0,5% BSA przed dodaniem kombinacji zawierających lub niezawierających 1nM HGF lub konkurującego przeciwciała OA5D5.v2. Po upływie 10 min. w temperaturze 37 C, podłoże usunięto a komórki poddano lizie buforem do lizy (150 mm NaCl, 1,5 mm MgC12, 1% Triton X-100, 1X koktajl inhibitorów proteazy, 1X koktajl inhibitorów fosfatazy (Sigma, St. Louis, MO)). Po odwirowaniu nadsącz lizatu inkubowano przez 1 godzinę w temperaturze 4 C z przeciwciałem poliklonalnym anty cmet-igg (c-28; Santa Cruz Biotechnology, Santa Cruz, CA, USA), związanym z białkiem G-Sepharose. Immunokompleksy przemyto i gotowano w 1X buforze do próbek przed rozdziałem techniką SDS-PAGE i technika elektroblotting na nitrocelulozie. Białka zawierające fosfotyrozynę wizualizowano przy użyciu przeciwciała antyfosfotyrozynowego (4G10; Upstate Biotechnology, Waltham, MA, USA) i następnie sprzężonego z HRP koziego anty-mysiego Fab (1:10 000; Jackson Labs, West Grove, PA, USA), zaś w przypadku całkowitego cmet przy użyciu przeciwciała c-28 (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) i następnie koziego anty-króliczego Fc-HRP (1:10 000; Jackson Labs, West Grove, PA, USA) z detekcją techniką chemiluminescencyjną. Badanie ksenograftu guza [0261] Atymiczne samice myszy zaszczepiono podskórnie 5 milionami komórek raka trzustki KP4. Kiedy guzy osiągały rozmiar mm 3, myszy przypisano do 2 grup po 10 osobników. Grupie 1 wstrzykiwano IP z substancją nośną dwa razy w tygodniu. Grupie 2 wstrzykiwano IP z OA5D5.v2 w dawce 30 mg/kg, dwa razy w tygodniu. Rozmiar guza mierzono dwa razy w tygodniu. Myszy poświęcano w momencie, gdy rozmiar guza osiągał dwukrotność wyjściowego rozmiaru guza lub gdy doszło do owrzodzenia guza. Wyniki i Dyskusja [0262] Humanizacja 5D5 - Ludzka akceptorowa sekwencja zrębowa stosowana do humanizacji 5D5 zawiera konsensusową ludzką sekwencję kappa I domeny VL oraz wariant ludzkiej konsensusowej sekwencji domeny VH z podgrupy III. Wariantowa domena VH charakteryzuje się 3 zmianami względem ludzkiej sekwencji konsensusowej: R71A, N73T i L78A. Domeny VL i VH mysiego przeciwciała 5D5 ułożono obok ludzkich domen kappa I i podgrupy III; każdy HVR identyfikowano i wszczepiano ludzkiej akceptorowej sekwencji zrębowej w celu wygenerowania graftu 5D5, który ulegałby ekspresji jako Fab w wektorze fagowym. Kiedy fag eksponujący graft 5D5 zbadano pod kątem wiązania immobilizowanego HGFR-Fc, wiązania nie zaobserwowano.
79 78 [0263] Utworzono bibliotekę, w której każdy z regionów HVR graftu 5D5 został miękko zrandomizowany (soft randomized). Bibliotekę panoramowano względem immobilizowanego HGFR- Fc w 4 rundy wyboru. Klony pobrano do sekwencjonowania DNA. Stwierdzono, że wybrano pojedynczy klon. Klon ten charakteryzował się pojedynczą zmianą w pozycji 94 (R94S) domeny VH tuż poza docelowym regionem mutagenezy HVR-H3. Analiza wybranego klonu w fagowym teście ELISA wskazywała, że miał on powinowactwo podobne do monowalencyjnego powinowactwa mysiego przeciwciała 5D5. Po ekspresji w postaci Fab i analizie techniką Biacore wyznaczono wartość Kd na poziomie 9.8 nm w porównaniu z wartością 8.3 nm dla monowalencyjnego powinowactwa mysiego przeciwciała 5D5. Tak więc nieoczekiwana substytucja prowadzi do odtworzenia pełnego powinowactwa wiązania dla graftu 5D5, zaś graft 5D5 plus R94S (hu5d5.v1) jest przeciwciałem w pełni humanizowanym. Co ciekawe, w tej samej pozycji w przeciwciele mysim znajduje się homologiczny aminokwas - treonina. MacCallum et al. MacCallum et al. J. Mol. Biol. 262: (1996)) analizowali struktury krystalograficzne kompleksów przeciwciało-antygen i stwierdzili, że pozycja 93 i 94 łańcucha ciężkiego są częścią regionu kontaktowego, w związku z czym zasadne wydaje się uwzględnienie tych pozycji w definicji hiperzmiennego regionu H3 (HVR-H3) w procesie humanizowania przeciwciał. [0264] Dojrzewanie powinowactwa hu5d5.v1 - W celu poprawy powinowactwa hu5d5.v1, wygenerowano sześć bibliotek fagowych wektorów ekspresyjnych na tle hu5d5.v1, z których każda miała na celu wprowadzenie miękkiej randomizacji w pojedynczym regionie HVR. W celu uniknięcia ponownej selekcji hu5d5.v1 z potencjalnie silnego tła szablonu dzikiego typu, przed wygenerowaniem każdej z bibliotek do regionu HVR mającego ulec mutacji wprowadzono kodony stop. Do zwiększenia skuteczności procesu selekcji faga w oparciu o powinowactwo wykorzystano metodę sortowania roztworów. Zmieniając stężenia biotynylowanego białka docelowego, zmniejszając czas wychwytu faga w celu osłabienia tła i dodając niebiotynylowane białko docelowe w celu wyeliminowania klonów o większych stałych dysocjacji można sprawnie wybrać klony o wysokim powinowactwie. Lee et al., supra. Począwszy od pierwszej rundy selekcji obserwowano wzbogacenie mieszanin (wychwyt faga zależny od cząsteczki docelowej), co wskazywało, że w każdej z bibliotek znajdowała się duża liczba klonów o dość dużym powinowactwie do HGFR-Fc. W kolejnych rundach zwiększano surowość selekcji (patrz sekcja Metody powyżej), a w trzeciej rundzie wszystkie 6 bibliotek połączono, tworząc siódmą pulę bibliotek. Po czterech rundach selekcji klony z wszystkich siedmiu pul poddano analizie. Wszystkie klony w bibliotekach kierowanych na modyfikacje HVR-L1 i HVR-L3 były identyczne z hu5d5.v1; zaobserwowano natomiast nowe sekwencje w bibliotekach kierowanych na modyfikacje HVR-L2, HVR-H1, HVR-H2 i HVR-H3 (Rycina 2). W puli obejmującej połączone 6 bibliotek dominowały sekwencje z biblioteki skierowanej na modyfikacje HVR-H3, co wskazywało, że sekwencje z tej biblioteki wykazywały najlepszą poprawę powinowactwa względem HGFR-Fc (Rycina 3). Wybrane klony poddano badaniu przesiewowemu w fagowym teście ELISA, a następnie eksponowano w postaci białka Fab protein. Powinowactwo białek oznaczano techniką Biacore. Kilka klonów z połączonej biblioteki, charakteryzujących się zmianami w HVR-H3 wykazywało poprawę powinowactwa w porównaniu z monowalencyjnym powinowactwem hu5d5.v1 lub mysiego przeciwciała 5D5 (Rycina 4). Klony te posiadały aminokwasy S/T w pozycji 94, R/S w pozycji 96 i T/S w pozycji 100, a także P/S/A w pozycji 100a. Najlepszy klon, klon 78 (hu5d5.v2) charakteryzował się 3 zmianami w porównaniu z hu5d5.v1 (94T, 96R i 100T) i 13-krotną poprawą powinowactwa. [0265] Tak więc, przy wyjściu od graftów 6 regionów HVR mysiego przeciwciała 5D5 HVRs w ludzkich akceptorowych sekwencjach zrębowych, wydłużenie HVR-H3 do pozycji 94 (treonina) i dodanie 2 zmian w HVR-H3 prowadzi do uzyskania a w pełni ludzkiego przeciwciała 5D5 z 13-krotną poprawą powinowactwa do HGFR. Ponadto stwierdzono, że opisane tu wybrane przeciwciała