(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
|
|
|
- Joanna Mucha
- 6 lat temu
- Przeglądów:
Transkrypt
 O udzieleniu patentu europejskiego ogłoszono: 06.04.16 Europejski Biuletyn Patentowy 16/14 EP 2907873 B1 (13) (1) T3 Int.Cl. C12N 1/ (06.01) C07K 16/00 (06.01) CB 0/06 (06.")
1 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: (97) O udzieleniu patentu europejskiego ogłoszono: Europejski Biuletyn Patentowy 16/14 EP B1 (13) (1) T3 Int.Cl. C12N 1/ (06.01) C07K 16/00 (06.01) CB 0/06 (06.01) (4) Tytuł wynalazku: Jednoczesna, zintegrowana selekcja i ewolucja wydajności przeciwciała/białka i wyrażanie w gospodarzu do wytwarzania () Pierwszeństwo: US P (43) Zgłoszenie ogłoszono: w Europejskim Biuletynie Patentowym nr 1/34 (4) O złożeniu tłumaczenia patentu ogłoszono: Wiadomości Urzędu Patentowego 16/ (73) Uprawniony z patentu: Bioatla LLC, San Diego, US (72) Twórca(y) wynalazku: PL/EP T3 JAY MILTON SHORT, Del Mar, US (74) Pełnomocnik: rzecz. pat. Magdalena Tagowska PATPOL KANCELARIA PATENTOWA SP. Z O.O. ul. Nowoursynowska 162 J Warszawa Uwaga: W ciągu dziewięciu miesięcy od publikacji informacji o udzieleniu patentu europejskiego, każda osoba może wnieść do Europejskiego Urzędu Patentowego sprzeciw dotyczący udzielonego patentu europejskiego. Sprzeciw wnosi się w formie uzasadnionego na piśmie oświadczenia. Uważa się go za wniesiony dopiero z chwilą wniesienia opłaty za sprzeciw (Art. 99 (1) Konwencji o udzielaniu patentów europejskich).
2 Opis DZIEDZINA WYNALAZKU [0001] W konkretnym aspekcie, niniejszy wynalazek dotyczy przeciwciał oraz ich optymalizacji poprzez ewolucję przeciwciała. Przeciwciała terapeutyczne są identyfikowane, poddawane ewolucji i wytwarzane w tym samym gospodarzu przy zastosowaniu tych samych systemów genetycznych TŁO WYNALAZKU [0002] Zaprojektowano, opracowano i wdrożono różne systemy dla przeciwciał i innych białek, które wytwarzają białka-kandydatów na cząsteczki terapeutyczne. Ostatnio, opracowano wiele systemów ewolucji w celu wzmocnienia funkcji białek. Odrębnie, opracowano ssacze systemy ekspresyjne dla wysokowydajnego wytwarzania przeciwciał i innych białek o zastosowaniach terapeutycznych. Do tej pory żadna grupa nie opracowała systemu dla umożliwienia wytwarzania, ewolucji przeciwciała lub białka i wytwarzania/produkcji białka w jednym wydajnym ssaczym systemie ekspresyjnym. [0003] WO dotyczy sposobów wytwarzania zbiorów lub bibliotek kwasów nukleinowych kodujących miejsca wiązania antygenów, takich jak przeciwciała, domeny przeciwciał lub inne fragmenty. Sposób ten odnosi się do wytwarzania wariantów miejsc wiązania antygenu przez zmienianie matrycowych kwasów nukleinowych, w tym przez mutagenezę saturacyjną, syntetyczne ponowne łączenie przez ligację lub ich kombinację. [0004] Rajpal i wsp., A general method for greatly improving the affinity of antibodies by using combinatorial libraries, Proc Natl Acad Sci USA., tom 2, strony , (0) ujawnia sposób polepszania powinowactwa przeciwciał przy użyciu tak zwanej mutagenezy przeglądowej, która jednocześnie ocenia i optymalizuje kombinatoryczne mutacje wybranych aminokwasów w przeciwciele. Proces koncentruje się na precyzyjnej dystrybucji w obrębie jednego lub więcej domen regionów determinujących dopasowanie i bada synergiczny udział właściwości chemicznych aminokwasowego łańcucha bocznego. [000] Iba i wsp., Expression vectors for the introduction of highly diverged sequences into the six complementarity-determining regions of an antibody, Gene, tom 194, strony 3-46, (1997) ujawnia trzy rodzaje wektorów fagmidowych, które pozwalają na jednoczesne wprowadzanie bardzo zróżnicowanych sekwencji do sześciu regionów determinujących dopasowanie przeciwciała przy zastosowaniu reakcji łańcuchowej polimerazy ze zdegenerowanymi starterami oligodeoksynukleotydowymi. Fagi wyrażają bądź Fv, jednołańcuchowy Fv (scfv) bądź postać Fab Ab połączonego z połową cząsteczki cpiii na powierzchni faga M13. [0006] WO 0/00334 ujawnia sposób mutagenezy, dzięki któremu określony uprzednio aminokwas jest wprowadzany do każdej i wszystkich pozycji w wybranym zestawie pozycji w wybranym uprzednio regionie (lub kilku różnych regionach) polipeptydu w celu wytworzenia biblioteki analogów polipeptydów. Wytworzone biblioteki mogą być stosowane do badania roli konkretnych aminokwasów w strukturze i funkcji polipeptydu oraz opracowania nowych lub ulepszonych
3 1 polipeptydów, takich jak przeciwciała, fragmenty przeciwciała, przeciwciała jednołańcuchowe, enzymy i ligandy. [0007] WO 03/0232 ujawnia sposób dla wysokoprzepustowej ukierunkowanej ewolucji peptydów i białek, szczególnie tych, które działają w złożonych układach biologicznych. Białka i peptydy obejmują, ale bez ograniczania się do tego, białka wewnątrzkomórkowe, białka przekaźnikowe/przekazujące sygnały/ hormonalne i białka wirusowe. [0008] Opracowano wiele przeciwciał stosując bakteryjne systemy prezentacji na fagach w bakteriach, natomiast wyrażanie przeciwciał pełnej długości odbywa się głównie w komórkach ssaczych. Ten brak podobieństwa czyni ewolucję lub selekcję klonów dla wyrażania niemożliwą. Dodatkowe bariery obejmują konwencjonalny wymóg dużej liczby wariantów, które mają być przeszukiwane z wykorzystaniem tradycyjnej technologii i ssacze systemy ekspresji na wysokim poziomie, które zostały zoptymalizowane do ekspresji, a nie do klonowania dużej liczby wariantów. Zintegrowana selekcja, ewolucja i ssaczy system ekspresyjny dla przeciwciała/białka nie został wcześniej zaprojektowany. Zastosowanie komórek eukariotycznych do manipulowania dużymi liczbami, łącznie z niestochastyczną ewolucją przeciwciała wewnątrz zoptymalizowanej eukariotycznej komórki gospodarza, zwiększa prawdopodobieństwo sukcesu i znacznie przyspiesza proces wytwarzania zoptymalizowanego przeciwciała, które będzie się wyrażać w komórkach eukariotycznych na wystarczająco wysokich poziomach wymaganych przy wytwarzaniu. STRESZCZENIE WYNALAZKU 2 3 [0009] Niniejsze ujawnienie dostarcza sposoby integracyjnego wytwarzania i/lub selekcji, ewolucji i wyrażania w gospodarzu eukariotycznym, takim jak gospodarz-komórka ssacza lub gospodarzkomórka drożdży, przeciwciał terapeutycznych, do wytwarzania w jednym systemie. Przeciwciała terapeutyczne uzyskuje się, optymalizuje i wytwarza w tym samym systemie gospodarza eukariotycznego. Ujawniony system kompleksowej, zintegrowanej optymalizacji przeciwciała (Comprehensive Integrated Antibody Optimization, CIAO! ) pozwala na jednoczesną ewolucję właściwości i optymalizację wyrażania przeciwciała. [00] W jednym wykonaniu wynalazek dostarcza sposobu selekcji, ewolucji i wyrażania przeciwciała w gospodarzu do wytwarzania-komórce eukariotycznej; gdzie sposób obejmuje etapy: wyselekcjonowania przeciwciała matrycowego; przeprowadzenia ewolucji przeciwciała matrycowego, aby wytworzyć zbiór przeciwciał zmutowanych w gospodarzu do wytwarzania-komórce eukariotycznej; przeszukiwania zmutowanych przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; wyselekcjonowania ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał w oparciu o optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności w porównaniu do tej samej właściwości, cechy lub aktywności przeciwciała matrycowego; i wyrażania ulepszonego zmutowanego przeciwciała w tym samym gospodarzu do wytwarzania-komórce eukariotycznej co stosowany w etapie ewolucji na skalę komercyjną. W jednym aspekcie etap selekcjonowania obejmuje wytwarzanie biblioteki dla przeciwciała anty-antygen w gospodarzu do wytwarzania-komórce eukariotycznej przy zastosowaniu 2
