Bioinformatyka: Wykład 5. Bioconductor
|
|
|
- Paweł Milewski
- 6 lat temu
- Przeglądów:
Transkrypt
1 Bioinformatyka: Wykład 5 Bioconductor
2 Pytanie z poprzedniego wkładu opisz dwie klasy R odnoszące się do czasu, podaj najważniejszą różnicę
3 BIOCONDUCTOR Zestaw ściśle ze sobą powiązanych pakietów (1104) do analizy danych pochodzących z eksperymentów biologii molekularnej Dwa wydania rocznie Społeczność użytkowników i developerów
4 INSTALACJA własny system instalacji, częściowo niezależny od standardowego mechanizmu R - czyli funkcji install.packages() umożliwia koordynację między pakietami i lepszą kontrolę błędów - (usuwa potencjalne niezgodności między wersjami R a i pakietów oraz pakietów między sobą). dostarczona przez Bioconductora funkcja bioclite() jest nakładką na install.packages() zapewniającą instalację poprawnej wersji pakietów zgodnej z wersją Bioconductora
5 INSTALACJA bieżąca wersja ( ) to Bioconductor 3.2 zgodny z R 3.2. Przed instalacją Bioconductora należy zainstalować właściwą wersję R. Instalacja Bioconductora:! source(" bioclite()! Instalacja pakietów Bioconductora (np pakietów GenomicFeatures and AnnotationDbi! bioclite(c("genomicfeatures", "AnnotationDbi"))
6 ZASTOSOWANIA Pakiety Bioconductora są najczęściej zorganizowane w potoki przetwarzania (workflows). Pakiety wykonują dobrze zdefiniowane zadania cząstkowe i łączą się z innymi używając wspólnych struktur danych. Przykładowe zastosowania Sequence Analysis Oligonucleotide Arrays Annotation Resources Annotating Genomic Ranges Annotating Genomic Variants
7 ZASTOSOWANIA Większość zastosowań Bioconductora dotyczy danych sekwencyjnych pochodzących z eksperymentów sekwencjonowania nowej generacji dla DNA i RNA Dlatego będziemy się dalej zajmować takimi właśnie przykładowymi zastosowaniami. Skoncentrujemy się na sekwencjonowaniu RNA w kontekście analizy poziomu ekspresji genów
8 RNA SEQ Dwa podstawowe zastosowania Analiza jakościowa: identyfikacja genów podlegających ekspresji, określenie granic intron/exon, miejsc startu transkrypcji. Analiza ilościowa: określenie różnic w poziomie transkrypcji, wariantów w splajsingu, startu transkrypcji dla różnych grup (np różnych fenotypów lub różnych terapii).
9 PROCESY Z UDZIAŁEM RNA Wikipedia: Dhorspool

10 ALTERNATYWNY SPLAJSING Narayanese at English Wikipedia National Human Genome Research Institute Wikipedia: Agathman

11 SEKWENCJONOWANIE RNA Przebieg eksperymentu sekwencjonowania RNA-seqlopedia Izolacja RNA z materiału biologicznego Oczyszczenie RNA Fragmentacja na kawałki odpowiedniej wielkości Synteza cdna na matrycy RNA Amplifikacja cdna metodą PCR Sekwencjonowanie Analiza sekwencji

12 SEKWENCJONOWANIE RNA Eksperyment sekwencjonowania RNA - przygotowanie biblioteki fragmentów cdna RNA-seqlopedia
13 SEKWNCJONOWANIE RNA W zależności od celu eksperymentu przeprowadzane są różne analizy i ważne są różne aspekty jakości eksperymentu. W nawiasach zaznaczamy ważność dla eksperymentów jakościowych/ilościowych wiele prób na różnych obiektach biologicznych (przydatne/kluczowe) stopień pokrycia transkryptu (ważne zwłaszcza dla identyfikacji izoform / niekonieczne - ważne jest istnienie unikatowych readów) krotność pokrycia (depth of sequencing) (wystarczająco wysoka by odkryć rzadkie transkrypty/kluczowe dla uzyskania dobrej oceny statystycznej) RNA-seqlopedia
14 ANALIZY ILOŚCIOWE RNA Replikacja - sposób na minimalizację błędów techniczna (wiele próbek materiału z tego samego obiektu) biologiczna (próby pochodzące od różnych obiektów biologicznych) Techniczna może pomóc zminimalizować błędy wynikające z niedoskonałości eksperymentu Biologiczna - pozwala na przebadanie efektu dla większej populacji Niemal zawsze lepszym wyborem jest zwiększenie liczby prób biologicznych!
15 ANALIZY ILOŚCIOWE RNA Testowanie hipotez H0 - hipoteza zerowa - różnice ekspresji genu X między obiema grupami są efektem losowym H1 - hipoteza alternatywna - różnica ekspresji genu X między grupami jest związana z badanym zjawiskiem. Algorytm (uproszczony) dla każdego genu policz średnią i wariancję ekspresji genu X w próbie kontrolnej E0(X) policz średnię i wariancję ekspresję genu X w próbie testowej E1(X) policz zmienną Z ze wzoru:!! Z = x 0 x 1 v 0 n0 + v 1 n1 sprawdź prawdopodobieństwo uzyskania takiego wyniku losowo. (n 0 i n 1 są liczbą obiektów w obu grupach pomniejszoną o 1)
16 Hipotetyczny średni rozkład liczby cząsteczek mrna w badanej populacji ŹRÓDŁA BŁĘDÓW
17 Pojedynczy osobnik z populacji - prawdziwy rozkład liczby cząstek mrna w dniu eksperymentu ŹRÓDŁA BŁĘDÓW
18 Pojedynczy osobnik z populacji - liczba cząstek mrna w próbie po procedurze eksperymentalnej ŹRÓDŁA BŁĘDÓW
19 Pojedynczy osobnik z populacji - liczba cząstek mrna zmierzona w sekwencjonowaniu ŹRÓDŁA BŁĘDÓW
20 ŹRÓDŁA BŁĘDÓW Dwa pomiary Hipotetyczny średni rozkład liczby cząsteczek mrna w badanej populacji Obiekt 1 Obiekt 2
21 Dwa pomiary ŹRÓDŁA BŁĘDÓW Realizacje dla dwóch obiektów cor(x1,y1) : Obiekt 1 Obiekt 2
22 Dwa pomiary Pomiary dla dwóch obiektów cor(x2,y2) : ŹRÓDŁA BŁĘDÓW Obiekt 1 Obiekt 2
23 Dwa pomiary ŹRÓDŁA BŁĘDÓW Procedura eksperymentalna dla dwóch obiektów cor(x3,y3) : Obiekt 1 Obiekt 2
24 Dwa pomiary ŹRÓDŁA BŁĘDÓW Procedura eksperymentalna dla dwóch obiektów cor(x3,y3) :
25 PRZYGOTOWANIE BIBLIOTEKI Synteza nici DNA na matrycy RNA potrzebny jest primer (fragment, do którego będą dodawane nukleotydy) oligo-dt (dołącza do ogona poli-a dojrzałego mrna) losowy primer (doczepia się w losowym miejscu) doklejanie do końca RNA sekwencji komplementarnej do przygotowanego primera Synteza drugiej nici DNA na pierwszej - również potrzebne są primery Doklejenie sekwencji adapterowych do dwuniciowego DNA. Adaptery są konieczne dla dalszych operacji - amplifikacji DNA dla zwiększenia liczby kopii.