humanizowane posiadają aktywność biologiczną co najmniej porównywalną z macierzystym przeciwciałem 5D5, na przykład w testach fosforylacji receptora itp. (danych nie przedstawiono). [0266] Charakteryzacja przeciwciał będących przedmiotem wynalazku - Jednoramienne (ang. one-arm, oznaczane również jako OA ) przeciwciała anty c-met poddano charakteryzacji. Zbadano dwa przeciwciała będące przedmiotem wynalazku. Konkretnie, przeciwciało OA-5D5.v2 zawierało pojedyncze ramię Fab zawierające sekwencje domen zmiennych przedstawione na Rycinie 13, przy czym ramię Fab było przyłączone do regionu Fc, gdzie region Fc było kompleksem jednego polipeptydu Fc o sekwencji Fc przedstawionej na Rycinie 13 i jednego polipeptydu Fc o sekwencji Fc przedstawionej na Rycinie 14. Przeciwciała charakteryzowano w następujący sposób: (1) W oznaczeniu zdolności przeciwciała OA-5D5.v2 do blokowania wiązania HGF z właściwym receptorem, przeciwciało OA-5D5.v2 było w stanie blokować wiązanie HGF z receptorem co najmniej tak dobrze, jak dwa przeciwciała porównawcze, a dokładniej chimeryczne jednoramienne
80 79 przeciwciało (zawierające ramię Fab z macierzystego mysiego przeciwciała 5D5 (domeny zmienne przedstawiono na Rycinie 7) połączone z ludzkim regionem Fc) oraz inne przeciwciało będące przedmiotem wynalazku (OA- 5D5.v1). W badaniu w zakresie stężeń przeciwciała od około 3333 do około 0,21 nm, w warunkach opisanych powyżej w sekcji Materiały i Metody, stwierdzono, że OA-5D5.v2 charakteryzuje się wartością IC50 mniejszą niż około połowa wartości IC50 porównawczego przeciwciała, np. chimerycznego przeciwciała jednoramiennego i OA-5D5.v1. Co interesujące, OA-5D5.v1 również wykazywało blokowanie z wartością IC50 lepszą niż referencyjne przeciwciało chimeryczne. Zob. Ryc. 8. (2) W oznaczeniu zdolności przeciwciała OA-5D5.v2 do hamowania aktywacji receptora HGF, przeciwciało OA-5D5.v2 hamowało aktywację receptora kinazy co najmniej tak dobrze, jak dwa przeciwciała porównawcze opisane wyżej w p. (1). W badaniu w zakresie stężeń przeciwciała od około 200 do około 0,19 nm, w warunkach opisanych powyżej w sekcji Materiały i Metody, stwierdzono, że OA-5D5.v2 charakteryzuje się wartością IC50 mniejszą niż około połowa wartości IC50 porównawczego przeciwciała, np. chimerycznego przeciwciała jednoramiennego i OA- 5D5.v1. Zob. Ryc. 9. (3) Przeciwciało OA-5D5.v2 zbadano również pod kątem krzyżowego wiązania w organizmach różnych gatunków, obejmujących człowieka, naczelne (makaka), psowate i myszowate (mysz). Stwierdzono, że przeciwciało OA-5D5.v2 wiąże się specyficznie receptorem HGF człowieka i naczelnych (makak), natomiast nie wiąże się z receptorem występującym u psowatych lub myszowatych (myszy) (danych nie przedstawiono). (4) Przeciwciało OA-5D5.v2 zbadano pod kątem zdolności hamowania proliferacji komórek w obecności HGF. Jak przedstawiono na Rycinie 10, OA-5D5.v2 hamowało proliferacje komórek co najmniej tak samo dobrze, jak jego odpowiedniki w postaci przeciwciała chimerycznego i OA- 5D5.v1 (zgodnie z opisem w p. (1) powyżej). W badaniu w zakresie stężeń przeciwciała od około 0,01 do około 100 nm, w warunkach opisanych powyżej w sekcji Materiały i Metody, stwierdzono, że OA-5D5.v2 charakteryzuje się wartością IC50 mniejszą niż około połowa wartości IC50 porównawczego przeciwciała, np. chimerycznego przeciwciała jednoramiennego i OA-5D5.v1. Zob. Ryc. 10. Specyficzne wiązanie OA-5D5.v2 do komórek transfekowanych Met potwierdzono w analizie FACs (danych nie przedstawiono). (5) Przeciwciało OA-5D5.v2 zbadano pod kątem zdolności hamowania fosforylacji reszt tyrozynowych receptora w obecności HGF. Jak przedstawiono na Rycinach 11A i B, przeciwciało OA-5D5.v2 hamowało fosforylację reszt tyrozynowych w badanych stężeniach od około 10 do około 1000 nm. Zob. Ryc. 11A i B. (6) Przeciwciało OA-5D5.v2 badano pod kątem skuteczności in vivo w modelu ksenograftu guza opartym o linię komórek raka trzustki (KP4). Wyniki rzeczonego badania skuteczności wykazały, że przeciwciało OA-5D5.v2 jest w stanie hamować rozwój guza i powodować jego regresję in vivo. Jak przedstawiono na Rycinie 12, u większości zwierząt leczonych przeciwciałem stwierdzono zanik guza.
81 80 CZĘŚCIOWA BIBLIOGRAFIA [0267] Angeloni, D., Danilkovitch-Miagkova, A., Miagkov, A., Leonard, E. J. i Lerman, M. I. (2003). The soluble sema domain of the Ron receptor inhibits MSP-induced receptor activation. J Biol Chem. Antipenko, A., Himanen, J. P., van Leyen, K., Nardi-Dei, V., Lesniak, J., Barton, W. A., Rajashankar, K. R., Lu, M., Hoemme, C., Puschel, A. W. i Nikolov, D. B. (2003). Structure of the semaphorin-3a receptor binding module. Neuron 39, Bardelli, A., Longati, P., Gramaglia, D., Stella, M. C. i Comoglio, P. M. (1997). Gab1 coupling to the HGF/Met receptor multifunctional docking site requires binding of Grb2 and correlates with the transforming potential. Oncogene 15, Bertotti, A. i Comoglio, P. M. (2003). Tyrosine kinase signal specificity: lessons from the HGF receptor. Trends Biochem Sci 28, Bladt, F., Riethmacher, D., Isenmann, S., Aguzzi, A. i Birchmeier, C. (1995). Essential role for the c- met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376, Blechman, J. M., Lev, S., Barg, J., Eisenstein, M., Vaks, B., Vogel, Z., Givol, D. i Yarden, Y. (1995). The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80, Boix, L, Rosa, J. L., Ventura, F., Castells, A., Bruix, J., Rodes, J. i Bartrons, R. (1994). c-met mrna overexpression in human hepatocellular carcinoma. Hepatology 19, Bottaro, D. P., Rubin, J. S., Faletto, D. L., Chan, A. M., Kmiecik, T. E., Vande Woude, G. F. i Aaronson, S. A. (1991). Identification of the hepatocyte growth factor receptor as the c-met protooncogene product. Science 251, Bussolino, F., Di Renzo, M. F., Ziche, M., Bocchietto, E., Olivero, M., Naldini, L., Gaudino, G., Tamagnone, L., Coffer, A. i Comoglio, P. M. (1992). Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 119, Coltella, N., Manara, M. C., Cerisano, V., Trusolino, L., Di Renzo, M. F., Scotlandi, K. i Ferracini, R. (2003). Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. Faseb J 17, Cooper, C. S., Park, M., Blair, D. G., Tainsky, M. A., Huebner, K., Croce, C. M. i Vande Woude, G. F. (1984). Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311, Di Renzo, M. F., Olivero, M., Giacomini, A., Porte, H., Chastre, E., Mirossay, L., Nordlinger, B., Bretti, S., Bottardi, S., Giordano, S. et al. (1995). Overexpression and amplification of the met/hgf receptor gene during the progression of colorectal cancer. Clin Cancer Res 1, Ferguson, K. M., Berger, M. B., Mendrola, J. M., Cho, H. S., Leahy, D. J. i Lemmon, M. A. (2003). EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell 11, Furge, K. A., Zhang, Y. W. i Vande Woude, G. F. (2000). Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene 19, Garrett, T. P., McKern, N. M., Lou, M., Elleman, T. C., Adams, T. E., Lovrecz, G. O., Zhu, H. J., Walker, F., Frenkel, M. J., Hoyne, P. A., et al. (2002). Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 110, Gherardi, E., Youles, M. E., Miguel, R. N., Blundell, T. L., Iamele, L., Gough, J., Bandyopadhyay, A., Hartmann, G. i Butler, P. J. (2003). Functional map and domain structure of MET, the product of the c- met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A. Giancotti, F. G. i Ruoslahti, E. (1999). Integrin signaling. Science 285, Giordano, S., Corso, S., Conrotto, P., Artigiani, S., Gilestro, G., Barberis, D., Tamagnone, L. i Comoglio, P. M. (2002). The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol 4,