4 1 2 3 prezentacji przeciwciała na powierzchni komórek; przeszukiwanie biblioteki pod kątem co najmniej jednej określonej właściwości, cechy lub aktywności; i selekcjonowania przeciwciała matrycowego z biblioteki. W innym aspekcie biblioteka dla przeciwciała anty-antygen jest biblioteką dla humanizowanego przeciwciała anty-antygen. [0011] W jednym aspekcie, gospodarz do wytwarzania-komórka eukariotyczna jest wybrany spośród mysich komórek fibroblastów 3T3; komórek fibroblastów chomika syryjskiego BHK21; psich komórek nabłonkowych MDCK; ludzkich komórek nabłonkowych Hela; komórek nabłonkowych kanguroszczura PtK1; mysich komórek plazmatycznych SP2/0; i mysich komórek plazmatycznych NS0; ludzkich komórek embrionalnych nerki HEK 293; komórek nerek małpy COS; komórek jajnika chomika chińskiego CHO, CHO-S; komórek embrionalnych myszy R1; komórek embrionalnych myszy E14.1; ludzkich komórek embrionalnych H1; ludzkich komórek embrionalnych H9; ludzkich komórek embrionalnych PER C.6, komórek drożdży S. cerevisiae; lub komórek drożdży Pichia. W jednym aspekcie, etapy przeszukiwania wykorzystują sortowanie komórek aktywowane fluorescencją (FACS, ang. fluorescence-activated cell sorting). [0012] W innym aspekcie, etap przeprowadzania ewolucji obejmuje wytworzenie zbioru zmutowanych przeciwciał wytworzonych z przeciwciała matrycowego mających m regionów determinujących dopasowanie (CDR, ang. complementary determining regions), gdzie m jest liczbą całkowitą wybraną spośród 1, 2, 3, 4, lub 6, gdzie każdy wymieniony CDR obejmuje n reszt aminokwasowych, sposób obejmuje wytwarzanie m x n oddzielnych zbiorów przeciwciał, każdy zbiór obejmuje elementyprzeciwciała mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji CDR; gdzie każdy zbiór przeciwciał różni się w pojedynczej określonej uprzednio pozycji; a liczba różnych wytwarzanych elementów-przeciwciał wynosi m x n x X. W konkretnym aspekcie, m wynosi 6. [0013] W jednym aspekcie, etap przeprowadzania ewolucji obejmuje wytwarzanie n-1 odrębnych zbiorów zmutowanych polipeptydów z przeciwciała matrycowego, gdzie każdy zbiór zawiera elementy-polipeptydy mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji; gdzie każdy zbiór polipeptydów różni się w pojedynczej określonej uprzednio pozycji; a liczba różnych wytwarzanych elementów-polipeptydów wynosi [n-1] x X. W konkretnym aspekcie, X wynosi 19. [0014] W innym aspekcie, etap przeszukiwania obejmuje testowanie każdego elementu-polipeptydu pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; identyfikowanie jakiejkolwiek zmiany w tej właściwości, cechy lub aktywności elementu-polipeptydu w stosunku do polipeptydu matrycowego; wytwarzanie mapy funkcyjnej, gdzie mapa funkcyjna jest wykorzystywana do identyfikacji pozycji i mutacji w zmutowanym polipeptydzie, które dają w rezultacie ulepszonego mutanta i/lub mutację cichą w porównaniu do polipeptydu matrycowego. [001] W innym aspekcie, fragment przeciwciała jest wybrany z łańcucha ciężkiego, łańcucha lekkiego, domeny zmiennej, domeny stałej, regionu hiperzmiennego, regionu determinującego dopasowanie 1 (CDR1), regionu determinującego dopasowanie 2 (CDR2) i regionu determinującego dopasowanie 3 (CDR3). 3
5 1 2 3 [0016] W innym aspekcie etap wytwarzania obejmuje poddanie polinukleotydu, zawierającego kodony kodujące taki polipeptyd matrycowy, amplifikacji opartej na polimerazie przy użyciu 64-krotnie zdegenerowanego oligonukleotydu dla każdego kodonu, który ma być mutagenizowany, gdzie każdy z tych 64-krotnie zdegenerowanych oligonukleotydów składa z pierwszej sekwencji homologicznej i zdegenerowanej sekwencji tripletu N, N, N, tak aby wytworzyć zbiór polinukleotydów potomnych; i poddanie tego zbioru polinukleotydów potomnych amplifikacji klonalnej tak, że wyrażane są polipeptydy kodowane przez polinukleotydy potomne. [0017] Również częścią wynalazku jest sposób według zastrzeżenia 1, przy czym określona uprzednio właściwość, cecha lub aktywność jest wybrana spośród zmniejszenia agregacji przeciwciało-białko, wzmocnienia stabilności przeciwciała, zwiększenia rozpuszczalności przeciwciała, wprowadzenia miejsc glikozylacji, wprowadzenia miejsc sprzęgania, zmniejszenia immunogenności, wzmocnienia wyrażania przeciwciała, wzrostu powinowactwa wobec antygenu, zmniejszenia powinowactwa wobec antygenu, zmiany w powinowactwie wiązania, zmiany w immunogenności lub wzmocnienia swoistości. [0018] W innym aspekcie, etap przeprowadzania ewolucji obejmuje techniki ewolucji wybrane spośród: kompleksowej ewolucji pozycyjnej (CPE, ang. comprehensive positional evolution); kompleksowej insercyjnej ewolucji pozycyjnej (CPI, ang. comprehensive positional insertion evolution); kompleksowej delecyjnej ewolucji pozycyjnej (CPD, ang. comprehensive positional deletion evolution); kompleksowej ewolucji pozycyjnej (CPE), po której następuje kombinatoryczna synteza białka (CPS, ang. combinatorial protein synthesis); kompleksowej delecyjnej ewolucji pozycyjnej (CPD), po której następuje kombinatoryczna synteza białka (CPS); lub kompleksowej delecyjnej ewolucji pozycyjnej (CPD), po której następuje kombinatoryczna synteza białka (CPS). [0019] Przeciwciało jest przeciwciałem pełnej długości. [00] W innym aspekcie co najmniej jedna określona uprzednio właściwość, cecha lub aktywność w etapie (e) obejmuje jeden lub więcej spomiędzy (1) przeszukiwania pod kątem mutacji cichej i (2) przeszukiwania pod kątem mutacji zmiany sensu; w porównaniu z przeciwciałem matrycowym. [0021] W innym aspekcie, jedna lub więcej części przeciwciała wybranych spośród Fc i Fv; regionu zrębowego; oraz jednego lub większej liczby CDR są zmodyfikowane w ulepszonym mutancie przeciwciała ludzkiego w porównaniu do matrycowego przeciwciała ludzkiego podlegającego ewolucji. [0022] W innym aspekcie etap przeszukiwania obejmuje przeszukiwanie zbioru zmutowanych ludzkich przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i przeszukiwania pod kątem zmodyfikowanego wyrażania jednocześnie. [0023] W innym aspekcie, komórką gospodarza do wytwarzania jest eukariotyczny gospodarz do wytwarzania i etap przeprowadzania ewolucji obejmuje poddanie ewolucji matrycowego ludzkiego przeciwciała, aby wytworzyć zbiór zmutowanych ludzkich przeciwciał w gospodarzu do wytwarzaniakomórce eukariotycznej przy zastosowaniu prezentacji na powierzchni komórki. [0024] W innym wykonaniu, ujawnienie dostarcza sposobu ewolucji dla wzmocnionego wyrażania i wytwarzania przeciwciała ludzkiego w gospodarzu do wytwarzania-komórce eukariotycznej; sposobu obejmującego selekcję ludzkiego przeciwciała matrycowego dla ewolucji; poddanie ewolucji przeciwciała matrycowego obejmujące wytwarzanie zmutowanych kodonów kodujących ludzkie 4
6 1 2 3 przeciwciało matrycowe, aby wytworzyć zbiór zmutowanych ludzkich przeciwciał w gospodarzu do wytwarzania; przeszukiwanie zbioru zmutowanych przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i przeszukiwania ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał pod kątem zwiększonego wyrażania w porównaniu z ludzkim przeciwciałem matrycowym; selekcję ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał w oparciu o (1) zachowanie lub optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i (2) zwiększone wyrażanie w porównaniu do przeciwciała matrycowego; i wytwarzanie ulepszonego zmutowanego ludzkiego przeciwciała obejmujące wyrażenie ulepszonego zmutowanego przeciwciała ludzkiego w tym samym gospodarzu do wytwarzania co w etapie przeprowadzania ewolucji. [002] W jednym aspekcie, zmutowane kodony ulepszonego zmutowanego przeciwciała ludzkiego skutkują co najmniej jedną mutacją cichą i/lub mutacją zmiany sensu. W innym aspekcie, zmutowane kodony ulepszonego zmutowanego przeciwciała ludzkiego skutkują co najmniej jedną mutacją cichą. [0026] W kolejnym aspekcie, matrycowe ludzkie przeciwciało jest zatwierdzonym etycznie lekiem przeciwciałem terapeutycznym, a zmutowane ludzkie przeciwciało jest biopodobne. [0027] W innym aspekcie etap selekcjonowania obejmuje wyselekcjonowanie ulepszonego zmutowanego przeciwciała ludzkiego ze zbioru zmutowanych przeciwciał ludzkich w oparciu o (1) optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i (2) zwiększone wyrażanie w porównaniu do matrycowego przeciwciała ludzkiego. [0028] W innym wykonaniu, ujawnienie dostarcza sposobu identyfikowania i wytwarzania docelowego przeciwciała ludzkiego, gdzie sposób obejmuje wytwarzanie biblioteki ludzkich przeciwciał w gospodarzu do wytwarzania-komórce eukariotycznej przy zastosowaniu prezentacji przeciwciała na powierzchni komórek; przeszukiwania biblioteki pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; identyfikowania docelowego przeciwciała ludzkiego z biblioteki w oparciu co najmniej jedną określoną uprzednio właściwość, cechę lub aktywność; i wyrażania docelowego ludzkiego przeciwciała w tym samym gospodarzu do wytwarzania-komórce eukariotycznej, co w etapie wytwarzania do wytwarzania ludzkiego przeciwciała docelowego. W jednym aspekcie, docelowym przeciwciałem ludzkim jest przeciwciało pełnej długości. [0029] W innym wykonaniu ujawnienie dostarcza sposobu ewolucji ludzkiego przeciwciała w gospodarzu do wytwarzania, gdzie sposób obejmuje mutowanie ludzkiego przeciwciała matrycowego, aby wytworzyć zbiór zmutowanych ludzkich przeciwciał w gospodarzu do wytwarzania; i przeszukanie zbioru zmutowanych przeciwciał potomnych pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności. W jednym aspekcie, sposób obejmuje ponadto wyselekcjonowanie ulepszonego zmutowanego przeciwciała ludzkiego ze zbioru zmutowanych przeciwciał ludzkich, w oparciu o co najmniej jedną określoną uprzednio właściwość, cechę lub aktywność. W innym aspekcie, sposób obejmuje także wytwarzanie ulepszonego zmutowanego przeciwciała ludzkiego, obejmujące wyrażanie ulepszonego zmutowanego przeciwciała ludzkiego w tym samym gospodarzu do wytwarzania, co w etapie dokonywania mutacji. W innym aspekcie etap selekcji obejmuje ponadto wyselekcjonowanie ulepszonego zmutowanego przeciwciała ludzkiego ze zbioru zmutowanych przeciwciał ludzkich w oparciu (1) optymalizację co najmniej jednej określonej uprzednio właściwości,