26 PRZYGOTOWANIE BIBLIOTEKI Synteza nici DNA na matrycy RNA RNA-seqlopedia

27 PRZYGOTOWANIE BIBLIOTEKI Doklejenie sekwencji adapterowych do dwuniciowego DNA. Adaptery są konieczne dla dalszych operacji - amplifikacji DNA dla zwiększenia liczby kopii. Doklejenie znaczników do sekwencjonowania RNA-seqlopedia
28 SEKWENCJONOWANIE Wiele firm rozwija metody sekwencjonowania RNA/DNA, oparte na różnych procesach chemicznych Illumina Affymetrix Pacific Bio
29 ILUMINA (SOLEXA) SEQUENCING Wikipedia: DMLapato
30 ILUMINA (SOLEXA) SEQUENCING Wikipedia: DMLapato
31 ILUMINA (SOLEXA) SEQUENCING Wikipedia: DMLapato
32 ILUMINA (SOLEXA) SEQUENCING Wikipedia: DMLapato
33 ILUMINA (SOLEXA) SEQUENCING Wikipedia: DMLapato
34 Etapy: ANALIZA SEKWENCJI Rozdzielenie eksperymentów Filtrowanie ze względu na jakość Obcięcie fragmentów znacznikowych Mapowanie (dopasowanie) fragmentów do referencyjnego genomu lub transkryptomu Annotacja transcryptów ze zmapowanymi fragmentami Zliczenie zmapowanych fragmentów w celu oceny ilości transkryptu Analiza statistyczna pozwalająca na zbadanie różnic w ekspresji dla pojedynczych transkryptów (genów lub wariantów) Wizualizacja i analiza wieloczynnikowa
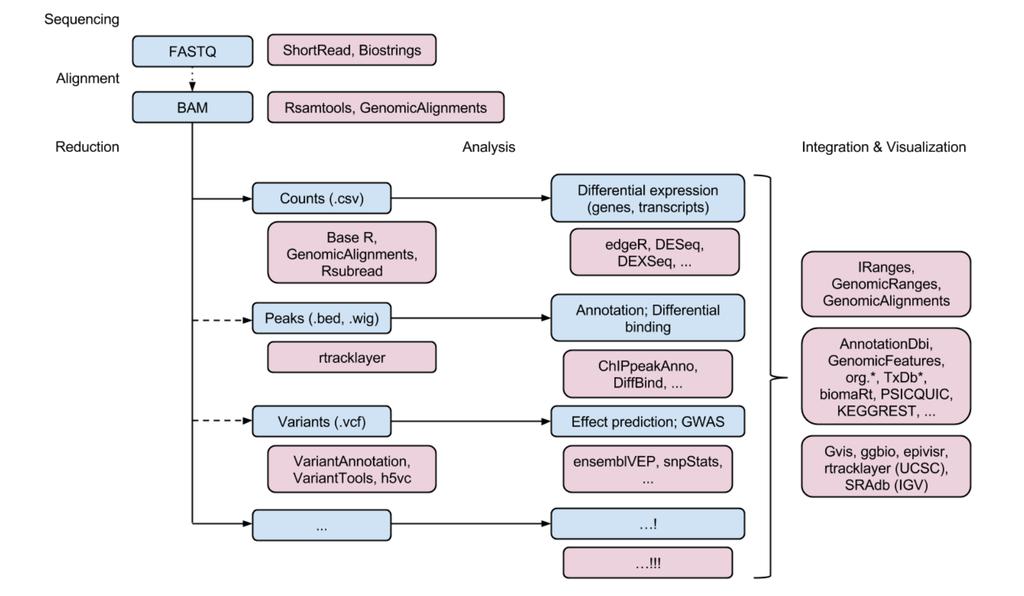
35 BIOCONDUCTOR
36 FASTQ Format do przechowywania danych z sekwencjonowania 4 linie na fragment 1. Linia zaczyna się od następnie identyfikator sekwencji i opcjonalnie opis 2. Linia zawiera wyłącznie sekwencję 3. Linia zaczyna się od znaku + i może opcjonalnie zawierać identyfikator i opis 4. Linia zawiera symbolicznie zapisaną ocenę jakości dla każdego nukleotydu. Musi zawierać dokładnie tyle samo znaków co sekwencja. Skala jakości (! najniższa ~ najwyższa):! #$%& ()*+,-./ :;<=>?@ABCDEFGHIJKLMNO! PQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{ }~ Q = 10log p Q = 10log p 1 p
37 Przykłady generyczny GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT!! +!!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65!! _SLXA-EAS1_s_7:5:1:817:345 length=36! GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC! +SRR _SLXA-EAS1_s_7:5:1:817:345 length=36! IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
38 SAM/BAM Sequence Alignment Map (BAM jest wersją binarną)
39 BIOSTRINGS Pakiet Biostrings służy do manipulacji sekwencjami biologicznymi (DNA/ RNA i białka instalacja! source(" bioclite("biostrings")! załadowanie do pamięci require(biostrings)! lub library(biostrings)
40 BIOSTRINGS > library(biostrings) Ładowanie wymaganego pakietu: BiocGenerics Ładowanie wymaganego pakietu: parallel Dołączanie pakietu: BiocGenerics Następujące obiekty zostały zakryte z package:parallel : clusterapply, clusterapplylb, clustercall, clusterevalq, clusterexport, clustermap, parapply, parcapply, parlapply, parlapplylb, parrapply, parsapply, parsapplylb Następujący obiekt został zakryty z package:stats : xtabs Następujące obiekty zostały zakryte z package:base : anyduplicated, append, as.data.frame, as.vector, cbind, colnames, do.call, duplicated, eval, evalq, Filter, Find, get, intersect, is.unsorted, lapply, Map, mapply, match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce, rep.int, rownames, sapply, setdiff, sort, table, tapply, union, unique, unlist, unsplit Ładowanie wymaganego pakietu: S4Vectors Ładowanie wymaganego pakietu: stats4 Creating a generic function for nchar from package base in package S4Vectors Ładowanie wymaganego pakietu: IRanges Ładowanie wymaganego pakietu: XVector
41 BIOSTRINGS Pakiet Biostrings służy do manipulacji sekwencjami biologicznymi (DNA/ RNA i białka Zawiera użyteczne klasy i obiekty Przykład - generowanie losowej sekwencji > letters [1] "a" "b" "c" "d" "e" "f" "g" "h" "i" "j" "k" "l" "m" "n" "o" "p" "q" "r" "s" "t" "u" "v" "w" "x" "y" "z" > LETTERS [1] "A" "B" "C" "D" "E" "F" "G" "H" "I" "J" "K" "L" "M" "N" "O" "P" "Q" "R" "S" "T" "U" "V" "W" "X" "Y" "Z" > sample(letters[c(1,3,7,20)],size=20,replace=true) [1] "G" "C" "T" "A" "C" "T" "A" "A" "T" "T" "C" "G" "G" "T" "C" "C" "G" "G" "C" "G" > DNA_ALPHABET [1] "A" "C" "G" "T" "M" "R" "W" "S" "Y" "K" "V" "H" "D" "B" "N" "-" "+" "." > seq<- sample(dna_alphabet[1:4],size=20,replace=true) > seq [1] "G" "T" "C" "C" "C" "A" "G" "C" "T" "G" "A" "G" "A" "G" "C" "A" "C" "A" "A" "G" > seq [1] "GTCCCAGCTGAGAGCACAAG"
42 BIOSTRINGS Klasa wirtualna XStrings nie jest dostępna bezpośrednio ale dostępne są jej podklasy: BString - ogólna klasa napisowa DNAString - sekwencja DNA RNAString - sekwencja RNA AAString - sekwencja aminikwasowa > bstring = BString("I am a BString object") > bstring 21-letter "BString" instance seq: I am a BString object > dnastring <- DNAString("I am a BString object") Błąd w poleceniu '.Call2("new_XString_from_CHARACTER", classname, x, start(solved_sew), ': key 73 (char 'I') not in lookup table > aastring <- AAString("I am a BString object") > aastring 21-letter "AAString" instance seq: I am a BString object
43 BIOSTRINGS obiekty klas pochodnych od XStrings nie są zwykłymi napisami: > dnastring = DNAString("TTGAAA-CTC-N") > dnastring 12-letter "DNAString" instance seq: TTGAAA-CTC-N > length(dnastring) [1] 12 > dnastring[1:4] 4-letter "DNAString" instance seq: TTGA > bstring[1:4] 4-letter "BString" instance seq: I am > napis<-"i am a BString object" > napis[1] [1] "I am a BString object" > napis[1:4] [1] "I am a BString object" NA NA NA!