82 81 Giordano, S., Di Renzo, M. F., Narsimhan, R. P., Cooper, C. S., Rosa, C. i Comoglio, P. M. (1989). Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 4, Giordano, S., Maffe, A., Williams, T. A., Artigiani, S., Gual, P., Bardelli, A., Basilico, C., Michieli, P. i Comoglio, P. M. (2000). Different point mutations in the met oncogene elicit distinct biological properties. Faseb J 14, Hamanoue, M., Takemoto, N., Matsumoto, K., Nakamura, T., Nakajima, K. i Kohsaka, S. (1996). Neurotrophic effect of hepatocyte growth factor on central nervous system neurons in vitro. J Neurosci Res 43, Hartmann, G., Weidner, K. M., Schwarz, H. i Birchmeier, W. (1994). The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase met requires intracellular action of Ras. J Biol Chem 269, Jeffers, M., Rong, S. i Vande Woude, G. F. (1996). Enhanced tumorigenicity and invasion-metastasis by hepatocyte growth factor/scatter factor-met signalling in human cells concomitant with induction of the urokinase proteolysis network. Mol Cell Biol 16, Jeffers, M., Schmidt, L., Nakaigawa, N., Webb, C. P., Weirich, G., Kishida, T., Zbar, B. i Vande Woude, G. F. (1997). Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A 94, Jin, L., Fuchs, A., Schnitt, S. J., Yao, Y., Joseph, A., Lamszus, K., Park, M., Goldberg, I. D. i Rosen, E. M. (1997). Expression of scatter factor and c-met receptor in benign and malignant breast tissue. Cancer 79, Kuniyasu, H., Yasui, W., Yokozaki, H., Kitadai, Y. i Tahara, E. (1993). Aberrant expression of c-met mrna in human gastric carcinomas. Int J Cancer 55, 72-75, Lev, S., Yarden, Y. i Givol, D. (1992). A recombinant ectodomain of the receptor for the stem cell factor (SCF) retains ligand-induced receptor dimerization and antagonizes SCF-stimulated cellular responses. J Biol Chem 267, Liu, C., Park, M. i Tsao, M. S. (1992). Overexpression of c-met proto-oncogene but not epidermal growth factor receptor or c-erbb-2 in primary human colorectal carcinomas. Oncogene 7, Lokker, N. A., Mark, M. R., Luis, E. A., Bennett, G. L., Robbins, K. A., Baker, J. B. i Godowski, P. J. (1992). Structure-function analysis of hepatocyte growth factor: identification of variants that lack mitogenic activity yet retain high affinity receptor binding. Embo J 11, Lorenzato, A., Olivero, M., Patane, S., Rosso, E., Oliaro, A., Comoglio, P. M. i Di Renzo, M. F. (2002). Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res 62, Love, C. A., Harlos, K., Mavaddat, N., Davis, S. J., Stuart, D. I., Jones, E. Y. i Esnouf, R. M. (2003). The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat Struct Biol 10, Maina, F., Casagranda, F., Audero, E., Simeone, A., Comoglio, P. M., Klein, R. i Ponzetto, C. (1996). Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 87, Matsumoto, K. i Nakamura, T. (1993). Roles of HGF as a pleiotropic factor in organ regeneration. Exs 65, Maulik, G., Shrikhande, A., Kijima, T., Ma, P. C., Morrison, P. T. i Salgia, R. (2002). Role of the hepatocyte growth factor receptor, c-met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 73, Meiners, S., Brinkmann, V., Naundorf, H. i Birchmeier, W. (1998). Role of morphogenetic factors in metastasis of mammary carcinoma cells. Oncogene 16, Morello, S., Olivero, M., Aimetti, M., Bernardi, M., Berrone, S., Di Renzo, M. F. i Giordano, S. (2001). MET receptor is overexpressed but not mutated in oral squamous cell carcinomas. J Cell Physiol 189,
83 82 Naka, D., Ishii, T., Yoshiyama, Y., Miyazawa, K., Hara, H., Hishida, T. i Kidamura, N. (1992). Activation of hepatocyte growth factor by proteolytic conversion of a single chain form to a heterodimer. J Biol Chem 267, Naldini, L., Weidner, K. M., Vigna, E., Gaudino, G., Bardelli, A., Ponzetto, C., Narsimhan, R. P., Hartmann, G., Zarnegar, R., Michalopoulos, G. K. et al. (1991). Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. Embo J 10, Natali, P. G., Prat, M., Nicotra, M. R., Bigotti, A., Olivero, M., Comoglio, P. M. i Di Renzo, M. F. (1996). Over-expression of the met/hgf receptor in renal cell carcinomas. Int J Cancer 69, Nguyen, L., Holgado-Madruga, M., Maroun, C., Fixman, E. D., Kamikura, D., Fournier, T., Charest, A., Tremblay, M. L., Wong, A. J. i Park, M. (1997). Association of the multisubstrate docking protein Gab1 with the hepatocyte growth factor receptor requires a functional Grb2 binding site involving tyrosine J Biol Chem 272, Nusrat, A., Parkos, C. A., Bacarra, A. E., Godowski, P. J., Delp-Archer, C., Rosen, E. M. i Madara, J. L. (1994). Hepatocyte growth factor/scatter factor effects on epithelia. Regulation of intercellular junctions in transformed and nontransformed cell lines, basolateral polarization of c-met receptor in transformed and natural intestinal epithelia i induction of rapid wound repair in a transformed model epithelium. J Clin Invest 93, Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., Kim, J. H., Saito, K., Sakamoto, A., Inoue, M., Shirouzu, M. i Yokoyama, S. (2002). Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, Olivero, M., Rizzo, M., Madeddu, R., Casadio, C., Pennacchietti, S., Nicotra, M. R., Prat, M., Maggi, G., Arena, N., Natali, P. G., et al. (1996). Overexpression and activation of hepatocyte growth factor/scatter factor in human non-small-cell lung carcinomas. Br J Cancer 74, Olivero, M., Valente, G., Bardelli, A., Longati, P., Ferrero, N., Cracco, C., Terrone, C., Rocca-Rossetti, S., Comoglio, P. M. i Di Renzo, M. F. (1999). Novel mutation in the ATP-binding site of the MET oncogene tyrosine kinase in a HPRCC family. Int J Cancer 82, Orian-Rousseau, V., Chen, L., Sleeman, J. P., Herrlich, P. i Ponta, H. (2002). CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev 16, Park, M., Dean, M., Cooper, C. S., Schmidt, M., O'Brien, S. J., Blair, D. G. i Vande Woude, G. F. (1986). Mechanism of met oncogene activation. Cell 45, Peek, M., Moran, P., Mendoza, N., Wickramasinghe, D. i Kirchhofer, D. (2002). Unusual proteolytic activation of pro-hepatocyte growth factor by plasma kallikrein and coagulation factor XIa. J Biol Chem 277, Pelicci, G., Giordano, S., Zhen, Z., Salcini, A. E., Lanfrancone, L., Bardelli, A., Panayotou, G., Waterfield, M. D., Ponzetto, C., Pelicci, P. G. et al. (1995). The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene 10, Plotnikov, A. N., Schlessinger, J., Hubbard, S. R. i Mohammadi, M. (1999). Structural basis for FGF receptor dimerization and activation. Cell 98, Ponzetto, C., Bardelli, A., Zhen, Z., Maina, F., dalla Zonca, P., Giordano, S., Graziani, A., Panayotou, G. i Comoglio, P. M. (1994). A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77, Ponzetto, C., Zhen, Z., Audero, E., Maina, F., Bardelli, A., Basile, M. L., Giordano, S., Narsimhan, R. i Comoglio, P. (1996). Specific uncoupling of GRB2 from the Met receptor. Differential effects on transformation and motility. J Biol Chem 271, Robertson, S. C., Tynan, J. A. i Donoghue, D. J. (2000). RTK mutations and human syndromeswhen good receptors turn bad. Trends Genet 16, Royal, I. i Park, M. (1995). Hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J Biol Chem 270, Schmidt, C., Bladt, F., Goedecke, S., Brinkmann, V., Zschiesche, W., Sharpe, M., Gherardi, E. i Birchmeier, C. (1995). Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373,
84 83 Schmidt, L., Duh, F. M., Chen, F., Kishida, T., Glenn, G., Choyke, P., Scherer, S. W., Zhuang, Z., Lubensky, I., Dean, M., et al. (1997). Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 16, Schmidt, L., Junker, K., Nakaigawa, N., Kinjerski, T., Weirich, G., Miller, M., Lubensky, I., Neumann, H. P., Brauch, H., Decker, J., et al. (1999). Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 18, Suzuki, K., Hayashi, N., Yamada, Y., Yoshihara, H., Miyamoto, Y., Ito, Y., Ito, T., Katayama, K., Sasaki, Y., Ito, A. et al. (1994). Expression of the c-met protooncogene in human hepatocellular carcinoma. Hepatology 20, Tamagnone, L., Artigiani, S., Chen, H., He, Z., Ming, G. I., Song, H., Chedotal, A., Winberg, M. L., Goodman, C. S., Poo, M., et al. (1999). Plexins are a large family of receptors for transmembrane, secreted i GPI-anchored semaphorins in vertebrates. Cell 99, Tempest, P. R., Stratton, M. R. i Cooper, C. S. (1988). Structure of the met protein and variation of met protein kinase activity among human tumour cell lines. Br J Cancer 58, 3-7. Trusolino, L., Bertotti, A. i Comoglio, P. M. (2001). A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 107, Uehara, Y., Minowa, O., Mori, C., Shiota, K., Kuno, J., Noda, T. i Kitamura, N. (1995). Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 373, Van Vactor, D. V. i Lorenz, L. J. (1999). Neural development: The semantics of axon guidance. Curr Biol 9, R Weidner, K. M., Di Cesare, S., Sachs, M., Brinkmann, V., Behrens, J. i Birchmeier, W. (1996). Interaction between Gab1 and the c-met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 384, Wiesmann, C., Fuh, G., Christinger, H. W., Eigenbrot, C., Wells, J. A. i de Vos, A. M. (1997). Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91, Wiesmann, C., Ultsch, M. H., Bass, S. H. i de Vos, A. M. (1999). Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 401,
85 84 Zastrzeżenia 1. Humanizowana postać mysiego przeciwciała anty c-met, zawierająca: (a) co najmniej jedną sekwencję HVR wybraną z grupy składającej się z: (i) HVR-L1 zawierający sekwencję A1-A17, gdzie sekwencja A1-A17 ma postać KSSQSLLYTSSQKNYLA, (ii) HVR-L2 zawierający sekwencję B1-B7, gdzie sekwencja B1-B7 ma postać WASTRES, (iii) HVR-L3 zawierający sekwencję C1-C9, gdzie sekwencja C1-C9 ma postać QQYYAYPWT, (iv) HVR-H1 zawierający sekwencję D1-D10, gdzie D1-D10 ma postać GYTFTSYWLH, oraz (v) HVR-H2 zawierający sekwencję E1-E18, gdzie E1-E18 ma postać GMIDPSNSOTRFNPNFKD; i (b) wariant HVR-H3 zawierający sekwencję TYRSYVTPLDY, SYRSYVTPLDY, TYSSYVTPLDY, SYSSYVTPLDY, TYSSYVTSLDY lub TYSSYVTALDY, w którym monowalencyjne powinowactwo rzeczonego przeciwciała do ludzkiego c-met jest większe niż monowalencyjne powinowactwo mysiego przeciwciała zawierającego zmienną sekwencję łańcucha lekkiego przedstawioną jako SEQ ID NO: 9 i zmienną sekwencję łańcucha ciężkiego przedstawioną jako SEQ ID NO: 10, oraz w którym rzeczone przeciwciało hamuje c- met-zależną proliferację komórek lepiej niż przeciwciało referencyjne w postaci chimerycznego przeciwciała anty c-met, zawierającego zmienne sekwencje łańcucha lekkiego i ciężkiego przedstawione odpowiednio jako SEQ ID NO_9 i Przeciwciało według zastrz. 1, w którym co najmniej część sekwencji zrębowej stanowi ludzka konsensusowa sekwencja zrębowa. 3. Przeciwciało według zastrz. 1 zawierające wariant HVR-L2 posiadający strukturę przedstawioną jako SEQ ID NO: Przeciwciało według zastrz. 1 zawierające wariant HVR-H2 posiadający strukturę przedstawioną jako SEQ ID NO: Przeciwciało według zastrz. 1 zawierające HVR-L1 posiadający strukturę przedstawioną jako SEQ ID NO: 1 6. Przeciwciało według zastrz. 1 zawierające HVR-L3 posiadający strukturę przedstawioną jako SEQ ID NO: 3 7. Przeciwciało według zastrz. 1, w którym wariant HVR-H3 zawiera sekwencję TYRSYVTPLDY, TYSSYVTPLDY, TYSSYVTSLDY lub TYSSYVTALDY. 8. Przeciwciało według zastrz. 1, w którym monowalencyjne powinowactwo przeciwciało do ludzkiego c-met jest co najmniej 3-krotnie większe niż monowalencyjne powinowactwo mysiego przeciwciała zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione na Fig. 7 jako SEQ ID NO: 9 i Humanizowane przeciwciało według zastrz. 1, w którym mysie przeciwciało wytwarzane jest przez linią komórek hybrydoma zdeponowaną w kolekcji kultur ATCC z oznaczeniem HB (hybrydoma 5D5.11.6). 10. Przeciwciało według zastrz. 1, w którym powinowactwo wiązania wyraża się wartością Kd. 11. Przeciwciało według zastrz. 1, 8 albo 9, w którym powinowactwo wiązania mierzone jest techniką Biacore lub w teście radioimmunologicznym. 12. Przeciwciało według zastrz. 1, zawierające ludzką konsensusową sekwencję zrębową κ z podgrupy I. 13. Przeciwciało według zastrz. 1, zawierające ludzką konsensusową sekwencję zrębową łańcucha ciężkiego z podgrupy III. 14. Przeciwciało według zastrz. 13, w którym sekwencja zrębowa zawiera substytucje aminokwasów w pozycjach 71, 73 i/lub 78.