7 1 2 cechy lub aktywności w porównaniu z ludzkim przeciwciałem matrycowym i (2) zmodyfikowane wyrażanie w porównaniu do wyrażania ludzkiego przeciwciała matrycowego. W jednym aspekcie wyrażaniem zmodyfikowanym jest zwiększone wyrażanie. [00] W innym wykonaniu ujawnienie dostarcza sposob ewolucji i wytwarzania ludzkiego przeciwciała w komórce gospodarza do wytwarzania, sposób obejmuje mutowanie ludzkiego przeciwciała matrycowego, aby wytworzyć zbiór zmutowanych ludzkich przeciwciał w gospodarzu do wytwarzania; przeszukiwanie zbioru zmutowanych przeciwciał potomnych pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i przeszukiwanie pod kątem zwiększonego wyrażania; selekcjonowanie ulepszonego zmutowanego przeciwciała ludzkiego ze zbioru zmutowanych przeciwciał ludzkich w oparciu o (1) optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności w porównaniu z ludzkim przeciwciałem matrycowym i (2) zmodyfikowane wyrażanie w porównaniu do wyrażania ludzkiego przeciwciała matrycowego; i wytwarzania ludzkiego przeciwciała obejmującego wyrażanie ulepszonego zmutowanego przeciwciała ludzkiego w tym samym gospodarzu do wytwarzania co w etapie dokonywania mutacji. [0031] W kolejnym wykonaniu, ujawnienie dostarcza sposobu ewolucji dla wzmocnionego wyrażania i wytwarzania przeciwciała ludzkiego w gospodarzu do wytwarzania-komórce eukariotycznej; sposób obejmuje mutowanie ludzkiego przeciwciała matrycowego, obejmujące wytwarzanie zmutowanych kodonów kodujących ludzkie przeciwciało matrycowe, aby wytworzyć zbiór zmutowanych ludzkich przeciwciał w gospodarzu do wytwarzania; przeszukiwanie zbioru zmutowanych przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i przeszukiwanie ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał pod kątem zwiększonego wyrażania w porównaniu z ludzkim przeciwciałem matrycowym; selekcję ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał w oparciu o (1) zachowanie lub optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i (2) zwiększone wyrażanie w porównaniu do przeciwciała matrycowego; i wytwarzania ulepszonego zmutowanego ludzkiego przeciwciała w tym samym gospodarzu do wytwarzania co w etapie mutowania. [0032] W jednym aspekcie etap przeszukiwania obejmuje przeszukiwanie zbioru zmutowanych ludzkich przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności i przeszukiwanie pod kątem zwiększonego wyrażania jednocześnie. KRÓTKI OPIS RYSUNKÓW [0033] 3 Fig. 1 przedstawia schemat metody CIAO kompleksowego wytwarzania i/lub selekcji, ewolucji i wyrażania przeciwciał terapeutycznych w gospodarzu eukariotycznym, takim jak gospodarzukomórkach ssaczych lub gospodarzu-komórkach drożdży, do wytwarzania w jednym systemie. Fig. 2 przedstawia jak kompleksowa ewolucja pozycyjna (CPE ) jest wykorzystywana do wytwarzania bazy danych swoistej dla cząsteczki (EvoMap ). 6
8 Fig. 3 pokazuje schemat kompleksowej syntezy pozycyjnej (CPS ), którą można stosować łącznie wobec ulepszonych mutantów z CPE. Fig. 4 pokazuje schemat kompleksowej inercyjnej ewolucji pozycyjnej (CPI ). Fig. przedstawia jedną kombinację metod ewolucji: wydłużony kwas nukleinowy z ewolucji CPI jest poddawany kompleksowej ewolucji pozycyjnej (CPE ) i stosowany do wytwarzania bazy danych swoistej dla cząsteczki (EvoMap ). Fig. 6 przedstawia schemat kompleksowej delecyjnej ewolucji pozycyjnej (CPD ). 1 Fig. 7 przedstawia kolejną kombinację metod ewolucji: skrócony kwas nukleinowy z ewolucji CPD jest poddawany kompleksowej ewolucji pozycyjnej (CPE ) i stosowany do wytwarzania bazy danych swoistej dla cząsteczki (EvoMap ). DEFINICJA TERMINÓW 2 3 [0034] W celu ułatwienia zrozumienia dostarczonych tu przykładów, opisane będą niektóre często występujące metody i/lub terminy. [003] Termin czynnik jest tu stosowany do oznaczenia polipeptydu, mieszaniny polipeptydów, zbioru zlokalizowanych przestrzennie związków (np., zbioru peptydowego VLSIPS, zbioru polinukleotydowego i/lub zbioru kombinatorycznego małych cząsteczek), makrocząsteczki biologicznej, bakteriofaga biblioteki peptydów prezentowanych na bakteriofagach, biblioteki przeciwciał (np. scfv) prezentowanych na bakteriofagach, biblioteki peptydów prezentowanych na polisomach lub ekstraktu wytworzonego z materiałów biologicznych, takich jak bakterie, rośliny, grzyby lub komórki lub tkanki zwierzęce (szczególnie ssacze). Czynniki są oceniane pod kątem ewentualnej aktywności jako środki przeciwnowotworowe, przeciwzapalne lub modulatory apoptozy przez włączenie do testów przesiewowych opisanych poniżej. Czynniki są oceniane pod względem potencjalnej aktywności jako swoiste inhibitory interakcji białka (to znaczy, czynnik, który wybiórczo hamuje interakcję wiązania między dwoma określonymi uprzednio polipeptydami, ale który zasadniczo nie zakłóca żywotności komórek) przez włączenie do testów przesiewowych opisanych poniżej. [0036] Termin aminokwas, stosowany w niniejszym opisie, odnosi się do dowolnego związku organicznego, który zawiera grupę aminową (--NH 2 ) i grupę karboksylową (--COOH); korzystnie bądź jako wolne grupy bądź, alternatywnie, po kondensacji jako część wiązań peptydowych. Dwadzieścia naturalnie zakodowanych alfa-aminokwasów tworzących polipeptyd jest zrozumiałe w tej dziedzinie i odnosi się do: alaniny (ala lub A), argininy (arg lub R), asparaginy (asn lub N), kwasu asparaginowego (asp lub D), cysteiny (cys lub C), kwasu gluataminowego (glu lub E), glutaminy (gln lub Q), glicyny (gly lub G), histydyny (his lub H), izoleucyny (ile lub I), leucyny (leu lub L), lizyny (lys lub K), metioniny 7
9 1 2 3 (met lub M), fenyloalaniny (phe lub F), proliny (pro lub P), seryny (ser lub S), treoniny (thr lub T), tryptofanu (trp lub W), tyrozyny (tyr lub Y) i waliny (val lub V). [0037] Termin amplifikacja oznacza, że liczba kopii polinukleotydu jest zwiększona. [0038] Termin przeciwciało, stosowany w niniejszym opisie, odnosi się do nienaruszonych cząsteczek immunoglobulin, jak również fragmentów cząsteczek immunoglobulin, takich jak Fab, Fab', (Fab')2, Fv i fragmentów SCA, które są zdolne do wiązania się z epitopem antygenu. Te fragmenty przeciwciał, które zachowują pewną zdolność do wybiórczego wiązania się z antygenem (np. antygenem polipeptydowym) przeciwciała, z którego pochodzą, można wytworzyć przy zastosowaniu dobrze znanych metod w tej dziedzinie (patrz np., Harlow i Lane, jak wyżej) i są opisane dalej poniżej. Przeciwciała te można stosować do izolowania preparatywnych ilości antygenu przy zastosowaniu chromatografii immunopowinowactwa. Różne inne zastosowania takich przeciwciał to diagnozowanie i/lub ocena stadium choroby (np., nowotworu) oraz terapeutyczne stosowanie do leczenia chorób, takich jak na przykład: nowotwór, choroba autoimmunologiczna, AIDS, choroba sercowo-naczyniowa, zakażenia i tym podobne. Przeciwciała chimeryczne, typu ludzkiego, humanizowane lub całkowicie ludzkie są szczególnie przydatne do podawania pacjentom-ludziom. [0039] Fragment Fab składa się z jednowartościowego fragmentu cząsteczki przeciwciała wiążącego antygen i może być wytwarzany przez trawienie całej cząsteczki przeciwciała enzymem papainą, z uzyskaniem fragmentu składającego się z nienaruszonego łańcucha lekkiego i części łańcucha ciężkiego. [00] Fragment Fab cząsteczki przeciwciała można otrzymać poprzez traktowanie całej cząsteczki przeciwciała pepsyną, a następnie redukcję, z uzyskaniem cząsteczki składającej się z nienaruszonego łańcucha lekkiego i części łańcucha