44 BIOSTRINGS klasy pochodnych od XStrings są klasami typu S4 - rzadziej występujące w R niż klasa typu S3. Inny jest sposób dostępu do elementów klasy > str(dnastring) Formal class 'DNAString' [package "Biostrings"] with 5 slots..@ shared :Formal class 'SharedRaw' [package "XVector"] with 2 slots......@ xp :<externalptr>......@.link_to_cached_object:<environment: 0x7f8b8aee9400>..@ offset : int 0..@ length : int 12..@ elementmetadata: NULL..@ metadata : list() > slotnames(dnastring) [1] "shared" "offset" "length" "elementmetadata" "metadata" > dnastring@length [1] 12 > dnastring$length Błąd w poleceniu 'dnastring$length':$ operator not defined for this S4 class >
Przetarg nieograniczony na zakup specjalistycznej aparatury laboratoryjnej Znak sprawy: DZ-2501/6/17
 Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Analizy wielkoskalowe w badaniach chromatyny
 Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing. Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
NGS ciąg dalszy. Bioinformatyczna analiza danych Wykład 9 Dr Wioleta Drobik-Czwarno
 NGS ciąg dalszy Bioinformatyczna analiza danych Wykład 9 Dr Wioleta Drobik-Czwarno Zalety NGS Sekwencjonowanie wysoko-przepustowe wiele próbek na raz Wiele genów w wielu próbkach co daje mniejszy koszt
NGS ciąg dalszy Bioinformatyczna analiza danych Wykład 9 Dr Wioleta Drobik-Czwarno Zalety NGS Sekwencjonowanie wysoko-przepustowe wiele próbek na raz Wiele genów w wielu próbkach co daje mniejszy koszt
Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
Testowanie hipotez statystycznych
 9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
Bioinformatyczna analiza danych. Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt
 Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
Bazy danych i R/Bioconductor
 Bazy danych i R/Bioconductor Praca z pakietem biomart Zagadnienia RStudio Oprogramowanie BioMart Pakiet biomart wprowadzenie funkcje biomart przykładowe zastosowania RStudio RStudio http://www.rstudio.com/
Bazy danych i R/Bioconductor Praca z pakietem biomart Zagadnienia RStudio Oprogramowanie BioMart Pakiet biomart wprowadzenie funkcje biomart przykładowe zastosowania RStudio RStudio http://www.rstudio.com/
BIOLOGICZNE BAZY DANYCH (1) GENOMY I ICH ADNOTACJE
 GENOMY I ICH ADNOTACJE") BIOLOGICZNE BAZY DANYCH (1) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 2 PODSTAWY BIOINFORMATYKI 2018/2019 MAGDA MIELCZAREK 1 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI
BIOLOGICZNE BAZY DANYCH (1) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 2 PODSTAWY BIOINFORMATYKI 2018/2019 MAGDA MIELCZAREK 1 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI
"Zapisane w genach, czyli Python a tajemnice naszego genomu."
 "Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
"Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
Metody analizy genomu
 Metody analizy genomu 1. Mapowanie restrykcyjne. 2. Sondy do rozpoznawania DNA 3. FISH 4. Odczytanie sekwencji DNA 5. Interpretacja sekwencji DNA genomu 6. Transkryptom 7. Proteom 1. Mapy restrykcyjne
Metody analizy genomu 1. Mapowanie restrykcyjne. 2. Sondy do rozpoznawania DNA 3. FISH 4. Odczytanie sekwencji DNA 5. Interpretacja sekwencji DNA genomu 6. Transkryptom 7. Proteom 1. Mapy restrykcyjne
BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE. Podstawy Bioinformatyki wykład 4
 GENOMY I ICH ADNOTACJE. Podstawy Bioinformatyki wykład 4") BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 4 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 2 GENOMY I ICH ADNOTACJE
BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 4 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 2 GENOMY I ICH ADNOTACJE
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE
 PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Database resources of the National Center for Biotechnology Information. Magdalena Malczyk
 Database resources of the National Center for Biotechnology Information Magdalena Malczyk NCBI NCBI = National Center for Biotechnology Information Założone w 1998 r. Cel: rozwijanie systemów informatycznych
Database resources of the National Center for Biotechnology Information Magdalena Malczyk NCBI NCBI = National Center for Biotechnology Information Założone w 1998 r. Cel: rozwijanie systemów informatycznych
Wykład 4: Wnioskowanie statystyczne. Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA
 Wykład 4: Wnioskowanie statystyczne Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA Idea wnioskowania statystycznego Celem analizy statystycznej nie jest zwykle tylko
Wykład 4: Wnioskowanie statystyczne Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA Idea wnioskowania statystycznego Celem analizy statystycznej nie jest zwykle tylko
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:
 w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:") Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Substancje stosowane do osadzania enzymu na stałym podłożu Biotyna (witamina H, witamina B 7 ) Tworzenie aktywnej powierzchni biosensorów
 Tworzenie aktywnej powierzchni biosensorów") SEKWECJWAIE ASTĘPEJ GEERACJI- GS Losowa fragmentacja nici DA Dołączanie odpowiednich linkerów (konstrukcja biblioteki) Amplifikacja biblioteki na podłożu szklanym lub plastikowym biosensory Wielokrotnie
SEKWECJWAIE ASTĘPEJ GEERACJI- GS Losowa fragmentacja nici DA Dołączanie odpowiednich linkerów (konstrukcja biblioteki) Amplifikacja biblioteki na podłożu szklanym lub plastikowym biosensory Wielokrotnie
Przybliżone algorytmy analizy ekspresji genów.