86 Przeciwciało według zastrz. 14, w którym substytucje mają postać R71A, N73T i/lub N78A. 16. Przeciwciało według zastrz. 1, w którym humanizowane przeciwciało hamuje wiązanie ludzkiego czynnika wzrostu hepatocytów przez właściwy receptor lepiej niż referencyjne przeciwciało w postaci chimerycznego przeciwciała anty c-met zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione odpowiednio jako SEQ ID NO: 9 i Przeciwciało według zastrz. 16, w którym przeciwciało hamuje wiązanie receptora z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. 18. Przeciwciało według zastrz. 17, w którym wartość IC50 oznaczana jest w zakresie stężeń przeciwciała od 0,01 nm do około 1000 nm. 19. Przeciwciało według zastrz. 1, w którym humanizowane przeciwciało hamuje aktywację receptora ludzkiego czynnika wzrostu hepatocytów (HGF) lepiej niż referencyjne przeciwciało w postaci chimerycznego przeciwciała anty c-met zawierającego zmienne sekwencje łańcucha lekkiego i łańcucha ciężkiego przedstawione odpowiednio jako SEQ ID NO: 9 i Przeciwciało według zastrz. 19, w którym przeciwciało hamuje aktywację receptora z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. 21. Przeciwciało według zastrz. 20, w którym wartość IC50 oznaczana jest w zakresie stężeń przeciwciała od 0,1 nm do około 100 nm. 22. Przeciwciało według zastrz. 1, w którym humanizowane przeciwciało hamuje proliferację komórek z wartością IC50 wynoszącą mniej niż połowę wartości właściwej dla przeciwciała chimerycznego. 23. Przeciwciało według zastrz. 22, w którym wartość IC50 oznaczana jest w zakresie stężeń przeciwciała od około 0,01 nm do około 100 nm. 24. Przeciwciało według któregokolwiek z powyższych zastrzeżeń, w którym zarówno przeciwciało humanizowane, jak i przeciwciało chimeryczne są przeciwciałami monowalencyjnymi. 25. Przeciwciało według powyższych zastrzeżeń, w którym zarówno przeciwciało humanizowane, jak i przeciwciało chimeryczne zawierają pojedynczy region Fab połączony z regionem Fc. 26. Przeciwciało według zastrz. 1, zawierające zmienną domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3-HC, przedstawione odpowiednio jako SEQ ID NO: Przeciwciało według zastrz. 26, w którym domena zmienna zawiera sekwencje FR1-HC, FR2-HC, FR3-HC i FR4-HC, przedstawione odpowiednio jako SEQ ID NO: Przeciwciało według zastrz. 26 albo 27, w którym przeciwciało zawiera sekwencję CH1 i/lub Fc, przedstawioną odpowiednio jako SEQ ID NO: 194 i Przeciwciało według zastrz. 1 zawierające zmienną domenę łańcucha lekkiego, zawierającą sekwencje HVR1-LC, HVR2-LC i HVR3-LC, przedstawione odpowiednio jako SEQ ID NO: Przeciwciało według zastrz. 29, w którym domena zmienna zawiera sekwencje FR1-LC, FR2-LC, FR3-LC i FR4-LC, przedstawione odpowiednio jako SEQ ID NO: Przeciwciało według zastrz. 29 albo 30, w którym przeciwciało zawiera sekwencję CL1, przedstawioną jako SEQ ID NO: Przeciwciało zawierające zmienną domenę łańcucha ciężkiego opisaną w którymkolwiek z zastrzeżeń od 26 do 28 oraz zmienną domenę łańcucha lekkiego opisaną w którymkolwiek z zastrzeżeń od 29 do Przeciwciało według zastrz. 32, zawierające zmienną domenę łańcucha ciężkiego, zawierającą sekwencje HVR1-HC, HVR2-HC i HVR3-HC, przedstawione odpowiednio jako SEQ ID NO: , sekwencje FR1-HC, FR2-HC, FR3-HC i FR4-HC, przedstawione odpowiednio jako SEQ ID NO: , oraz sekwencje CH1 i Fc przedstawione odpowiednio jako SEQ ID NO: 194 i 195, oraz zmienną domenę łańcucha lekkiego, zawierającą sekwencje HVR1-LC, HVR2-LC i HVR3- LC, przedstawione odpowiednio jako SEQ ID NO: , sekwencje FR1-LC, FR2-LC, FR3-LC i FR4-LC, przedstawione odpowiednio jako SEQ ID NO: , oraz sekwencję CL1 przedstawioną jako SEQ ID NO: 186.
87 Przeciwciało według zastrz. 32 albo 33, w którym przeciwciało jest monowalencyjne i zawiera region Fc. 35. Przeciwciało według zastrz. 34, w którym region Fc zawiera pierwszy i drugi polipeptyd, gdzie zarówno pierwszy, jak i drugi polipeptyd zawiera jedną lub większą liczbę mutacji ludzkiej sekwencji Fc dzikiego typu. 36. Przeciwciało według zastrz. 35, w którym pierwszy polipeptyd zawiera sekwencję Fc przedstawioną jako SEQ ID NO: 195 zaś drugi polipeptyd zawiera sekwencję przedstawioną jako SEQ ID NO: Przeciwciało według któregokolwiek z powyższych zastrzeżeń, przeznaczone do zastosowań leczniczych. 38. Przeciwciało według któregokolwiek z powyższych zastrzeżeń, przeznaczone do zastosowań w leczeniu raka. 39. Przeciwciało według zastrz. 38, znamienne tym, że rak jest rakiem płuca, rakiem mózgu, rakiem nerki, rakiem żołądka, rakiem okrężnicy i odbytu i/lub rakiem trzustki. 40. Przeciwciało według któregokolwiek z zastrzeżeń 1 do 36, przeznaczone do zastosowań w leczeniu chorób proliferacyjnych. 41. Zastosowanie przeciwciała według któregokolwiek z zastrzeżeń 1 do 36 w przygotowaniu substancji leczniczej do stosowania w leczeniu raka. 42. Zastosowanie według zastrz. 41, w którym rak jest rakiem płuca, rakiem mózgu, rakiem nerki, rakiem żołądka, rakiem okrężnicy i odbytu i/lub rakiem trzustki. 43. Zastosowanie przeciwciała opisanego w którymkolwiek z zastrzeżeń 1 do 36 w przygotowaniu substancji leczniczej do stosowania w leczeniu choroby proliferacyjnej. 44. Kwas nukleinowy kodujący przeciwciało opisane w którymkolwiek z zastrzeżeń 1 do Komórka gospodarza zawierająca kwas nukleinowy opisany w zastrzeżeniu Kompozycja zawierająca przeciwciało opisane w którymkolwiek z zastrzeżeń 1 do Kompozycja według zastrz. 46, w którym rzeczona kompozycja zawiera substancję nośną.