ciężkiego. Dwa fragmenty Fab są uzyskiwane na cząsteczkę traktowanego w ten sposób przeciwciała. [0041] Fragment (Fab )2 przeciwciała można otrzymać poprzez traktowanie całej cząsteczki przeciwciała pepsyną bez późniejszej redukcji. Fragment (Fab )2 jest dimerem dwóch fragmentów Fab, utrzymywanych razem przez dwa mostki disiarczkowe. [0042] Fragment Fv jest definiowany jako fragment uzyskany poprzez zabiegi inżynierii genetycznej, zawierający region zmienny łańcucha lekkiego i region zmienny łańcucha ciężkiego, wyrażone jako dwa łańcuchy. [0043] Przeciwciało jednołańcuchowe ( SCA, ang. single chain antibody), jest uzyskaną poprzez zabiegi inżynierii genetycznej cząsteczką o pojedynczym łańcuchu, zawierającą region zmienny łańcucha lekkiego i region zmienny łańcucha ciężkiego, połączone poprzez odpowiedni, elastyczny łącznik polipeptydowy. [0044] Termin biopodobny, nazywany również ang. follow-on biologic, odnosi się do oficjalnie zatwierdzonych nowych wersji innowacyjnych produktów biofarmaceutycznych, po wygaśnięciu patentu lub wyłączności. [004] Termin komórka gospodarza do wytwarzania lub gospodarz do wytwarzania odnosi się do linii komórkowej wykorzystywanej do produkcji lub wytwarzania przeciwciał. Termin ten obejmuje komórki eukariotyczne, takie jak komórki ssacze, w tym, ale bez ograniczania się do tego, linie komórkowe człowieka, myszy, chomika, szczura, małpy oraz linie komórkowe drożdży, owadów i 8
10 1 2 3 roślin. Alternatywnie, mogą być również wykorzystane komórki prokariotyczne. W jednym aspekcie, gospodarz do wytwarzania-komórka ssaka jest wybrany spośród przedstawicieli grupy składającej się z komórek fibroblastów myszy 3T3; komórek fibroblastów chomika syryjskiego BHK21; komórek nabłonkowych psa MDCK; komórek nabłonkowych człowieka Hela; komórek nabłonkowych kanguroszczura PtK1; komórek plazmatycznych myszy SP2/0; i komórek plazmatycznych myszy NS0; komórek embrionalnych nerki człowieka HEK 293; komórek nerek małpy COS; komórek jajnika chomika chińskiego CHO, CHO-S; komórek embrionalnych myszy R1; komórek embrionalnych myszy E14.1; komórek embrionalnych człowieka H1; komórek embrionalnych człowieka H9; komórek embrionalnych człowieka PER C.6. W innym aspekcie, komórką gospodarza do wytwarzania jest linia komórkowa GS-NS0 lub GS-CHOK1. W innym aspekcie, komórkę gospodarza do wytwarzania wybiera się spośród komórek drożdży S. cerevisiae i komórek drożdży Pichia. [0046] Cząsteczką, która ma właściwości chimeryczne jest cząsteczka, która jest: 1) w części homologiczna i w części heterologiczna do pierwszej cząsteczki odnośnikowej; podczas gdy 2) w tym samym czasie będąc w części homologiczną i w części heterologiczną do drugiej cząsteczki odnośnikowej; bez 3) wykluczania możliwości bycia samym czasie w części homologiczną i w części heterologiczną do jeszcze jednej lub więcej dodatkowych cząsteczek odnośnikowych. W nieograniczającym wykonaniu, cząsteczkę chimeryczną można wytwarzać przez łączenie asortymentu częściowych sekwencji cząsteczek. W nieograniczającym wykonaniu polinukleotydową cząsteczkę chimeryczną można wytworzyć poprzez zsyntetyzowanie chimerycznego polinukleotydu przy zastosowaniu wielu matryc molekularnych, tak, że powstający w rezultacie chimeryczny polinukleotyd ma właściwości wielu matryc. [0047] Termin spokrewniony, stosowany w niniejszym opisie, odnosi się do sekwencji genowej, która jest ewolucyjnie i funkcjonalnie spokrewniona pomiędzy gatunkami. Na przykład, ale bez ograniczania, w ludzkim genomie ludzki gen CD4 jest genem spokrewnionym z genem 3d4 u myszy, ponieważ sekwencje i struktury tych dwóch genów wskazują, że są one wysoce homologiczne i oba geny kodują białko, które pełni funkcję w przekazywaniu sygnałów dla aktywacji komórek T przez rozpoznawanie antygenu przez MHC ograniczone do klasy II. [0048] Termin skala komercyjna oznacza wytwarzanie przeciwciał w skali odpowiedniej do odsprzedaży. [0049] Okno porównywania, stosowane w niniejszym opisie, odnosi się do pojęciowego odcinka co najmniej sąsiadujących pozycji nukleotydowych, gdzie sekwencję polinukleotydową można porównać z sekwencją odnośnikową złożoną z co najmniej sąsiadujących nukleotydów, gdzie część sekwencji polinukleotydowej w oknie porównywania może zawierać addycje lub delecje (tj. przerwy) wynoszące procent lub mniej w porównaniu z sekwencją odnośnikową (która nie zawiera addycji lub delecji) w celu optymalnego przyrównania dwóch sekwencji. Optymalne przyrównanie sekwencji do ustawiania okna porównywania można przeprowadzić przy zastosowaniu algorytmu homologii lokalnej według Smitha i Watermana (1981) Adv. Appl. Math. 2: 482, przy zastosowaniu algorytmu przyrównywania homologii według Needlemen i Wuncsch J. Mol. Biol. 48: 443 (1970), przez poszukiwanie podobieństwa metodą według Pearson i Lipman, Proc. Natl. Acad. Sci. (USA) 8: 2444 (1988), poprzez skomputeryzowane implementacje tych algorytmów (GAP, BESTFIT, FASTA i 9
11 1 2 3 TFASTA w Wisconsin Genetics Software Package Release 7.0, Genetics Computer Group, 7 Science Dr., Madison, Wis.) lub przez przeglądanie, i wybiera się najlepsze przyrównanie (tj., dające w rezultacie najwyższy procent homologii w oknie porównywania) wytworzone przez różne metody. [000] Stosowany tu termin region determinujący dopasowanie i CDR odnosi się do terminu uznanego w tej dziedzinie, jak przedstawiono przez Kabat i Chothia. Definicje CDR są generalnie znane jako regiony superzmienne albo pętle hiperzmienne (Chothia i Leks, 1987; Clothia i wsp., 1989;. Kabat i wsp., 1987; oraz Tramontano i wsp., 1990). Domeny regionu zmiennego zawierają typowo około -11 amino-końcowych aminokwasów z naturalnie występującego łańcucha immunoglobuliny (np., aminokwasy 1-1), chociaż domeny zmienne do pewnego stopnia krótsze lub dłuższe nadają się także do wytwarzania przeciwciał jednołańcuchowych. CDR są częściami immunoglobulin, które wyznaczają swoistość tych cząsteczek i tworzą kontakt ze swoistym ligandem. CDR są najbardziej zmienną częścią cząsteczki i przyczyniają się do różnorodności tych cząsteczek. Istnieją trzy regiony CDR, CDR1, CDR2 i CDR3, w każdej domenie V. CDR-H przedstawia region CDR zmiennego łańcucha ciężkiego, a CDR-L odnosi się do regionu CDR zmiennego łańcucha lekkiego. H oznacza zmienny łańcuch ciężki, a L oznacza zmienny łańcuch lekki. Regiony CDR regionu pochodzącego z Ig mogą być określane w sposób opisany w Kabat (1991)., Sequences of Proteins of Immunological Interest, wyd.., Publikacja NIH nr U.S. Department of Health and Human Services, Chothia (1987) J. Mol. Biol. 196, oraz Chothia (1989) Nature, 342, [001] Termin kompleksowy jest tu stosowany w odniesieniu do techniki ewolucji, gdzie każda możliwa zmiana jest dokonywana w każdej pozycji matrycowego polinukleotydu lub matrycowego polipeptydu i polinukleotyd lub polipeptyd jest testowany w celu potwierdzenia, że zamierzone zmiany zostały dokonane. [002] Konserwatywne podstawienia aminokwasowe odnoszą się do wzajemnej wymienności reszt mających podobne łańcuchy boczne. Na przykład, grupą aminokwasów mających alifatyczne łańcuchy boczne jest glicyna, alanina, walina, leucyna i izoleucyna; grupą aminokwasów mających alifatyczno-hydroksylowe łańcuchy boczne jest seryna i treonina; grupą aminokwasów posiadających łańcuchy boczne zawierające amidy jest asparagina i glutamina; grupą aminokwasów mających aromatyczne łańcuchy boczne jest fenyloalanina, tyrozyna i tryptofan; grupą aminokwasów mających zasadowe łańcuchy boczne jest lizyna, arginina i histydyna; a grupą aminokwasów mających łańcuchy boczne zawierające siarkę jest cysteina i metionina. Korzystnymi grupami konserwatywnych podstawień aminokwasowych są: walina-leucyna-izoleucyna, fenyloalanina-tyrozyna, lizyna-arginina, alanina-walina i asparagina-glutamina. [003] Termin odpowiada jest używany tu w znaczeniu, że sekwencja polinukleotydowa jest homologiczna (tj. jest identyczna, nie będąc ściśle ewolucyjnie pokrewna) do całości lub części odnośnikowej sekwencji polinukleotydowej, lub że sekwencja polipeptydowa jest identyczna z odnośnikową sekwencją polipeptydową. W przeciwieństwie do tego, termin komplementarny do jest używany tu w znaczeniu, że sekwencja komplementarna jest homologiczna do całej lub części odnośnikowej sekwencji polinukleotydowej. Dla ilustracji, sekwencja nukleotydowa TATAC odpowiada odnośnikowi TATAC i jest komplementarna do sekwencji odnośnikowej GTATA.