 Przybliżone algorytmy analizy ekspresji genów. Opracowanie i implementacja algorytmu analizy danych uzyskanych z eksperymentu biologicznego. 20.06.04 Seminarium - SKISR 1 Wstęp. Dane wejściowe dla programu
Przybliżone algorytmy analizy ekspresji genów. Opracowanie i implementacja algorytmu analizy danych uzyskanych z eksperymentu biologicznego. 20.06.04 Seminarium - SKISR 1 Wstęp. Dane wejściowe dla programu
Przeglądanie bibliotek
 Przeglądanie bibliotek Czyli jak złapać (i sklonować) ciekawy gen? Klonowanie genów w oparciu o identyczność lub podobieństwo ich sekwencji do znanego już DNA Sonda homologiczna (komplementarna w 100%)
Przeglądanie bibliotek Czyli jak złapać (i sklonować) ciekawy gen? Klonowanie genów w oparciu o identyczność lub podobieństwo ich sekwencji do znanego już DNA Sonda homologiczna (komplementarna w 100%)
weryfikacja hipotez dotyczących parametrów populacji (średnia, wariancja)
") PODSTAWY STATYSTYKI. Teoria prawdopodobieństwa i elementy kombinatoryki. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5. Testy parametryczne (na
PODSTAWY STATYSTYKI. Teoria prawdopodobieństwa i elementy kombinatoryki. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5. Testy parametryczne (na
Analiza danych pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego. - część I -
 pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego - część I - Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Plan wykładów --------------------------------------------------------
pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego - część I - Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Plan wykładów --------------------------------------------------------
1. Analiza asocjacyjna. Cechy ciągłe. Cechy binarne. Analiza sprzężeń. Runs of homozygosity. Signatures of selection
 BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Ekologia molekularna. wykład 11
 Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
STATYSTYKA MATEMATYCZNA
 STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
UML a kod w C++ i Javie. Przypadki użycia. Diagramy klas. Klasy użytkowników i wykorzystywane funkcje. Związki pomiędzy przypadkami.
 UML a kod w C++ i Javie Projektowanie oprogramowania Dokumentowanie oprogramowania Diagramy przypadków użycia Przewoznik Zarzadzanie pojazdami Optymalizacja Uzytkownik Wydawanie opinii Zarzadzanie uzytkownikami
UML a kod w C++ i Javie Projektowanie oprogramowania Dokumentowanie oprogramowania Diagramy przypadków użycia Przewoznik Zarzadzanie pojazdami Optymalizacja Uzytkownik Wydawanie opinii Zarzadzanie uzytkownikami
Biologia medyczna, materiały dla studentów
 Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
UNIWERSYTET RZESZOWSKI KATEDRA INFORMATYKI
 UNIWERSYTET RZESZOWSKI KATEDRA INFORMATYKI LABORATORIUM TECHNOLOGIA SYSTEMÓW INFORMATYCZNYCH W BIOTECHNOLOGII Pakiet R: Cz. II Strona 1 z 7 OBIEKTY Faktory (factors) Faktor jest specjalną strukturą, przechowującą
UNIWERSYTET RZESZOWSKI KATEDRA INFORMATYKI LABORATORIUM TECHNOLOGIA SYSTEMÓW INFORMATYCZNYCH W BIOTECHNOLOGII Pakiet R: Cz. II Strona 1 z 7 OBIEKTY Faktory (factors) Faktor jest specjalną strukturą, przechowującą
Dane dotyczące wartości zmiennej (cechy) wprowadzamy w jednej kolumnie. W przypadku większej liczby zmiennych wprowadzamy każdą w oddzielnej kolumnie.
 wprowadzamy w jednej kolumnie. W przypadku większej liczby zmiennych wprowadzamy każdą w oddzielnej kolumnie.") STATISTICA INSTRUKCJA - 1 I. Wprowadzanie danych Podstawowe / Nowy / Arkusz Dane dotyczące wartości zmiennej (cechy) wprowadzamy w jednej kolumnie. W przypadku większej liczby zmiennych wprowadzamy każdą
STATISTICA INSTRUKCJA - 1 I. Wprowadzanie danych Podstawowe / Nowy / Arkusz Dane dotyczące wartości zmiennej (cechy) wprowadzamy w jednej kolumnie. W przypadku większej liczby zmiennych wprowadzamy każdą
SCENARIUSZ LEKCJI BIOLOGII Z WYKORZYSTANIEM FILMU Transkrypcja RNA
 SCENARIUSZ LEKCJI BIOLOGII Z WYKORZYSTANIEM FILMU Transkrypcja RNA SPIS TREŚCI: I. Wprowadzenie. II. Części lekcji. 1. Część wstępna. 2. Część realizacji. 3. Część podsumowująca. III. Karty pracy. 1. Karta
SCENARIUSZ LEKCJI BIOLOGII Z WYKORZYSTANIEM FILMU Transkrypcja RNA SPIS TREŚCI: I. Wprowadzenie. II. Części lekcji. 1. Część wstępna. 2. Część realizacji. 3. Część podsumowująca. III. Karty pracy. 1. Karta
Sekwencjonowanie, przewidywanie genów
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
Sylabus Biologia molekularna
 Sylabus Biologia molekularna 1. Metryczka Nazwa Wydziału Program kształcenia Wydział Farmaceutyczny z Oddziałem Medycyny Laboratoryjnej Analityka Medyczna, studia jednolite magisterskie, studia stacjonarne
Sylabus Biologia molekularna 1. Metryczka Nazwa Wydziału Program kształcenia Wydział Farmaceutyczny z Oddziałem Medycyny Laboratoryjnej Analityka Medyczna, studia jednolite magisterskie, studia stacjonarne
Searching for SNPs with cloud computing
 Ben Langmead, Michael C Schatz, Jimmy Lin, Mihai Pop and Steven L Salzberg Genome Biology November 20, 2009 April 7, 2010 Problem Cel Problem Bardzo dużo krótkich odczytów mapujemy na genom referencyjny
Ben Langmead, Michael C Schatz, Jimmy Lin, Mihai Pop and Steven L Salzberg Genome Biology November 20, 2009 April 7, 2010 Problem Cel Problem Bardzo dużo krótkich odczytów mapujemy na genom referencyjny
TECHNIKI ANALIZY RNA TECHNIKI ANALIZY RNA TECHNIKI ANALIZY RNA