88 87
89 88
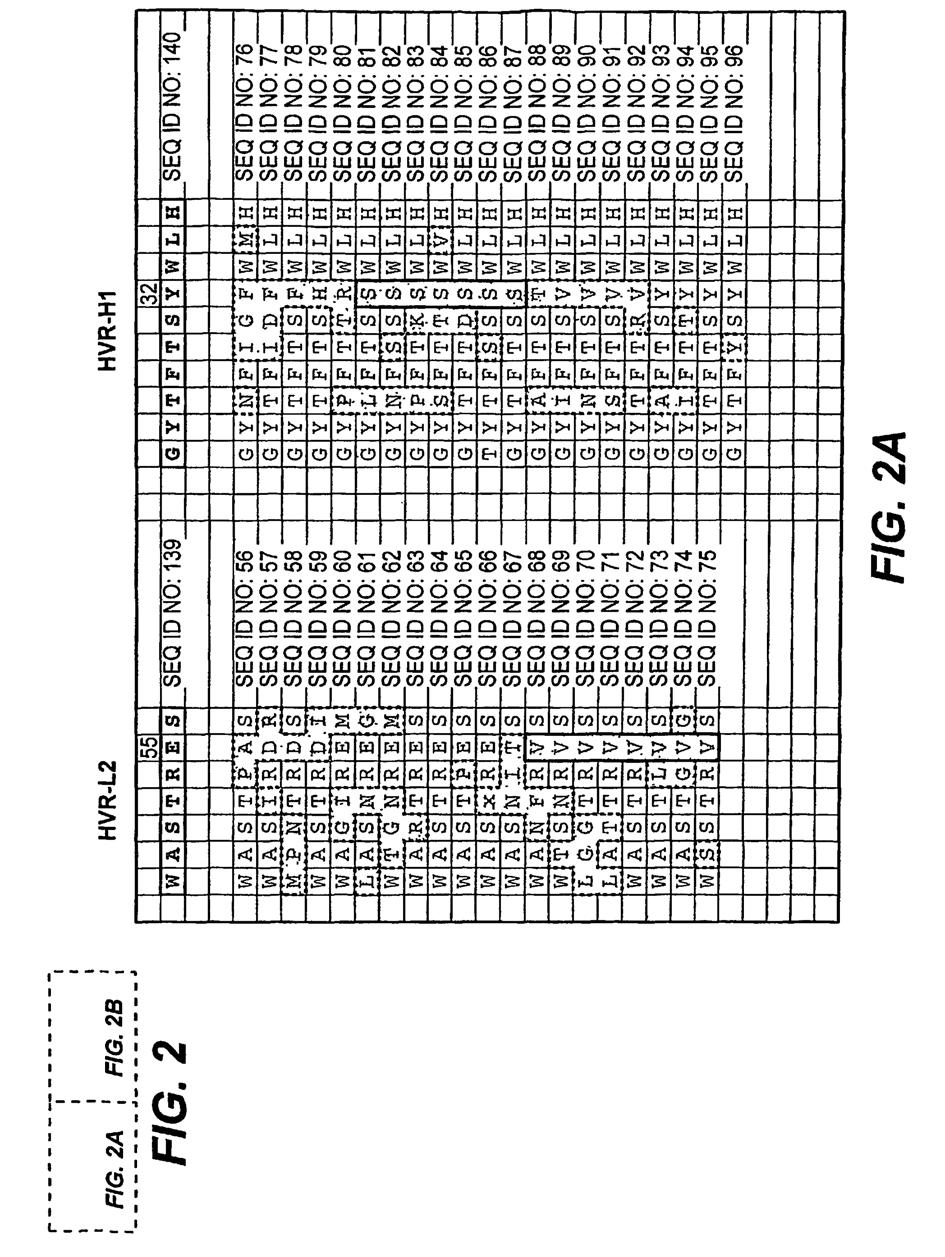
90 89

91 90
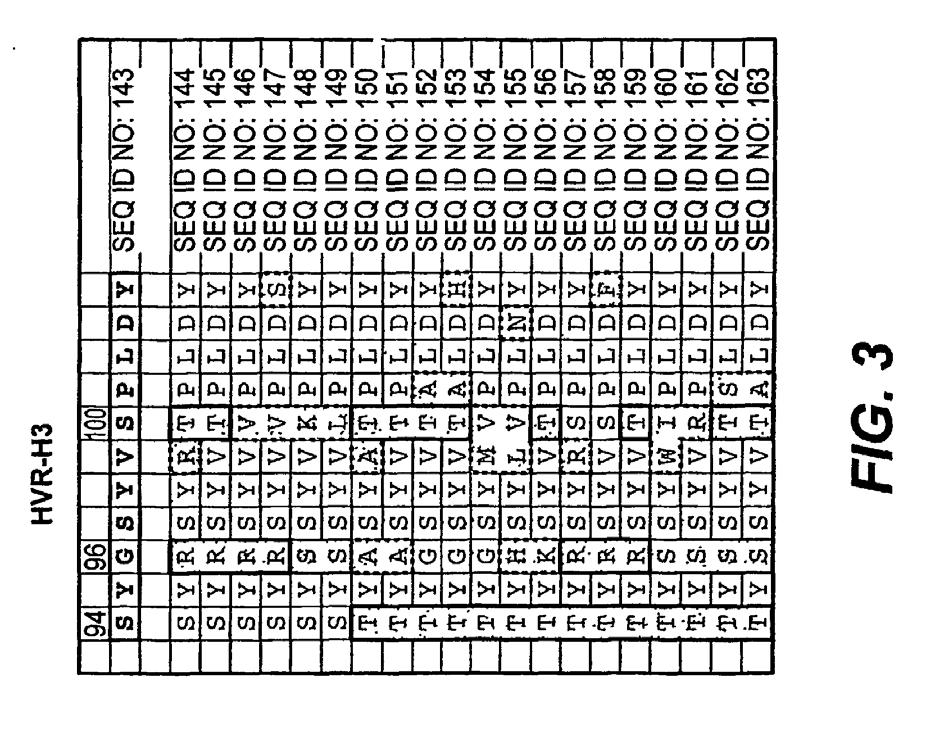
92 91
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE. Ewa Waszkowska ekspert UPRP
 WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152748 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.06.2008 08768087.2
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152748 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.06.2008 08768087.2
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1671552 (96) Data i numer zgłoszenia patentu europejskiego: 02.12.2005 05026319.3 (13) T3 (51) Int. Cl. A23L1/305 A23J3/16
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1671552 (96) Data i numer zgłoszenia patentu europejskiego: 02.12.2005 05026319.3 (13) T3 (51) Int. Cl. A23L1/305 A23J3/16
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
(96) Data i numer zgłoszenia patentu europejskiego:
 Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886585 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.07.2006 06291197.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886585 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.07.2006 06291197.9
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii
 Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1659297 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 12.10.2005 05354036.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1659297 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 12.10.2005 05354036.5
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1712702 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 28.03.2006 06006359.1 (51) Int. Cl. E04F15/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1712702 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 28.03.2006 06006359.1 (51) Int. Cl. E04F15/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1449961 (96) Data i numer zgłoszenia patentu europejskiego: 14.04.2004 04405227.2 (13) T3 (51) Int. Cl. E01B9/14 F16B13/00
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1449961 (96) Data i numer zgłoszenia patentu europejskiego: 14.04.2004 04405227.2 (13) T3 (51) Int. Cl. E01B9/14 F16B13/00
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
INICJACJA ELONGACJA TERMINACJA
 INICJACJA ELONGACJA TERMINACJA 2007 by National Academy of Sciences Kornberg R D PNAS 2007;104:12955-12961 Struktura chromatyny pozwala na różny sposób odczytania informacji zawartej w DNA. Możliwe staje
INICJACJA ELONGACJA TERMINACJA 2007 by National Academy of Sciences Kornberg R D PNAS 2007;104:12955-12961 Struktura chromatyny pozwala na różny sposób odczytania informacji zawartej w DNA. Możliwe staje
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1816307 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:.07.06 060114.3 (1) Int. Cl. E06B9/68 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1816307 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:.07.06 060114.3 (1) Int. Cl. E06B9/68 (06.01) (97) O udzieleniu
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
Rozmnażanie i wzrost komórek sąściśle kontrolowane. Genetyczne podłoże nowotworzenia
 Rozmnażanie i wzrost komórek sąściśle kontrolowane Genetyczne podłoże nowotworzenia Rozmnażanie i wzrost komórek sąściśle kontrolowane Rozmnażanie i wzrost komórek sąściśle kontrolowane Połączenia komórek
Rozmnażanie i wzrost komórek sąściśle kontrolowane Genetyczne podłoże nowotworzenia Rozmnażanie i wzrost komórek sąściśle kontrolowane Rozmnażanie i wzrost komórek sąściśle kontrolowane Połączenia komórek
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1755549 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2005 05780098.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1755549 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2005 05780098.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.") PL/EP 1809944 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1809944 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.4 (51) Int. Cl.
PL/EP 1809944 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1809944 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.4 (51) Int. Cl.
Leczenie biologiczne co to znaczy?