12 1 2 3 [004] Stosowany tu termin zdefiniowana sekwencja zrębowa odnosi się do zbioru określonych sekwencji, które zostały wybrane w nieprzypadkowy sposób, zwykle na podstawie danych eksperymentalnych lub danych strukturalnych; na przykład, zdefiniowana sekwencja zrębowa może obejmować zbiór sekwencji aminokwasowych, które, jak się przewiduje, tworzą strukturę β-kartki lub może zawierać powtórzenia motywu heptad suwaka leucynowego, domenę palca cynkowego, wśród innych wariacji. Zdefiniowany kernel to zbiór sekwencji, które obejmują ograniczony zakres zmienności. Podczas gdy (1) całkowicie losowa sekwencja -merowa z typowych aminokwasów może być dowolną z () sekwencji, a (2) pseudolosowa sekwencja -merowa z zwykłych aminokwasów może być dowolną z () sekwencji, ale będzie wykazywać odchylenie dla pewnych reszt w pewnych pozycjach i/lub całkowite, (3) zdefiniowany kernel sekwencji jest podzbiorem sekwencji, jeśli pozycją dla każdej reszty może być dowolny z dopuszczalnych typowych aminokwasów (i lub dopuszczalne niekonwencjonalne amino/iminokwasy). Zdefiniowany kernel sekwencji generalnie zawiera zmienne lub niezmienne pozycje reszt w i/lub zawiera zmienne pozycje reszt, które mogą zawierać resztę wybraną z określonego podzbioru reszt aminokwasowych), i tym podobne, bądź odcinkowo, bądź na całej długości danej wybranej sekwencji będącej przedstawicielem biblioteki. Zdefiniowane kernele sekwencji mogą odnosić się bądź do sekwencji aminokwasowej, bądź sekwencji polinukleotydowej. Jako ilustracja, a nie ograniczenie, sekwencje (NNK) i (NNM), gdzie N oznacza A, T, G lub C; K oznacza G lub T; a M oznacza lub C, są zdefiniowanymi kernelami sekwencji. [00] Termin deimmunizacja, tak jak jest tu stosowany, odnosi się do wytwarzania wariantu matrycowej cząsteczki wiążącej, która jest zmodyfikowana w porównaniu do wyjściowej cząsteczki typu dzikiego z uzyskaniem wariantu nieimmunogennego lub mniej immunogennego u ludzi. Cząsteczki deimmunizowane według wynalazku odnoszą się do przeciwciał lub ich części (takich jak zręby i/lub CDR) pochodzenia innego niż ludzkie. Odpowiednimi przykładami są przeciwciała lub ich fragmenty opisane w US 4,361,49. Termin deimmunizowane dotyczy także cząsteczek, które wykazują obniżoną tendencję do wytwarzania epitopów komórek T. Zgodnie z tym wynalazkiem, termin obniżona tendencja do wytwarzania epitopów komórek T odnosi się do usuwania epitopów komórek T, prowadzących do swoistej aktywacji komórek T. [006] Ponadto, zmniejszona tendencja do wytwarzania epitopów komórek T oznacza podstawienie aminokwasowe uczestniczące w tworzeniu epitopów komórek T, to znaczy podstawienie aminokwasowe, które jest niezbędne dla utworzenia epitopu komórek T. Innymi słowy, zmniejszona tendencja do wytwarzania epitopów komórek T dotyczy zmniejszonej immunogenności lub zmniejszonej zdolności do indukowania proliferacji komórek T niezależnej od antygenu. Ponadto, zmniejszona tendencja do wytwarzania epitopów komórek T odnosi się do deimmunizacji, co oznacza utratę lub zmniejszenie potencjalnych epitopów komórek T w sekwencji aminokwasowej antygenu indukującą proliferację komórek T niezależną od antygenu. [007] Termin epitop komórki T, tak jak jest tu stosowany, odnosi się do krótkich sekwencji peptydowych, które mogą być uwalniane w czasie degradacji peptydów, polipeptydów lub białek w komórkach, a następnie prezentowane przez cząsteczki głównego kompleksu zgodności tkankowej (MHC) w celu wywołania aktywacji komórek T; patrz m. in. WO 02/ Dla peptydów 11
13 1 2 3 prezentowanych przez MHC klasy II, taka aktywacja komórek T może następnie wywołać odpowiedź przeciwciał poprzez bezpośrednią stymulację komórek B do wytwarzania tych przeciwciał. [008] Trawienie DNA dotyczy cięcia katalitycznego DNA enzymem restrykcyjnym, który działa tylko w miejscu określonych sekwencji w DNA. Używane tu rozmaite enzymy restrykcyjne są dostępne komercyjnie i ich warunki reakcji, kofaktory i inne wymagania używano w taki sposób, jak jest to wiadome specjaliście w tej dziedzinie. Do celów analitycznych, zazwyczaj stosuje się 1 g plazmidu lub fragmentu DNA z około 2 jednostkami enzymu w około l roztworu buforu. Dla celów izolowania fragmentów DNA do konstrukcji plazmidu, zwykle od do 0 g DNA trawi się przy zastosowaniu do jednostek enzymu w dużej objętości. Odpowiednie bufory i ilości substratów dla poszczególnych enzymów restrykcyjnych są określone przez producenta. Zwykle stosuje się czasy inkubacji około 1 godzinę w 37 C, ale mogą się one zmieniać zgodnie z instrukcjami dostawcy. Po strawieniu, mieszaninę reakcyjną poddaje się elektroforezie bezpośrednio na żelu w celu wyizolowania pożądanego fragmentu. [009] Termin tasowanie DNA jest tu stosowany dla wskazania rekombinacji pomiędzy zasadniczo homologicznymi, ale nieidentycznymi sekwencjami. W niektórych wykonaniach tasowanie DNA może obejmować crossing over poprzez rekombinację niehomologiczną, taką jak przy zastosowaniu systemów cer/lox i/lub flp/frt i tym podobnych. Tasowanie DNA może być losowe lub nielosowe. [0060] Jak stosowany w niniejszym wynalazku, termin epitop oznacza determinantę antygenową na antygenie, takim jak polipeptyd fitazy, z którą wiąże się paratop przeciwciała, takiego jak przeciwciało swoiste wobec fitazy. Determinanty antygenowe zazwyczaj składają się z chemicznie aktywnych powierzchniowych ugrupowań cząsteczek, takich jak aminokwasy lub cukrowe łańcuchy boczne, i mogą mieć specyficzne trójwymiarowe cechy strukturalne, jak również specyficzną charakterystykę ładunku. Tak jak jest tu stosowany, termin epitop oznacza tę część antygenu lub innej makrocząsteczki, zdolnej do tworzenia oddziaływania wiążącego, która oddziałuje z częścią wiążącą regionu zmiennego przeciwciała. Typowo, takie oddziaływanie wiążące przejawia się jako kontakt międzycząsteczkowy z jedną lub więcej reszt aminokwasowych w CDR. [0061] Termin ewolucja odnosi się do zmian w co najmniej jednej właściwości, cesze lub aktywności zmodyfikowanego genetycznie lub syntetycznie przeciwciała w porównaniu z przeciwciałem matrycowym. [0062] Terminy fragment, pochodna i analog, kiedy odnoszą się do polipeptydu odnośnikowego obejmują polipeptyd, który zachowuje co najmniej jedną funkcję lub aktywność biologiczną, która jest co najmniej zasadniczo taka sama, jak polipeptydu odnośnikowego. Ponadto, przykładem terminów fragment, pochodna i analog jest cząsteczka w postaci proformy, na przykład probiałka o niskiej aktywności, które może być modyfikowane przez cięcie w celu wytworzenia dojrzałej postaci o znacznie wyższej aktywności. [0063] Dostarczony jest tu sposób wytwarzania z polipeptydu matrycowego zbioru polipeptydów potomnych, w których w każdej pozycji aminokwasowej reprezentowany jest pełen zakres podstawień pojedynczego aminokwasu. Stosowany tu termin pełen zakres podstawień pojedynczego aminokwasu jodnosi się do naturalnie kodowanych naturalnie kodowanych alfaaminokwasów tworzących polipeptyd, jak tu opisano. 12
14 1 2 3 [0064] Termin gen oznacza fragment DNA uczestniczący w wytwarzaniu łańcucha polipeptydowego; obejmuje regiony poprzedzające i następujące po regionie kodującym ( UTR i 3 UTR, ang. leader and trailer), jak również sekwencje przerywnikowe (introny) pomiędzy poszczególnymi odcinkami kodującymi (eksonami). [006] Niestabilność genetyczna, stosowana w niniejszym opisie, odnosi się do naturalnej tendencji wysoce powtarzalnych sekwencji do utraty w procesie zdarzeń redukcyjnych generalnie obejmujących uproszczenie sekwencji poprzez utratę sekwencji powtarzających się. Delecje zazwyczaj mają tendencję do obejmowania utraty jednej kopii powtórzenia i wszystkiego pomiędzy powtórzeniami. [0066] Termin heterologiczna oznacza, że jedna sekwencja jednoniciowego kwasu nukleinowego nie ma zdolności do hybrydyzowania z inną sekwencją jednoniciowego kwasu nukleinowego lub jej dopełnieniem. A zatem regiony heterologii oznaczają, że obszary polinukleotydów lub polinukleotydy mają obszary lub regiony w swojej sekwencji, które nie ma zdolności do hybrydyzowania z innym kwasem nukleinowym lub polinukleotydem. Takimi regionami lub obszarami są na przykład regiony mutacji. [0067] Termin homologiczny (ang. homologous lub homeologous ) oznacza, że jedna sekwencja jednoniciowego kwasu nukleinowego może hybrydyzować z komplementarną sekwencją jednoniciowego kwasu nukleinowego. Stopień hybrydyzacji może zależeć od wielu czynników, w tym od ilości identyczności sekwencji i warunków hybrydyzacji, takich jak temperatura i stężenie soli, jak omówiono później. Korzystniej, jeżeli region identyczności jest większy niż par zasad (bp), korzystniej w region identyczności jest większy niż bp. [0068] Termin humanizowane jest stosowany do opisu przeciwciał, gdzie regiony determinujące dopasowanie (CDR) ze zwierzęcia-ssaka, na przykład myszy, są połączone z ludzkim regionem zrębowym. Często polinukleotydy kodujące wyizolowane CDR będą wszczepiane do polinukleotydów kodujących odpowiedni region zrębowy regionu zmiennego (i ewentualnie regiony stałe) z wytworzeniem polinukleotydów kodujących całe przeciwciała (np., humanizowane lub całkowicie ludzkie), fragmenty przeciwciał i tym podobne. W innym aspekcie, oprócz przeciwciał mysich, mogą być humanizowane inne gatunki, takie jak, na przykład, inny gryzoń, wielbłąd, królik, pies, kot, świnia, koń, krowa, ryba, lama i rekin. W szerokim aspekcie, dowolny gatunek, który wytwarza przeciwciała można zastosować do wytwarzania przeciwciał humanizowanych. Ponadto, przeciwciałami według wynalazku mogą być przeciwciała chimeryczne, typu ludzkiego, humanizowane lub całkowicie ludzkie, w celu zmniejszenia ich potencjalnej antygenowości bez zmniejszania ich powinowactwa do ich celu. Przeciwciała chimeryczne, typu ludzkiego, jak i humanizowane są generalnie opisane tej dziedzinie. Poprzez wprowadzenie tak mało obcej sekwencji jak to możliwe, antygenowość hybrydowego przeciwciała jest zmniejszana. Wytwarzanie takich przeciwciał hybrydowych można przeprowadzić przy zastosowaniu metod znanych w tej dziedzinie. [0069] W alternatywnym aspekcie, przeciwciała ludzkie lub mysie są przystosowane do innego gatunku biorcy, takiego jak gatunek zagrożony, w celu dostarczenia środków leczniczych dla gatunku zwierzęcia, chroniąc je od negatywnej odpowiedzi immunologicznej. W tym aspekcie, regiony zrębowe z gatunku biorcy są stosowane w połączeniu z CDR z przeciwciał znanego lub drugiego gatunku. 13