 DNA 28SRNA 18/16S RNA 5SRNA mrna Ilościowa analiza mrna aktywność genów w zależności od wybranych czynników: o rodzaju tkanki o rodzaju czynnika zewnętrznego o rodzaju upośledzenia szlaku metabolicznego
DNA 28SRNA 18/16S RNA 5SRNA mrna Ilościowa analiza mrna aktywność genów w zależności od wybranych czynników: o rodzaju tkanki o rodzaju czynnika zewnętrznego o rodzaju upośledzenia szlaku metabolicznego
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Sylabus Biologia molekularna
 Sylabus Biologia molekularna 1. Metryczka Nazwa Wydziału Wydział Farmaceutyczny z Oddziałem Medycyny Laboratoryjnej Program kształcenia Farmacja, jednolite studia magisterskie, forma studiów: stacjonarne
Sylabus Biologia molekularna 1. Metryczka Nazwa Wydziału Wydział Farmaceutyczny z Oddziałem Medycyny Laboratoryjnej Program kształcenia Farmacja, jednolite studia magisterskie, forma studiów: stacjonarne
STATYSTYKA MATEMATYCZNA
 STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez statystycznych
STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez statystycznych
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Błędy przy testowaniu hipotez statystycznych. Decyzja H 0 jest prawdziwa H 0 jest faszywa
 Weryfikacja hipotez statystycznych Hipotezą statystyczną nazywamy każde przypuszczenie dotyczące nieznanego rozkładu badanej cechy populacji, o prawdziwości lub fałszywości którego wnioskuje się na podstawie
Weryfikacja hipotez statystycznych Hipotezą statystyczną nazywamy każde przypuszczenie dotyczące nieznanego rozkładu badanej cechy populacji, o prawdziwości lub fałszywości którego wnioskuje się na podstawie
CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1
 CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1 PRACOWNIA INFORMATYCZNA PROWADZĄCY: Dr Magda Mielczarek (biolog) Katedra Genetyki, pokój nr 21
CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1 PRACOWNIA INFORMATYCZNA PROWADZĄCY: Dr Magda Mielczarek (biolog) Katedra Genetyki, pokój nr 21
Testowanie hipotez statystycznych.
 Bioinformatyka Wykład 4 Wrocław, 17 października 2011 Temat. Weryfikacja hipotez statystycznych dotyczących wartości oczekiwanej w dwóch populacjach o rozkładach normalnych. Model 3. Porównanie średnich
Bioinformatyka Wykład 4 Wrocław, 17 października 2011 Temat. Weryfikacja hipotez statystycznych dotyczących wartości oczekiwanej w dwóch populacjach o rozkładach normalnych. Model 3. Porównanie średnich
Programowanie proceduralne INP001210WL rok akademicki 2018/19 semestr letni. Wykład 6. Karol Tarnowski A-1 p.
 Programowanie proceduralne INP001210WL rok akademicki 2018/19 semestr letni Wykład 6 Karol Tarnowski karol.tarnowski@pwr.edu.pl A-1 p. 411B Plan prezentacji Wskaźnik do pliku Dostęp do pliku: zapis, odczyt,
Programowanie proceduralne INP001210WL rok akademicki 2018/19 semestr letni Wykład 6 Karol Tarnowski karol.tarnowski@pwr.edu.pl A-1 p. 411B Plan prezentacji Wskaźnik do pliku Dostęp do pliku: zapis, odczyt,
VII WYKŁAD STATYSTYKA. 30/04/2014 B8 sala 0.10B Godz. 15:15
 VII WYKŁAD STATYSTYKA 30/04/2014 B8 sala 0.10B Godz. 15:15 WYKŁAD 7 (c.d) WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI Weryfikacja hipotez ( błędy I i II rodzaju, poziom istotności,
VII WYKŁAD STATYSTYKA 30/04/2014 B8 sala 0.10B Godz. 15:15 WYKŁAD 7 (c.d) WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI Weryfikacja hipotez ( błędy I i II rodzaju, poziom istotności,
Uogólniony model liniowy
 Uogólniony model liniowy Ogólny model liniowy y = Xb + e Każda obserwacja ma rozkład normalny Każda obserwacja ma tą samą wariancję Dane nienormalne Rozkład binomialny np. liczba chorych krów w stadzie
Uogólniony model liniowy Ogólny model liniowy y = Xb + e Każda obserwacja ma rozkład normalny Każda obserwacja ma tą samą wariancję Dane nienormalne Rozkład binomialny np. liczba chorych krów w stadzie
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA
 Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Testowanie hipotez. Marcin Zajenkowski. Marcin Zajenkowski () Testowanie hipotez 1 / 25
 Testowanie hipotez 1 / 25") Testowanie hipotez Marcin Zajenkowski Marcin Zajenkowski () Testowanie hipotez 1 / 25 Testowanie hipotez Aby porównać ze sobą dwie statystyki z próby stosuje się testy istotności. Mówią one o tym czy uzyskane
Testowanie hipotez Marcin Zajenkowski Marcin Zajenkowski () Testowanie hipotez 1 / 25 Testowanie hipotez Aby porównać ze sobą dwie statystyki z próby stosuje się testy istotności. Mówią one o tym czy uzyskane
Statystyczna analiza danych w programie STATISTICA (wykład 2) Dariusz Gozdowski
 Dariusz Gozdowski") Statystyczna analiza danych w programie STATISTICA (wykład ) Dariusz Gozdowski Katedra Doświadczalnictwa i Bioinformatyki Wydział Rolnictwa i Biologii SGGW Weryfikacja (testowanie) hipotez statystycznych
Statystyczna analiza danych w programie STATISTICA (wykład ) Dariusz Gozdowski Katedra Doświadczalnictwa i Bioinformatyki Wydział Rolnictwa i Biologii SGGW Weryfikacja (testowanie) hipotez statystycznych
Informacje dotyczące pracy kontrolnej
 Informacje dotyczące pracy kontrolnej Słuchacze, którzy z przyczyn usprawiedliwionych nie przystąpili do pracy kontrolnej lub otrzymali z niej ocenę negatywną zobowiązani są do dnia 06 grudnia 2015 r.
Informacje dotyczące pracy kontrolnej Słuchacze, którzy z przyczyn usprawiedliwionych nie przystąpili do pracy kontrolnej lub otrzymali z niej ocenę negatywną zobowiązani są do dnia 06 grudnia 2015 r.