 Leczenie biologiczne co to znaczy? lek med. Anna Bochenek Centrum Badawcze Współczesnej Terapii C B W T 26 Październik 2006 W oparciu o materiały źródłowe edukacyjnego Grantu, prezentowanego na DDW 2006
Leczenie biologiczne co to znaczy? lek med. Anna Bochenek Centrum Badawcze Współczesnej Terapii C B W T 26 Październik 2006 W oparciu o materiały źródłowe edukacyjnego Grantu, prezentowanego na DDW 2006
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 182634 (96) Data i numer zgłoszenia patentu europejskiego: 19.04.07 070963.1 (13) T3 (1) Int. Cl. F16H/17 F16H7/04 (06.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 182634 (96) Data i numer zgłoszenia patentu europejskiego: 19.04.07 070963.1 (13) T3 (1) Int. Cl. F16H/17 F16H7/04 (06.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1859720. (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2007 07003173.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1859720. (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2007 07003173.") PL/EP 1859720 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1859720 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2007 07003173.7 (13) (51) T3 Int.Cl. A47L
PL/EP 1859720 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1859720 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2007 07003173.7 (13) (51) T3 Int.Cl. A47L
CHOROBY NOWOTWOROWE. Twór składający się z patologicznych komórek
 CHOROBY NOWOTWOROWE Twór składający się z patologicznych komórek Powstały w wyniku wielostopniowej przemiany zwanej onkogenezą lub karcinogenezą Morfologicznie ma strukturę zbliżoną do tkanki prawidłowej,
CHOROBY NOWOTWOROWE Twór składający się z patologicznych komórek Powstały w wyniku wielostopniowej przemiany zwanej onkogenezą lub karcinogenezą Morfologicznie ma strukturę zbliżoną do tkanki prawidłowej,
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1754519 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2006 06016676.6 (51) Int. Cl. A62C13/66 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1754519 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2006 06016676.6 (51) Int. Cl. A62C13/66 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1668001 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.09.2004 04784968.2
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1668001 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.09.2004 04784968.2
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1614553 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 01.07.2005 05014326.2 (51) Int. Cl. B60C27/06 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1614553 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 01.07.2005 05014326.2 (51) Int. Cl. B60C27/06 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618. (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618. (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618 (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.0 (13) (1) T3 Int.Cl. A41B 11/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618 (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.0 (13) (1) T3 Int.Cl. A41B 11/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1661542 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 12.08.2004 04762070.3 (51) Int. Cl. A61G7/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1661542 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 12.08.2004 04762070.3 (51) Int. Cl. A61G7/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1658064 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.6 (51) Int. Cl. A61K31/37 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1658064 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.6 (51) Int. Cl. A61K31/37 (2006.01)
(96) Data i numer zgłoszenia patentu europejskiego: 28.07.2004 04017866.7
 Data i numer zgłoszenia patentu europejskiego: 28.07.2004 04017866.7") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1504998 (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2004 04017866.7 (13) T3 (51) Int. Cl. B65C9/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1504998 (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2004 04017866.7 (13) T3 (51) Int. Cl. B65C9/04 (2006.01)
Agencja Oceny Technologii Medycznych
 Agencja Oceny Technologii Medycznych Rada Przejrzystości Stanowisko Rady Przejrzystości nr 182/2013 z dnia 9 września 2013 r. w sprawie oceny leku Iressa (gefitynib) we wskazaniu leczenie niedrobnokomórkowego
Agencja Oceny Technologii Medycznych Rada Przejrzystości Stanowisko Rady Przejrzystości nr 182/2013 z dnia 9 września 2013 r. w sprawie oceny leku Iressa (gefitynib) we wskazaniu leczenie niedrobnokomórkowego
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
Agenda: Wynalazek jako własnośd intelektualna. Co może byd wynalazkiem. Wynalazek biotechnologiczny. Ochrona patentowa
 dr Bartosz Walter Agenda: Wynalazek jako własnośd intelektualna Co może byd wynalazkiem Wynalazek biotechnologiczny Ochrona patentowa Procedura zgłoszenia patentowego Gdzie, kiedy i jak warto patentowad
dr Bartosz Walter Agenda: Wynalazek jako własnośd intelektualna Co może byd wynalazkiem Wynalazek biotechnologiczny Ochrona patentowa Procedura zgłoszenia patentowego Gdzie, kiedy i jak warto patentowad
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1648484 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 01.06.04 047366.4 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1648484 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 01.06.04 047366.4 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2468142. (96) Data i numer zgłoszenia patentu europejskiego: 21.12.2011 11194996.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2468142. (96) Data i numer zgłoszenia patentu europejskiego: 21.12.2011 11194996.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2468142 (96) Data i numer zgłoszenia patentu europejskiego: 21.12.2011 11194996.2 (13) (51) T3 Int.Cl. A47C 23/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2468142 (96) Data i numer zgłoszenia patentu europejskiego: 21.12.2011 11194996.2 (13) (51) T3 Int.Cl. A47C 23/00 (2006.01)
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2814723 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2013 13704452.5 (13) (51) T3 Int.Cl. B63G 8/39 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2814723 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2013 13704452.5 (13) (51) T3 Int.Cl. B63G 8/39 (2006.01)
Ocena ekspresji genów proangiogennych w komórkach nowotworowych OVP-10 oraz transfektantach OVP-10/SHH i OVP-10/VEGF
 Agnieszka Gładysz Ocena ekspresji genów proangiogennych w komórkach nowotworowych OVP-10 oraz transfektantach OVP-10/SHH i OVP-10/VEGF Katedra i Zakład Biochemii i Chemii Klinicznej Akademia Medyczna Prof.
Agnieszka Gładysz Ocena ekspresji genów proangiogennych w komórkach nowotworowych OVP-10 oraz transfektantach OVP-10/SHH i OVP-10/VEGF Katedra i Zakład Biochemii i Chemii Klinicznej Akademia Medyczna Prof.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1957539 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.12.2006 06848508.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1957539 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.12.2006 06848508.5
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1689214 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 19.01.06 06091.4 (1) Int. Cl. H0B37/02 (06.01) (97) O
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1689214 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 19.01.06 06091.4 (1) Int. Cl. H0B37/02 (06.01) (97) O
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 172874 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 04.0.2006 0611312. (1) Int. Cl. B23B31/28 (2006.01) (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 172874 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 04.0.2006 0611312. (1) Int. Cl. B23B31/28 (2006.01) (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1802536 (96) Data i numer zgłoszenia patentu europejskiego: 20.09.2004 04774954.4 (13) T3 (51) Int. Cl. B65D77/20 B65D85/72
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1802536 (96) Data i numer zgłoszenia patentu europejskiego: 20.09.2004 04774954.4 (13) T3 (51) Int. Cl. B65D77/20 B65D85/72
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
Ocena ekspresji genu ABCG2 i białka oporności raka piersi (BCRP) jako potencjalnych czynników prognostycznych w raku jelita grubego
 jako potencjalnych czynników prognostycznych w raku jelita grubego") Aleksandra Sałagacka Ocena ekspresji genu ABCG2 i białka oporności raka piersi (BCRP) jako potencjalnych czynników prognostycznych w raku jelita grubego Pracownia Biologii Molekularnej i Farmakogenomiki
Aleksandra Sałagacka Ocena ekspresji genu ABCG2 i białka oporności raka piersi (BCRP) jako potencjalnych czynników prognostycznych w raku jelita grubego Pracownia Biologii Molekularnej i Farmakogenomiki
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
Materiał i metody. Wyniki
 Abstract in Polish Wprowadzenie Selen jest pierwiastkiem śladowym niezbędnym do prawidłowego funkcjonowania organizmu. Selen jest wbudowywany do białek w postaci selenocysteiny tworząc selenobiałka (selenoproteiny).
Abstract in Polish Wprowadzenie Selen jest pierwiastkiem śladowym niezbędnym do prawidłowego funkcjonowania organizmu. Selen jest wbudowywany do białek w postaci selenocysteiny tworząc selenobiałka (selenoproteiny).