15 1 2 3 [0070] Region zmienny łańcucha lekkiego lub ciężkiego immunoglobuliny składa się z regionu zrębowego przerywanego przez trzy regiony hiperzmienne, zwane również CDR. Zakres regionu zrębowego i CDR zostały wyraźnie zdefiniowane (patrz Sequences of Proteins of Immunological Interest, Kabat i wsp., 1987). Sekwencje regionów zrębowych różnych łańcuchów lekkich lub ciężkich są względnie konserwowane w obrębie gatunku. Termin ludzki region zrębowy, stosowany w niniejszym opisie, to region zrębowy, który jest zasadniczo identyczny (8 lub więcej, zazwyczaj 90-9 lub więcej) z regionem zrębowym naturalnie występującej immunoglobuliny ludzkiej. Region zrębowy przeciwciała, które jest połączony z regionami zrębowymi składowych łańcuchów lekkich i ciężkich, służy do usytuowania i ustawienia CDR. Regiony CDR są przede wszystkim odpowiedzialne za wiązanie się z epitopem antygenu. Zgodnie z tym wynalazkiem, region zrębowy odnosi się do regionu w domenie V (domenie VH lub VL) immunoglobuliny, która dostarcza szkieletu białkowego dla hiperzmiennych regionów determinujących dopasowanie (CDR), które stykają się z antygenem. W każdej domenie V istnieją cztery regiony zrębowe oznaczone jako FR1, FR2, FR3 i FR4. Region zrębowy 1 obejmuje region od końca N domeny V do początku CDR1, region zrębowy 2 odnosi się do regionu pomiędzy CDR1 i CDR2, region zrębowy 3 obejmuje region między CDR2 i CDR3, a region zrębowy 4 oznacza region od końca CDR3 do końca C domeny V; patrz, między innymi, Janeway, Immunobiology, Garland Publishing, 01, wyd.. A zatem, regiony zrębowe obejmują wszystkie regiony poza regionami CDR w domenach VH lub VL. [0071] Osoba biegła w dziedzinie jest w stanie łatwo wydedukować z danej sekwencji regiony zrębowe i CDR; patrz, Kabat (1991) Sequences of Proteins of Immunological Interest, th edit., Publikacja NIH nr U.S. Department of Health and Human Services, Chothia (1987) J. Mol. Biol. 196, oraz Chothia (1989) Nature, 342, [0072] Korzyści z tego wynalazku rozszerzają się na zastosowania przemysłowe (lub procesy przemysłowe), który to termin jest stosowany aby objąć zastosowania właściwe - przemysłowe komercyjne (lub po prostu przemysłowe), jak i niekomercyjne przemysłowe (np. badania biomedyczne w instytucjach non-profit). Odpowiednie zastosowania obejmują te w zakresie diagnozowania, medycyny, rolnictwa, wytwarzania i nauki. [0073] Termin identyczny lub identyczność oznacza, że dwie sekwencje kwasów nukleinowych mają taką samą sekwencję lub sekwencję komplementarną. A zatem obszary identyczności oznaczają, że regiony lub obszary polinukleotydu lub cały polinukleotyd są identyczne lub komplementarne do obszarów innego polinukleotydu lub danego polinukleotydu. [0074] Termin wyizolowany oznacza, że materiał jest pobierany ze swojego wyjściowego środowiska (na przykład środowiska naturalnego, jeżeli występuje naturalnie). Na przykład, naturalnie występujący polinukleotyd lub enzym obecny w żywym zwierzęciu nie jest wyizolowany, ale ten sam polinukleotyd lub enzym, oddzielony od niektórych lub wszystkich współistniejących materiałów w systemie naturalnym, jest wyizolowany. Takie polinukleotydy mogą być częścią wektora i/lub takie polinukleotydy lub enzymy mogą być częścią kompozycji i nadal być wyizolowane z tego względu, że taki wektor lub kompozycja nie jest częścią ich naturalnego środowiska. [007] Wyizolowany kwas nukleinowy ma oznaczać, kwas nukleinowy, np., cząsteczkę DNA lub RNA, która nie sąsiaduje bezpośrednio z sekwencjami flankującymi po stronie i 3, z którymi 14
16 1 2 3 normalnie sąsiaduje bezpośrednio, gdy jest obecna w naturalnie występującym genomie organizmu, z którego pochodzi. Termin ten opisuje zatem, na przykład, kwas nukleinowy, który jest włączony do wektora, takiego jak plazmid lub wektor wirusowy; kwas nukleinowy, który jest włączony do genomu komórki heterologicznej (lub genomu komórki homologicznej, ale w miejscu innym niż to, w którym naturalnie występuje); oraz kwas nukleinowy, który istnieje jako oddzielna cząsteczka, na przykład, fragment DNA otrzymany przez amplifikację w reakcji PCR bądź trawienie enzymem restrykcyjnym, lub cząsteczkę RNA wytworzoną przez transkrypcję in vitro. Termin opisuje również rekombinowany kwas nukleinowy, który stanowi część hybrydowego genu kodującego dodatkowe sekwencje polipeptydowe, które mogą być stosowane, na przykład, do wytwarzania fuzyjnego polipeptydu. [0076] Ligacja odnosi się do procesu tworzenia wiązań fosfodiestrowych między dwoma fragmentami dwuniciowego kwasu nukleinowego (Maniatis i wsp., 1982, str. 146). O ile nie wskazano inaczej, ligację można przeprowadzić stosując znane bufory i warunki, z jednostkami ligazy DNA T4 ( ligazy ) na 0, g w przybliżeniu równomolowych ilości fragmentów DNA, które podlegają ligacji. [0077] Termin łącznik lub odstępnik, stosowany w niniejszym opisie, odnosi się do cząsteczki lub grupy cząsteczek, które łączą dwie cząsteczki, takie jak białko wiążące DNA i losowy peptyd, i służą do umieszczania dwóch cząsteczek w korzystnej konfiguracji, na przykład tak, że peptyd losowy może wiązać się z receptorem z minimalną przeszkodą steryczną od białka wiążącego DNA. [0078] Termin prezentacja na powierzchni komórki ssaczej odnosi się do techniki, dzięki której przeciwciało lub fragment przeciwciała jest wyrażany i prezentowany na powierzchni komórki ssakagospodarza do celów przeszukiwania; na przykład poprzez przeszukiwanie pod kątem swoistego wiązania antygenów przez kombinację kulek magnetycznych i sortowania komórek aktywowanego fluorescencją. W jednym aspekcie, ssacze wektory ekspresyjne stosuje się do jednoczesnej ekspresji immunoglobulin zarówno wydzielanych, jak i w postaci związanej z powierzchnią komórek, jak w DuBridge i wsp., US 09/ W innym aspekcie, techniki Gao i wsp. stosuje się dla wektora wirusowego kodującego bibliotekę przeciwciał lub fragmentów przeciwciał prezentowanych na błonach komórkowych, po wyrażaniu w komórce, tak jak w Gao i wsp., US 07/ Znana jest powierzchniowa prezentacja całych IgG na komórkach ssaków. Na przykład, Akamatsuu i wsp. opracowali wektor do prezentacji na powierzchni komórki ssaczej, odpowiedni do bezpośredniego izolowania cząsteczek IgG na podstawie ich powinowactwa do wiązania antygenu i aktywności biologicznej. Stosując wektor episomalny pochodzący z wirusa Epsteina-Barra, dokonano prezentacji bibliotek przeciwciał jako całych cząsteczek IgG na powierzchni komórek i przeszukiwano pod kątem swoistego wiązania antygenu poprzez kombinację kulek magnetycznych i sortowanie komórek aktywowane fluorescencją. Plazmidy kodujące przeciwciała o pożądanych właściwościach wiązania odzyskano z sortowanych komórek i przekształcono w postać do wytwarzania rozpuszczalnej IgG. Akamatsuu i wsp. J. Immunol. Methods (1-2):-2. Ho i wsp. zastosowali ludzkie embrionalne komórki nerkowe 293T, które są powszechnie stosowane do przejściowego wyrażania białka w celu prezentacji na powierzchni komórki jednołańcuchowych przeciwciał Fv w celu dojrzewania powinowactwa. Komórki wyrażające rzadkie zmutowane przeciwciało o wyższym powinowactwie wzbogacono 2-krotnie poprzez jeden przebieg sortowania komórek spośród dużego nadmiaru komórek wyrażających przeciwciało WT z nieznacznie niższym powinowactwem. Ponadto 1