Techniki biologii molekularnej Kod przedmiotu
 Techniki biologii molekularnej - opis przedmiotu Informacje ogólne Nazwa przedmiotu Techniki biologii molekularnej Kod przedmiotu 13.9-WB-BMD-TBM-W-S14_pNadGenI2Q8V Wydział Kierunek Wydział Nauk Biologicznych
Techniki biologii molekularnej - opis przedmiotu Informacje ogólne Nazwa przedmiotu Techniki biologii molekularnej Kod przedmiotu 13.9-WB-BMD-TBM-W-S14_pNadGenI2Q8V Wydział Kierunek Wydział Nauk Biologicznych
Nowoczesne systemy ekspresji genów
 Nowoczesne systemy ekspresji genów Ekspresja genów w organizmach żywych GEN - pojęcia podstawowe promotor sekwencja kodująca RNA terminator gen Gen - odcinek DNA zawierający zakodowaną informację wystarczającą
Nowoczesne systemy ekspresji genów Ekspresja genów w organizmach żywych GEN - pojęcia podstawowe promotor sekwencja kodująca RNA terminator gen Gen - odcinek DNA zawierający zakodowaną informację wystarczającą
Podstawy bioinformatyki sekwencjonowanie nowej generacji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Testy nieparametryczne
 Testy nieparametryczne 1 Wybrane testy nieparametryczne 1. Test chi-kwadrat zgodności z rozkładem oczekiwanym 2. Test chi-kwadrat niezależności dwóch zmiennych kategoryzujących 3. Test U Manna-Whitney
Testy nieparametryczne 1 Wybrane testy nieparametryczne 1. Test chi-kwadrat zgodności z rozkładem oczekiwanym 2. Test chi-kwadrat niezależności dwóch zmiennych kategoryzujących 3. Test U Manna-Whitney
Glimmer umożliwia znalezienie regionów kodujących
 Narzędzia ułatwiające identyfikację właściwych genów GLIMMER TaxPlot Narzędzia ułatwiające amplifikację tych genów techniki PCR Primer3, Primer3plus PrimerBLAST Reverse Complement Narzędzia ułatwiające
Narzędzia ułatwiające identyfikację właściwych genów GLIMMER TaxPlot Narzędzia ułatwiające amplifikację tych genów techniki PCR Primer3, Primer3plus PrimerBLAST Reverse Complement Narzędzia ułatwiające
Dane mikromacierzowe. Mateusz Markowicz Marta Stańska
 Dane mikromacierzowe Mateusz Markowicz Marta Stańska Mikromacierz Mikromacierz DNA (ang. DNA microarray) to szklana lub plastikowa płytka (o maksymalnych wymiarach 2,5 cm x 7,5 cm) z naniesionymi w regularnych
Dane mikromacierzowe Mateusz Markowicz Marta Stańska Mikromacierz Mikromacierz DNA (ang. DNA microarray) to szklana lub plastikowa płytka (o maksymalnych wymiarach 2,5 cm x 7,5 cm) z naniesionymi w regularnych
Wykład 2 Hipoteza statystyczna, test statystyczny, poziom istotn. istotności, p-wartość i moc testu
 Wykład 2 Hipoteza statystyczna, test statystyczny, poziom istotności, p-wartość i moc testu Wrocław, 01.03.2017r Przykład 2.1 Właściciel firmy produkującej telefony komórkowe twierdzi, że wśród jego produktów
Wykład 2 Hipoteza statystyczna, test statystyczny, poziom istotności, p-wartość i moc testu Wrocław, 01.03.2017r Przykład 2.1 Właściciel firmy produkującej telefony komórkowe twierdzi, że wśród jego produktów
LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI
 LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI WERYFIKACJA HIPOTEZ Hipoteza statystyczna jakiekolwiek przypuszczenie dotyczące populacji generalnej- jej poszczególnych
LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI WERYFIKACJA HIPOTEZ Hipoteza statystyczna jakiekolwiek przypuszczenie dotyczące populacji generalnej- jej poszczególnych
Biologiczne podstawy ewolucji. Informacja genetyczna. Co to jest ewolucja.
 Biologiczne podstawy ewolucji. Informacja genetyczna. Co to jest ewolucja. Historia } Selekcja w hodowli zwierząt, co najmniej 10 000 lat temu } Sztuczne zapłodnienie (np. drzewa daktylowe) 1000 lat temu
Biologiczne podstawy ewolucji. Informacja genetyczna. Co to jest ewolucja. Historia } Selekcja w hodowli zwierząt, co najmniej 10 000 lat temu } Sztuczne zapłodnienie (np. drzewa daktylowe) 1000 lat temu
Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online
 Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
października 2013: Elementarz biologii molekularnej. Wykład nr 2 BIOINFORMATYKA rok II
 10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
Testowanie hipotez statystycznych
 Agenda Instytut Matematyki Politechniki Łódzkiej 2 stycznia 2012 Agenda Agenda 1 Wprowadzenie Agenda 2 Hipoteza oraz błędy I i II rodzaju Hipoteza alternatywna Statystyka testowa Zbiór krytyczny Poziom
Agenda Instytut Matematyki Politechniki Łódzkiej 2 stycznia 2012 Agenda Agenda 1 Wprowadzenie Agenda 2 Hipoteza oraz błędy I i II rodzaju Hipoteza alternatywna Statystyka testowa Zbiór krytyczny Poziom
Statystyka matematyczna dla leśników
 Statystyka matematyczna dla leśników Wydział Leśny Kierunek leśnictwo Studia Stacjonarne I Stopnia Rok akademicki 03/04 Wykład 5 Testy statystyczne Ogólne zasady testowania hipotez statystycznych, rodzaje
Statystyka matematyczna dla leśników Wydział Leśny Kierunek leśnictwo Studia Stacjonarne I Stopnia Rok akademicki 03/04 Wykład 5 Testy statystyczne Ogólne zasady testowania hipotez statystycznych, rodzaje
STATYSTYKA MATEMATYCZNA WYKŁAD 4. Testowanie hipotez Estymacja parametrów
 STATYSTYKA MATEMATYCZNA WYKŁAD 4 Testowanie hipotez Estymacja parametrów WSTĘP 1. Testowanie hipotez Błędy związane z testowaniem hipotez Etapy testowana hipotez Testowanie wielokrotne 2. Estymacja parametrów
STATYSTYKA MATEMATYCZNA WYKŁAD 4 Testowanie hipotez Estymacja parametrów WSTĘP 1. Testowanie hipotez Błędy związane z testowaniem hipotez Etapy testowana hipotez Testowanie wielokrotne 2. Estymacja parametrów
VI WYKŁAD STATYSTYKA. 9/04/2014 B8 sala 0.10B Godz. 15:15
 VI WYKŁAD STATYSTYKA 9/04/2014 B8 sala 0.10B Godz. 15:15 WYKŁAD 6 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI Weryfikacja hipotez ( błędy I i II rodzaju, poziom istotności, zasady
VI WYKŁAD STATYSTYKA 9/04/2014 B8 sala 0.10B Godz. 15:15 WYKŁAD 6 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI Weryfikacja hipotez ( błędy I i II rodzaju, poziom istotności, zasady
Przykładowe zadania. przygotowujące do egzaminu maturalnego
 Przykładowe zadania z BIOLOGii przygotowujące do egzaminu maturalnego Prezentowane zadania są zgodne z podstawą programową kształcenia ogólnego w zakresie rozszerzonym i podstawowym. Prócz zadań, które
Przykładowe zadania z BIOLOGii przygotowujące do egzaminu maturalnego Prezentowane zadania są zgodne z podstawą programową kształcenia ogólnego w zakresie rozszerzonym i podstawowym. Prócz zadań, które
Podstawy programowania. Wykład 7 Tablice wielowymiarowe, SOA, AOS, itp. Krzysztof Banaś Podstawy programowania 1
 Podstawy programowania. Wykład 7 Tablice wielowymiarowe, SOA, AOS, itp. Krzysztof Banaś Podstawy programowania 1 Tablice wielowymiarowe C umożliwia definiowanie tablic wielowymiarowych najczęściej stosowane
Podstawy programowania. Wykład 7 Tablice wielowymiarowe, SOA, AOS, itp. Krzysztof Banaś Podstawy programowania 1 Tablice wielowymiarowe C umożliwia definiowanie tablic wielowymiarowych najczęściej stosowane
SYLABUS DOTYCZY CYKLU KSZTAŁCENIA
 SYLABUS DOTYCZY CYKLU KSZTAŁCENIA 2015-2021 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Techniki biologii molekularnej Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej
SYLABUS DOTYCZY CYKLU KSZTAŁCENIA 2015-2021 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Techniki biologii molekularnej Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej
GENOMIKA FUNKCJONALNA. Jak działają geny i genomy? Poziom I: Analizy transkryptomu
 GENOMIKA FUNKCJONALNA Jak działają geny i genomy? Poziom I: Analizy transkryptomu Adnotacja (ang. annotation) pierwszy etap po uzyskaniu kompletnej sekwencji nukleotydyowej genomu analiza bioinformatyczna
GENOMIKA FUNKCJONALNA Jak działają geny i genomy? Poziom I: Analizy transkryptomu Adnotacja (ang. annotation) pierwszy etap po uzyskaniu kompletnej sekwencji nukleotydyowej genomu analiza bioinformatyczna
Bioinformatyka, edycja 2016/2017, laboratorium
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: dr Jacek Śmietański Mikromacierze 1. Mikromacierze wprowadzenie Mikromacierze to technologia pozwalająca na pomiar aktywności genów w komórce.