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1624265 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2005 05106119.0 (13) T3 (51) Int. Cl. F25D23/06 F25D25/02
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1624265 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2005 05106119.0 (13) T3 (51) Int. Cl. F25D23/06 F25D25/02
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych
 Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1495737 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 19.06.2004 04014424.8 (51) Int. Cl. A61F2/18 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1495737 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 19.06.2004 04014424.8 (51) Int. Cl. A61F2/18 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2074843. (96) Data i numer zgłoszenia patentu europejskiego: 27.09.2007 07818485.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2074843. (96) Data i numer zgłoszenia patentu europejskiego: 27.09.2007 07818485.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 74843 (96) Data i numer zgłoszenia patentu europejskiego: 27.09.07 0781848.0 (13) (1) T3 Int.Cl. H04W 4/12 (09.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 74843 (96) Data i numer zgłoszenia patentu europejskiego: 27.09.07 0781848.0 (13) (1) T3 Int.Cl. H04W 4/12 (09.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 16234 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 18..0 0022716.4 (1) Int. Cl. B6D71/00 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 16234 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 18..0 0022716.4 (1) Int. Cl. B6D71/00 (06.01) (97) O udzieleniu
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2307863. (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2009 09790873.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2307863. (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2009 09790873.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2307863 (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2009 09790873.5 (13) (51) T3 Int.Cl. G01J 3/44 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2307863 (96) Data i numer zgłoszenia patentu europejskiego: 28.07.2009 09790873.5 (13) (51) T3 Int.Cl. G01J 3/44 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2210706 (96) Data i numer zgłoszenia patentu europejskiego: 21.01.2010 10000580.0 (13) (51) T3 Int.Cl. B24B 21/20 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2210706 (96) Data i numer zgłoszenia patentu europejskiego: 21.01.2010 10000580.0 (13) (51) T3 Int.Cl. B24B 21/20 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2913207 (96) Data i numer zgłoszenia patentu europejskiego: 08.05.2014 14167514.0 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2913207 (96) Data i numer zgłoszenia patentu europejskiego: 08.05.2014 14167514.0 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2353894 (96) Data i numer zgłoszenia patentu europejskiego: 19.02.2010 10001703.7 (13) (51) T3 Int.Cl. B60D 5/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2353894 (96) Data i numer zgłoszenia patentu europejskiego: 19.02.2010 10001703.7 (13) (51) T3 Int.Cl. B60D 5/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1700812 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.03.2006 06004461.7 (51) Int. Cl. B66B9/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1700812 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.03.2006 06004461.7 (51) Int. Cl. B66B9/08 (2006.01)
Biologia komórki i biotechnologia w terapii schorzeń narządu ruchu
 Biologia komórki i biotechnologia w terapii schorzeń Ilość godzin: 40h seminaria Ilość grup: 2 Forma zaliczenia: zaliczenie z oceną Kierunek: Fizjoterapia ścieżka neurologiczna Rok: II - Lic Tryb: stacjonarne
Biologia komórki i biotechnologia w terapii schorzeń Ilość godzin: 40h seminaria Ilość grup: 2 Forma zaliczenia: zaliczenie z oceną Kierunek: Fizjoterapia ścieżka neurologiczna Rok: II - Lic Tryb: stacjonarne
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1614512 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 28.06.2005 05013915.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1614512 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 28.06.2005 05013915.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1786660 (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9 (13) T3 (51) Int. Cl. B62D25/08 B60G15/06
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1786660 (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9 (13) T3 (51) Int. Cl. B62D25/08 B60G15/06
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2259949 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2009 09727379.1 (13) (51) T3 Int.Cl. B60L 11/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2259949 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2009 09727379.1 (13) (51) T3 Int.Cl. B60L 11/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2828428 (96) Data i numer zgłoszenia patentu europejskiego: 12.03.13 13731877.0 (13) (1) T3 Int.Cl. D0B 19/12 (06.01) D0B
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2828428 (96) Data i numer zgłoszenia patentu europejskiego: 12.03.13 13731877.0 (13) (1) T3 Int.Cl. D0B 19/12 (06.01) D0B
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2946811 (96) Data i numer zgłoszenia patentu europejskiego: 21.04.2015 15164439.0 (13) (51) T3 Int.Cl. A62C 2/12 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2946811 (96) Data i numer zgłoszenia patentu europejskiego: 21.04.2015 15164439.0 (13) (51) T3 Int.Cl. A62C 2/12 (2006.01)
Promotor: prof. dr hab. Katarzyna Bogunia-Kubik Promotor pomocniczy: dr inż. Agnieszka Chrobak
 INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ IM. LUDWIKA HIRSZFELDA WE WROCŁAWIU POLSKA AKADEMIA NAUK mgr Milena Iwaszko Rola polimorfizmu receptorów z rodziny CD94/NKG2 oraz cząsteczki HLA-E w patogenezie
INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ IM. LUDWIKA HIRSZFELDA WE WROCŁAWIU POLSKA AKADEMIA NAUK mgr Milena Iwaszko Rola polimorfizmu receptorów z rodziny CD94/NKG2 oraz cząsteczki HLA-E w patogenezie
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2326237 (96) Data i numer zgłoszenia patentu europejskiego: 07.07.2009 09780285.4 (13) (51) T3 Int.Cl. A47L 15/50 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2326237 (96) Data i numer zgłoszenia patentu europejskiego: 07.07.2009 09780285.4 (13) (51) T3 Int.Cl. A47L 15/50 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
Mechanochemiczny przełącznik między wzrostem i różnicowaniem komórek
 Mechanochemiczny przełącznik między wzrostem i różnicowaniem komórek Model tworzenia mikrokapilar na podłożu fibrynogenowym eksponencjalny wzrost tempa proliferacji i syntezy DNA wraz ze wzrostem stężenia
Mechanochemiczny przełącznik między wzrostem i różnicowaniem komórek Model tworzenia mikrokapilar na podłożu fibrynogenowym eksponencjalny wzrost tempa proliferacji i syntezy DNA wraz ze wzrostem stężenia
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
Profil metaboliczny róŝnych organów ciała
 Profil metaboliczny róŝnych organów ciała Uwaga: tkanka tłuszczowa (adipose tissue) NIE wykorzystuje glicerolu do biosyntezy triacylogliceroli Endo-, para-, i autokrynna droga przekazu informacji biologicznej.
Profil metaboliczny róŝnych organów ciała Uwaga: tkanka tłuszczowa (adipose tissue) NIE wykorzystuje glicerolu do biosyntezy triacylogliceroli Endo-, para-, i autokrynna droga przekazu informacji biologicznej.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1837003 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2007 07005252.7 (51) Int. Cl. A61G5/10 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1837003 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2007 07005252.7 (51) Int. Cl. A61G5/10 (2006.01)
Stanowisko Rady Przejrzystości nr 262/2013 z dnia 17 grudnia 2013 r. w sprawie oceny leku Perjeta (pertuzumab) we wskazaniu zaawansowanego raka piersi
 we wskazaniu zaawansowanego raka piersi") Agencja Oceny Technologii Medycznych Rada Przejrzystości Stanowisko Rady Przejrzystości nr 262/2013 z dnia 17 grudnia 2013 r. w sprawie oceny leku Perjeta (pertuzumab) we wskazaniu zaawansowanego raka
Agencja Oceny Technologii Medycznych Rada Przejrzystości Stanowisko Rady Przejrzystości nr 262/2013 z dnia 17 grudnia 2013 r. w sprawie oceny leku Perjeta (pertuzumab) we wskazaniu zaawansowanego raka
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2949485 (96) Data i numer zgłoszenia patentu europejskiego: 06.10.2014 14187774.6 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2949485 (96) Data i numer zgłoszenia patentu europejskiego: 06.10.2014 14187774.6 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
Dane mikromacierzowe. Mateusz Markowicz Marta Stańska
 Dane mikromacierzowe Mateusz Markowicz Marta Stańska Mikromacierz Mikromacierz DNA (ang. DNA microarray) to szklana lub plastikowa płytka (o maksymalnych wymiarach 2,5 cm x 7,5 cm) z naniesionymi w regularnych
Dane mikromacierzowe Mateusz Markowicz Marta Stańska Mikromacierz Mikromacierz DNA (ang. DNA microarray) to szklana lub plastikowa płytka (o maksymalnych wymiarach 2,5 cm x 7,5 cm) z naniesionymi w regularnych
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1740398 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 1.03.200 071703.9 (1) Int. Cl. B60C1/06 (2006.01) (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1740398 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 1.03.200 071703.9 (1) Int. Cl. B60C1/06 (2006.01) (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2084461 (96) Data i numer zgłoszenia patentu europejskiego: 27.11.2007 07847411.1 (13) (51) T3 Int.Cl. F24C 3/10 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2084461 (96) Data i numer zgłoszenia patentu europejskiego: 27.11.2007 07847411.1 (13) (51) T3 Int.Cl. F24C 3/10 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1701111 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 11.03.2005 05090064.6 (51) Int. Cl. F24H9/20 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1701111 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 11.03.2005 05090064.6 (51) Int. Cl. F24H9/20 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2068922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2007 07868510.4
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2068922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2007 07868510.4
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 27.05.2005 05746418.2
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 27.05.2005 05746418.2") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1817186 (96) Data i numer zgłoszenia patentu europejskiego: 27.05.2005 05746418.2 (13) T3 (51) Int. Cl. B60G21/055 F16D1/06
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1817186 (96) Data i numer zgłoszenia patentu europejskiego: 27.05.2005 05746418.2 (13) T3 (51) Int. Cl. B60G21/055 F16D1/06
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1588845 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2004 04405247.0
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1588845 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2004 04405247.0
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2207808. (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2008 08843849.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2207808. (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2008 08843849.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 27808 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27..08 08843849.4 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 27808 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27..08 08843849.4 (97)