17 1 2 3 uzyskano wysoko wzbogaconego mutanta ze zwiększonym powinowactwem wiązania do CD22 po pojedynczej selekcji kombinatorycznej biblioteki, randomizując wewnętrzne gorące miejsce przeciwciała. Ho i wsp. Isolation of anti-cd22 Fv with high affinity by Fv display on human cells, Proc Natl Acad Sci USA 06, czerwiec ; 3(2): [0079] Beerli i wsp. zastosowali komórki B swoiste dla antygenu będącego przedmiotem zainteresowania, które były bezpośrednio wyizolowane z komórek jednojądrzastych krwi obwodowej (PBMC, ang. peripheral blood mononuclear cells) od dawców ludzkich. Z puli komórek B wytwarza się rekombinowane, swoiste dla antygenu biblioteki jednołańcuchowych Fv (scfv, ang. single-chain Fv) i przeszukuje przy zastosowaniu prezentacji na powierzchni komórek ssaczych przy użyciu systemu ekspresyjnego w oparciu o wirusa Sindbis. Metoda ta pozwala na izolowanie przeciwciał swoistych dla antygenu poprzez jedną rundę FACS. Z pozytywnych klonów izolowano regiony zmienne (VR, ang. variable region) łańcuchów ciężkich (HC, ang. heavy chain) i łańcuchów lekkich (LS, ang. light chain) i wytworzono rekombinowane w pełni ludzkie przeciwciała jako całe IgG lub fragmenty Fab. W ten sposób, wyizolowano kilka hiperzmutowanych przeciwciał o wysokim powinowactwie, wiążących cząstki wirusopodobne (VLP, ang. virus like particle) Qβ, modelowy wirusowy antygen, jak również przeciwciała swoiste dla nikotyny. Wszystkie przeciwciała wykazywały wysoki poziom wyrażania w hodowlach komórkowych. Ludzkie mab (przeciwciała monoklonalne, ang. monoclonal antibodies) swoiste dla nikotyny poddano walidacji w modelu mysim. Beerli i wsp., Isolation of human monoclonal antibodies by mammalian cell display, Proc Natl Acad Sci USA września; (38): [0080] Znana jest również prezentacja na powierzchni komórek drożdży, patrz na przykład, Kondo i Ueda 04, Yeast cell-surface display-applications of molecular display, Appl. Microbiol. Biotechnol., 64(1): 28-, dokument ten opisuje na przykład system oparty o inżynierię powierzchni komórki z użyciem drożdży Saccharomyces cerevisiae. Kilka reprezentatywnych systemów prezentacji do ekspresji w drożdżach S. cerevisiae jest opisanych w Lee i wsp., 03, Microbial cell-surface display, TRENDS in Bitechnol. 21(1): 4-2. Również Boder i Wittrup 1997, Yeast surface display for screening combinatorial polypeptide libraries, Nature Biotechnol., 1(6): 3. [0081] Termin wytwarzanie odnosi się do wytwarzania przeciwciała w wystarczającej ilości, aby umożliwić co najmniej badania kliniczne fazy I terapeutycznego przeciwciała lub ilości wystarczającej do zatwierdzenia regulacyjnego przeciwciała diagnostycznego. [0082] Termin mutacja zmiany sensu dotyczy mutacji punktowej, gdzie zmieniony jest pojedynczy nukleotyd, co skutkuje kodonem, który koduje inny aminokwas. Mutacje, które zmieniają aminokwas w kodon stop są nazywane mutacjami nonsensownymi. [0083] Stosowana w niniejszym opisie, właściwość molekularna, która ma podlegać ewolucji obejmuje odniesienie do cząsteczek składających się z sekwencji polinukleotydowej, cząsteczek składających się z sekwencji polipeptydowej i cząsteczek składających się w części z sekwencji polinukleotydowej i w części z sekwencji polipeptydowej. Szczególnie odpowiednie -- ale w żaden sposób nie ograniczające -- przykłady właściwości molekularnych, które mają podlegać ewolucji, obejmują aktywności enzymatyczne w określonych warunkach, takich jak te dotyczące temperatury; soli; ciśnienia; ph; i stężenia glicerolu, DMSO, detergentów, i/lub innych rodzajów cząsteczek, z 16
18 1 2 3 którymi dochodzi do kontaktu w środowisku reakcji. Dodatkowe szczególnie odpowiednie, ale w żaden sposób nieograniczające przykłady właściwości molekularnych, które mają podlegać ewolucji, obejmują stabilność np. ilość pozostającej właściwości cząsteczki, która jest obecna po określonym czasie ekspozycji na określone środowisko, takie, jakie można spotkać podczas przechowywania. [0084] Termin mutowanie odnosi się do dokonywania mutacji w sekwencji kwasu nukleinowego; w przypadku, gdy mutacja występuje w regionie kodującym genu, będzie to prowadzić do zmiany kodonu, która może prowadzić lub nie prowadzić do zmiany aminokwasowej. [008] Termin mutacje oznacza zmiany w sekwencji kwasu nukleinowego typu dzikiego lub zmiany w sekwencji peptydu lub polipeptydów. Takimi mutacjami mogą być mutacje punktowe, takie jak tranzycje lub transwersje. Mutacjami mogą być delecje, insercje lub duplikacje. [0086] Stosowany w niniejszym opisie zdegenerowana sekwencja nukleotydowa N, N, G/T reprezentuje 32 możliwe tryplety, gdzie N może być A, C, G lub T. [0087] Stosowany w niniejszym opisie zdegenerowana sekwencja nukleotydowa N, N, N reprezentuje 64 możliwe tryplety, gdzie N może być A, C, G lub T. [0088] Termin występujący naturalnie, tak jak tu używany przy stosowaniu względem obiektu odnosi się do faktu, że obiekt może znajdować się w naturze. Na przykład, sekwencja polipeptydowa lub polinukleotydowa, która jest obecna w organizmie (w tym wirusach), która może być wyizolowana ze źródła w naturze i która nie została celowo zmodyfikowana przez człowieka w laboratorium jest sekwencją występującą naturalnie. Generalnie, termin występujący naturalnie odnosi się do takiego obiektu, jak obecny u osobnika niepatologicznego (niechorego), tak jak byłoby to typowe dla danego gatunku. [0089] Stosowana w niniejszym opisie, cząsteczka kwasu nukleinowego składa się z co najmniej jednej zasady lub jednej pary zasad, w zależności od tego, czy jest, odpowiednio, jednoniciowa czy dwuniciowa. Ponadto, cząsteczka kwasu nukleinowego może należeć wyłącznie lub chimerycznie do dowolnej grupy cząsteczek zawierających nukleotydy, czego przykładem są, ale bez ograniczania się do tego, następujące grupy cząsteczek kwasu nukleinowego: RNA, DNA, genomowe kwasy nukleinowe, niegenomowe kwasy nukleinowe, kwasy nukleinowe występujące naturalnie i niewystępujące naturalnie oraz kwasy nukleinowe syntetyczne. Obejmuje to, jako nieograniczający przykład, kwasy nukleinowe związane z dowolnym organellum, takim jak mitochondria, rybosomalny RNA oraz cząsteczki kwasów nukleinowych składające się chimerycznie z jednego lub więcej składników, które nie są naturalnie występującymi wraz ze składnikami występującymi naturalnie. [0090] Dodatkowo, cząsteczka kwasu nukleinowego może zawierać w części jeden lub więcej składników nienukleotydowych, czego przykładem są, ale bez ograniczania się do tego, aminokwasy i cukry. A zatem, jako przykład, ale nie ograniczenie, rybozym, która jest w części oparty na nukleotydach, a w części oparty na białkach uważa się za cząsteczkę kwasu nukleinowego. [0091] Ponadto, jako przykład, ale nie ograniczenie, cząsteczka kwasu nukleinowego, która jest wyznakowana wykrywalnym ugrupowaniem, takim jak radioaktywny lub, alternatywnie, nieradioaktywny znacznik, jest również uważana za cząsteczkę kwasu nukleinowego. [0092] Terminy sekwencja kwasu nukleinowego kodująca lub sekwencja kodująca DNA lub nukleotydowa sekwencja kodująca konkretnego przeciwciała -- jak również inne terminy synonimowe 17
19 1 2 3 odnoszą się do sekwencji DNA, która podlega transkrypcji i translacji do przeciwciała po umieszczeniu pod kontrolą odpowiednich sekwencji regulacyjnych. Sekwencja promotorowa to region regulacyjny DNA zdolny do wiązania polimerazy RNA w komórce i inicjowania transkrypcji leżącej poniżej (w kierunku 3') sekwencji kodującej. Promotor jest częścią sekwencji DNA. Ten region sekwencji ma kodon start na swoim końcu 3'. Sekwencja promotorowa zawiera minimalną liczbę zasad, gdzie elementy są niezbędne do inicjacji transkrypcji na poziomie wykrywalnym powyżej tła. Jednak po związaniu się polimerazy RNA z sekwencją i zainicjowaniu transkrypcji od kodonu start (koniec 3' promotora), transkrypcja przebiega w dół w kierunku 3'. W obrębie sekwencji promotora będzie znajdywać się miejsce inicjacji transkrypcji (dogodnie definiowane przez mapowanie przy użyciu nukleazy SI), a także, domeny wiążące białka (sekwencje konsensusowe) odpowiedzialne za wiązanie polimerazy RNA. [0093] Terminy kwas nukleinowy kodujący przeciwciało lub DNA kodujący przeciwciało lub polinukleotyd kodujący przeciwciało i inne terminy synonimowe obejmują polinukleotyd, który zawiera tylko sekwencję kodującą dla przeciwciała, jak również polinukleotyd, który zawiera dodatkowo sekwencję kodującą i/lub sekwencję niekodującą. [0094] W jednym korzystnym wykonaniu, specyficzny rodzaj cząsteczki kwasu nukleinowego jest definiowany przez jego strukturę chemiczną, czego przykładem jest, ale bez ograniczania się do tego, jego sekwencja pierwszorzędowa. W innym korzystnym wykonaniu, specyficzny rodzaj cząsteczki kwasu nukleinowego jest definiowany przez funkcję danego rodzaju kwasu nukleinowego lub funkcję produktu pochodzącego z danego rodzaju kwasu nukleinowego. A zatem, jako nieograniczający przykład, specyficzny rodzaj cząsteczki kwasu nukleinowego może być zdefiniowany przez jedną lub więcej aktywności lub właściwości, które można mu przypisać, w tym aktywności lub właściwości, które można przypisać jego wyrażanemu produktowi. [009] Obecna definicja składania roboczej próbki kwasu nukleinowego w bibliotekę kwasu nukleinowego obejmuje proces włączania próbki kwasu nukleinowego do kolekcji opartej na wektorze, na przykład poprzez ligację do wektora i transformację gospodarza. Opis odpowiednich wektorów, gospodarzy i innych odczynników, jak również ich konkretne, nieograniczające przykłady są podane poniżej. Obecna definicja składania roboczej próbki kwasu nukleinowego w bibliotekę kwasu nukleinowego obejmuje proces włączania próbki kwasu nukleinowego do kolekcji nieopartej na wektorze, na przykład przez ligację do adaptorów. Korzystnie adaptory mogą przyłączać startery PCR dla ułatwienia amplifikacji poprzez reakcję PCR. [0096] Zgodnie z tym, w nieograniczającym wykonaniu, biblioteka kwasów nukleinowych składa się z kolekcji opartej na wektorach jednej lub większej liczby cząsteczek kwasu nukleinowego. W innym korzystnym wykonaniu biblioteka kwasu nukleinowego składa się z kolekcji cząsteczek kwasu nukleinowego nieopartej na wektorach. W jeszcze innym korzystnym wykonaniu biblioteka kwasu nukleinowego składa się z kombinowanej kolekcji cząsteczek kwasu nukleinowego, która jest częściowo oparta na wektorze, a częściowo nieoparta na wektorze. Korzystnie, jeżeli kolekcję cząsteczek stanowiących bibliotekę można przeszukiwać i rozdzielać na indywidualne rodzaje cząsteczek kwasu nukleinowego. 18