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: dr Jacek Śmietański Mikromacierze 1. Mikromacierze wprowadzenie Mikromacierze to technologia pozwalająca na pomiar aktywności genów w komórce.
Zaawansowane programowanie w języku C++ Biblioteka standardowa
 Zaawansowane programowanie w języku C++ Biblioteka standardowa Prezentacja jest współfinansowana przez Unię Europejską w ramach Europejskiego Funduszu Społecznego w projekcie pt. Innowacyjna dydaktyka
Zaawansowane programowanie w języku C++ Biblioteka standardowa Prezentacja jest współfinansowana przez Unię Europejską w ramach Europejskiego Funduszu Społecznego w projekcie pt. Innowacyjna dydaktyka
Podstawy biologii. Informacja genetyczna. Co to jest ewolucja.
 Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Materiał genetyczny Materiałem genetycznym są kwasy nukleinowe Materiałem genetycznym organizmów komórkowych jest kwas deoksyrybonukleinowy
Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Materiał genetyczny Materiałem genetycznym są kwasy nukleinowe Materiałem genetycznym organizmów komórkowych jest kwas deoksyrybonukleinowy
Metody: PCR, MLPA, Sekwencjonowanie, PCR-RLFP, PCR-Multiplex, PCR-ASO
 Diagnostyka molekularna Dr n.biol. Anna Wawrocka Strategia diagnostyki genetycznej: Aberracje chromosomowe: Metody:Analiza kariotypu, FISH, acgh, MLPA, QF-PCR Gen(y) znany Metody: PCR, MLPA, Sekwencjonowanie,
Diagnostyka molekularna Dr n.biol. Anna Wawrocka Strategia diagnostyki genetycznej: Aberracje chromosomowe: Metody:Analiza kariotypu, FISH, acgh, MLPA, QF-PCR Gen(y) znany Metody: PCR, MLPA, Sekwencjonowanie,
BIOLOGICZNE BAZY DANYCH GENOMY I ICH ADNOTACJE. Pracownia Informatyczna 2
 BIOLOGICZNE BAZY DANYCH GENOMY I ICH ADNOTACJE Pracownia Informatyczna 2 WYBRANE BIOLOGICZNE BAZY DANYCH GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION NCBI Utworzone
BIOLOGICZNE BAZY DANYCH GENOMY I ICH ADNOTACJE Pracownia Informatyczna 2 WYBRANE BIOLOGICZNE BAZY DANYCH GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION NCBI Utworzone
LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI
 LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI WERYFIKACJA HIPOTEZ Hipoteza statystyczna jakiekolwiek przypuszczenie dotyczące populacji generalnej- jej poszczególnych
LABORATORIUM 8 WERYFIKACJA HIPOTEZ STATYSTYCZNYCH PARAMETRYCZNE TESTY ISTOTNOŚCI WERYFIKACJA HIPOTEZ Hipoteza statystyczna jakiekolwiek przypuszczenie dotyczące populacji generalnej- jej poszczególnych
Zadania ze statystyki, cz.7 - hipotezy statystyczne, błąd standardowy, testowanie hipotez statystycznych
 Zadania ze statystyki, cz.7 - hipotezy statystyczne, błąd standardowy, testowanie hipotez statystycznych Zad. 1 Średnia ocen z semestru letniego w populacji studentów socjologii w roku akademickim 2011/2012
Zadania ze statystyki, cz.7 - hipotezy statystyczne, błąd standardowy, testowanie hipotez statystycznych Zad. 1 Średnia ocen z semestru letniego w populacji studentów socjologii w roku akademickim 2011/2012
Zadania ze statystyki cz. 8 I rok socjologii. Zadanie 1.
 Zadania ze statystyki cz. 8 I rok socjologii Zadanie 1. W potocznej opinii pokutuje przekonanie, że lepsi z matematyki są chłopcy niż dziewczęta. Chcąc zweryfikować tę opinię, przeprowadzono badanie w
Zadania ze statystyki cz. 8 I rok socjologii Zadanie 1. W potocznej opinii pokutuje przekonanie, że lepsi z matematyki są chłopcy niż dziewczęta. Chcąc zweryfikować tę opinię, przeprowadzono badanie w
GENOMIKA FUNKCJONALNA. Jak działają geny i genomy? Poziom I: Analizy transkryptomu
 GENOMIKA FUNKCJONALNA Jak działają geny i genomy? Poziom I: Analizy transkryptomu Adnotacja (ang. annotation) pierwszy etap po uzyskaniu kompletnej sekwencji nukleotydyowej genomu analiza bioinformatyczna
GENOMIKA FUNKCJONALNA Jak działają geny i genomy? Poziom I: Analizy transkryptomu Adnotacja (ang. annotation) pierwszy etap po uzyskaniu kompletnej sekwencji nukleotydyowej genomu analiza bioinformatyczna
Wykład 3 Testowanie hipotez statystycznych o wartości średniej. średniej i wariancji z populacji o rozkładzie normalnym
 Wykład 3 Testowanie hipotez statystycznych o wartości średniej i wariancji z populacji o rozkładzie normalnym Wrocław, 08.03.2017r Model 1 Testowanie hipotez dla średniej w rozkładzie normalnym ze znaną
Wykład 3 Testowanie hipotez statystycznych o wartości średniej i wariancji z populacji o rozkładzie normalnym Wrocław, 08.03.2017r Model 1 Testowanie hipotez dla średniej w rozkładzie normalnym ze znaną
MIKROMACIERZE. dr inż. Aleksandra Świercz dr Agnieszka Żmieńko
 MIKROMACIERZE dr inż. Aleksandra Świercz dr Agnieszka Żmieńko Informacje ogólne Wykłady będą częściowo dostępne w formie elektronicznej http://cs.put.poznan.pl/aswiercz aswiercz@cs.put.poznan.pl Godziny
MIKROMACIERZE dr inż. Aleksandra Świercz dr Agnieszka Żmieńko Informacje ogólne Wykłady będą częściowo dostępne w formie elektronicznej http://cs.put.poznan.pl/aswiercz aswiercz@cs.put.poznan.pl Godziny
Weryfikacja hipotez statystycznych, parametryczne testy istotności w populacji
 Weryfikacja hipotez statystycznych, parametryczne testy istotności w populacji Dr Joanna Banaś Zakład Badań Systemowych Instytut Sztucznej Inteligencji i Metod Matematycznych Wydział Informatyki Politechniki
Weryfikacja hipotez statystycznych, parametryczne testy istotności w populacji Dr Joanna Banaś Zakład Badań Systemowych Instytut Sztucznej Inteligencji i Metod Matematycznych Wydział Informatyki Politechniki
Klonowanie molekularne Kurs doskonalący. Zakład Geriatrii i Gerontologii CMKP
 Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej
 Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej rozdzielczości jest sekwencja nukleotydowa -mapowanie fizyczne genomu
Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej rozdzielczości jest sekwencja nukleotydowa -mapowanie fizyczne genomu
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów:
 Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Podstawy programowania. Podstawy C# Tablice
 Podstawy programowania Podstawy C# Tablice Tablica to indeksowany zbiór elementów Tablica jest typem referencyjnym (deklaracja tworzy tylko referencję, sama tablica musi być utworzona oddzielnie, najprościej
Podstawy programowania Podstawy C# Tablice Tablica to indeksowany zbiór elementów Tablica jest typem referencyjnym (deklaracja tworzy tylko referencję, sama tablica musi być utworzona oddzielnie, najprościej
Weryfikacja hipotez statystycznych
 Weryfikacja hipotez statystycznych Hipoteza Test statystyczny Poziom istotności Testy jednostronne i dwustronne Testowanie równości wariancji test F-Fishera Testowanie równości wartości średnich test t-studenta
Weryfikacja hipotez statystycznych Hipoteza Test statystyczny Poziom istotności Testy jednostronne i dwustronne Testowanie równości wariancji test F-Fishera Testowanie równości wartości średnich test t-studenta
Analiza wariancji - ANOVA
 Analiza wariancji - ANOVA Analiza wariancji jest metodą pozwalającą na podział zmienności zaobserwowanej wśród wyników eksperymentalnych na oddzielne części. Każdą z tych części możemy przypisać oddzielnemu
Analiza wariancji - ANOVA Analiza wariancji jest metodą pozwalającą na podział zmienności zaobserwowanej wśród wyników eksperymentalnych na oddzielne części. Każdą z tych części możemy przypisać oddzielnemu
Elementy statystyki STA - Wykład 5
 STA - Wykład 5 Wydział Matematyki i Informatyki Uniwersytet im. Adama Mickiewicza 1 ANOVA 2 Model jednoczynnikowej analizy wariancji Na model jednoczynnikowej analizy wariancji możemy traktować jako uogólnienie
STA - Wykład 5 Wydział Matematyki i Informatyki Uniwersytet im. Adama Mickiewicza 1 ANOVA 2 Model jednoczynnikowej analizy wariancji Na model jednoczynnikowej analizy wariancji możemy traktować jako uogólnienie
Analiza wariancji. dr Janusz Górczyński
 Analiza wariancji dr Janusz Górczyński Wprowadzenie Powiedzmy, że badamy pewną populację π, w której cecha Y ma rozkład N o średniej m i odchyleniu standardowym σ. Powiedzmy dalej, że istnieje pewien czynnik
Analiza wariancji dr Janusz Górczyński Wprowadzenie Powiedzmy, że badamy pewną populację π, w której cecha Y ma rozkład N o średniej m i odchyleniu standardowym σ. Powiedzmy dalej, że istnieje pewien czynnik
Wstęp do programowania INP001213Wcl rok akademicki 2017/18 semestr zimowy. Wykład 12. Karol Tarnowski A-1 p.
 Wstęp do programowania INP001213Wcl rok akademicki 2017/18 semestr zimowy Wykład 12 Karol Tarnowski karol.tarnowski@pwr.edu.pl A-1 p. 411B Plan prezentacji (1) Obsługa łańcuchów znakowych getchar(), putchar()
Wstęp do programowania INP001213Wcl rok akademicki 2017/18 semestr zimowy Wykład 12 Karol Tarnowski karol.tarnowski@pwr.edu.pl A-1 p. 411B Plan prezentacji (1) Obsługa łańcuchów znakowych getchar(), putchar()
Systemy uczące się Lab 4
 Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
Testowanie hipotez statystycznych.
 Statystyka Wykład 10 Wrocław, 22 grudnia 2011 Testowanie hipotez statystycznych Definicja. Hipotezą statystyczną nazywamy stwierdzenie dotyczące parametrów populacji. Definicja. Dwie komplementarne w problemie
Statystyka Wykład 10 Wrocław, 22 grudnia 2011 Testowanie hipotez statystycznych Definicja. Hipotezą statystyczną nazywamy stwierdzenie dotyczące parametrów populacji. Definicja. Dwie komplementarne w problemie
Podstawy biologii. Informacja genetyczna. Co to jest ewolucja.
 Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Zarys biologii molekularnej genu Podstawowe procesy genetyczne Replikacja powielanie informacji Ekspresja wyrażanie (realizowanie funkcji)
Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Zarys biologii molekularnej genu Podstawowe procesy genetyczne Replikacja powielanie informacji Ekspresja wyrażanie (realizowanie funkcji)
W2. Zmienne losowe i ich rozkłady. Wnioskowanie statystyczne.
 W2. Zmienne losowe i ich rozkłady. Wnioskowanie statystyczne. dr hab. Jerzy Nakielski Katedra Biofizyki i Morfogenezy Roślin Plan wykładu: 1. Etapy wnioskowania statystycznego 2. Hipotezy statystyczne,
W2. Zmienne losowe i ich rozkłady. Wnioskowanie statystyczne. dr hab. Jerzy Nakielski Katedra Biofizyki i Morfogenezy Roślin Plan wykładu: 1. Etapy wnioskowania statystycznego 2. Hipotezy statystyczne,