20 1 2 3 [0097] Niniejszy wynalazek dostarcza konstruktu kwasu nukleinowego lub alternatywnie konstruktu nukleotydowego lub alternatywnie konstruktu DNA. Termin konstrukt jest tu stosowany do opisu cząsteczki, takiej jak polinukleotyd (np., polinukleotyd fitazy), który ewentualnie może być chemicznie związany z jednym lub więcej dodatkowych ugrupowań cząsteczkowych, takich jak wektor lub części wektora. W konkretnym -- ale w żaden sposób nieograniczającym -- aspekcie, przykładem konstruktu nukleotydowego są konstrukty do ekspresji DNA nadające się do transformacji komórki gospodarza. [0098] Termin oligonukleotyd (lub synonimowo oligo ) odnosi się bądź do jednoniciowego polideoksynukleotydu, bądź dwóch komplementarnych nici polideoksynukleotydowych, które mogą być zsyntetyzowane chemicznie. Takie syntetyczne oligonukleotydy mogą mieć lub mogą nie mieć - fosforanu. Te, które nie mają, nie będą podlegać ligacji z innym oligonukleotydem bez dodania fosforanu wraz z ATP w obecności kinazy. Syntetyczny oligonukleotyd będzie podlegał ligacji z fragmentem, który nie został zdefosforylowany. W celu uzyskania amplifikacji opartej na polimerazie (na przykład przy zastosowaniu reakcji PCR), można wspomnieć 32-krotnie zdegenerowany oligonukleotyd, który jest złożony, kolejno, z co najmniej pierwszej sekwencji homologicznej, zdegenerowanej sekwencji N, N, G/T i drugiej sekwencji homologicznej. Jak się stosuje w tym kontekście, homologiczny jest w odniesieniu do homologii pomiędzy oligo i polinukleotydem rodzicielskim, który jest poddawany amplifikacji opartej na polimerazie. [0099] Stosowany w niniejszym opisie termin połączony funkcjonalnie odnosi się do połączenia elementów polinukleotydowych w związek funkcjonalny. Kwas nukleinowy jest połączony funkcjonalnie, gdy jest umieszczony w funkcjonalnej zależności z inną sekwencją kwasu nukleinowego. Na przykład, promotor lub wzmacniacz jest połączony funkcjonalnie z sekwencją kodującą, jeżeli wpływa na transkrypcję sekwencji kodującej. Połączony funkcjonalnie oznacza, że połączone sekwencje DNA zazwyczaj sąsiadują ze sobą, i tam, gdzie niezbędne jest połączenie dwóch regionów kodujących białko, sąsiadują i są w ramce odczytu. [00] Sekwencja kodująca jest połączona funkcjonalnie z inną sekwencją kodującą, gdy polimeraza RNA będzie dokonywać transkrypcji dwóch sekwencji kodujących w jedną cząsteczkę mrna, który podlega następnie translacji do pojedynczego polipeptydu mającego aminokwasy pochodzące z obydwu sekwencji kodujących. Sekwencje kodujące nie muszą sąsiadować ze sobą tak długo, jak ulegające ekspresji sekwencje podlegają ostatecznie obróbce z wytworzeniem pożądanego przeciwciała. [01] Stosowany w niniejszym opisie termin warunki fizjologiczne odnosi się do temperatury, ph, siły jonowej, lepkości i tym podobnych parametrów biochemicznych, które są kompatybilne z żywym organizmem i/lub które zazwyczaj występują wewnątrzkomórkowo w żywych hodowanych komórkach drożdży lub komórkach ssaków. Na przykład, warunki wewnątrzkomórkowe w komórkach drożdży hodowanych w typowych warunkach hodowli laboratoryjnej są warunkami fizjologicznymi. Odpowiednie warunki reakcji in vitro dla koktajli do transkrypcji in vitro są na ogół warunkami fizjologicznymi. Generalnie, warunki fizjologiczne in vitro obejmują 0-0 mm NaCl lub KCl, ph 6,- 8,, -4 C i 0,001- mm kationu dwuwartościowego (np., Mg++, Ca++); korzystnie, mm NaCl lub KCl, ph 7,2-7,6, mm kationu dwuwartościowego i często zawierają 0,01-1,0 procent nieswoistego białka (na przykład, BSA). Często może być obecny niejonowy detergent (Tween, NP- 19
21 1 2 3, Triton X-0), zwykle od 0,001 do 2%, zazwyczaj od 0,0-0,2% (obj./obj.). Konkretne warunki wodne mogą być wybrane przez specjalistę zgodnie z konwencjonalnymi metodami. Jako ogólna wskazówka mogą być stosowane następujące buforowane warunki wodne: - mm NaCl, -0 mm Tris HCl, o ph -8, z ewentualnym dodatkiem kationu(ów) dwuwartościowego(ych) i/lub metali chelatujących i/lub detergentów niejonowych lub scyntylatorów. [02] Termin populacja, stosowany w niniejszym opisie, oznacza zbiór składników, takich jak polinukleotydy, części lub polinukleotydów lub przeciwciał. Populacja mieszana oznacza zbiór składników, które należą do tej samej rodziny kwasów nukleinowych lub przeciwciał (tj., są spokrewnione), ale różnią się sekwencją (tj., nie są identyczne), a zatem aktywnością biologiczną. [03] Cząsteczka mająca pro-postać odnosi się do cząsteczki, która ulega dowolnej kombinacji jednej lub więcej kowalencyjnych i niekowalencyjnych modyfikacji chemicznych (na przykład, glikozylacji, cięciu proteolitycznemu, dimeryzacji lub oligomeryzacji, zmianie konformacyjnej indukowanej temperaturą lub indukowanej ph, łączeniu z kofaktorem, etc.) na drodze do osiągnięcia bardziej dojrzałej postaci cząsteczki mającej różnicę we właściwości (np. wzrost aktywności) w porównaniu z odnośnikową pro-postacią cząsteczki. Gdy dwie lub więcej modyfikacje chemiczne (np. dwa cięcia proteolityczne lub cięcie proteolityczne i deglikozylację) można odróżnić na drodze do wytwarzania dojrzałej cząsteczki, odnośnikowa cząsteczka prekursorowa może być określana jako pre-pro- postać cząsteczki. [04] Właściwość można opisać jako dowolną cechę charakterystyczną, w tym charakterystyczną właściwość fizyczną, chemiczną lub aktywność przeciwciała, która ma być zoptymalizowana. Na przykład, w pewnych aspektach, określona uprzednio właściwość, cecha lub aktywność, która ma być zoptymalizowana, może być wybrana spośród zmniejszania agregacji przeciwciało-białko, wzmocnienia stabilności przeciwciała, zwiększonej rozpuszczalności przeciwciała, zwiększonej stabilność przeciwciała względem ph, podwyższonej stabilności temperaturowej przeciwciała, zwiększonej stabilności przeciwciała w rozpuszczalniku, zwiększonej swoistości, zmniejszonej swoistości, wprowadzenia miejsc glikozylacji, wprowadzenia miejsc sprzęgania, zmniejszenia immunogenności, wzmocnienia wyrażania białka, zwiększania powinowactwa względem antygenu, zmniejszania powinowactwa względem antygenu, zmiany w powinowactwie wiązania, zmiany w immunogenności, zmiany w aktywności katalitycznej, optymalizacji ph lub wzmocnienia swoistości. Właściwość zoptymalizowana odnosi się do pożądanej zmiany w danej właściwości przeciwciała zmutowanego w porównaniu z przeciwciałem matrycowym. [0] Stosowany w niniejszym opisie termin pseudolosowy odnosi się do zbioru sekwencji, które mają ograniczoną zmienność, taką jak, na przykład, stopień zmienności reszt w innej pozycji, ale dowolna pozycja pseudolosowa dopuszcza pewien stopień zmienności reszt, jednak ograniczony. [06] Jednostki quasi-powtarzane, stosowane w niniejszym opisie, odnoszą się do powtórzeń, które mają być ponownie dobrane i z definicji nie są identyczne. W istocie proponowany jest sposób nie tylko dla praktycznie identycznych jednostek kodujących wytwarzanych poprzez mutagenezę identycznej sekwencji startowej, ale także ponowny zbiór podobnych lub pokrewnych sekwencji, które mogą znacznie odbiegać w niektórych regionach. Niemniej jednak, jeśli sekwencje zawierają
22 1 2 3 wystarczającą homologię dla potwierdzenia przy zastosowaniu tego podejścia, mogą być określane jako jednostki quasi-powtarzane. [07] Stosowany w niniejszym opisie termin losowa biblioteka peptydowa odnosi się do zbioru sekwencji polinukleotydowych, który koduje zbiór losowych peptydów oraz zbiór losowych peptydów kodowanych przez te sekwencje polinukleotydowe, jak również fuzje białkowe zawierające te losowe peptydy. [08] Stosowany w niniejszym opisie termin losowa sekwencja peptydowa odnosi się do sekwencji aminokwasowej składającej się z dwóch lub większej liczby monomerów aminokwasowych i skonstruowana jest przy zastosowaniu procesu stochastycznego lub losowego. Peptyd losowy może zawierać region zrębowy lub motywy szkieletowe, które mogą zawierać sekwencje niezmienne. [09] Stosowany w niniejszym opisie, receptor odnosi się do cząsteczki, która ma powinowactwo do danego liganda. Receptorami mogą być cząsteczki występujące naturalnie lub syntetyczne. Receptory mogą być stosowane w stanie niezmienionym lub jako agregaty z innymi rodzajami. Receptory mogą być dołączone kowalencyjnie lub niekowalencyjnie do elementu wiążącego, bądź bezpośrednio, bądź za pośrednictwem swoistej substancji wiążącej. Przykłady receptorów obejmują, ale nie są do tego ograniczone, przeciwciała, w tym przeciwciała monoklonalne i surowice odpornościowe reaktywne ze swoistymi determinantami antygenowymi (takimi jak wirusy, komórki lub inne materiały). [01] Przeciwciała rekombinowane odnoszą się do przeciwciał wytwarzanych przy zastosowaniu technik rekombinowania DNA, tj. wytwarzane z komórek transformowanych egzogennym konstruktem DNA kodującym pożądane przeciwciało. Przeciwciała syntetyczne są tymi wytwarzanymi poprzez syntezę chemiczną. [0111] Termin polinukleotydy spokrewnione oznacza, że regiony lub obszary polinukleotydów są identyczne, a regiony lub obszary polinukleotydów heterologiczne. [0112] Redukcyjny ponowny dobór, stosowany w niniejszym opisie, odnosi się do wzrostu zróżnicowania cząsteczkowego który jest uzyskiwany poprzez zdarzenia delecji (i/lub insercji), w których pośredniczą sekwencje powtórzone. [0113] Następujące terminy są stosowane do opisania zależności w odniesieniu do sekwencji, pomiędzy dwoma lub więcej polinukleotydami: sekwencja odnośnikowa, okno porównywania, identyczność sekwencji, procent identyczności sekwencji oraz zasadnicza identyczność. [0114] Sekwencja odnośnikowa jest zdefiniowaną sekwencją stosowaną jako podstawa do porównywania sekwencji; sekwencja odnośnikowa może być fragmentem dłuższej sekwencji, na przykład, jako odcinek sekwencji cdna lub genu pełnej długości podanej w wykazie sekwencji, lub może zawierać kompletną sekwencję cdna lub genu. Generalnie, sekwencja odnośnikowa ma długość co najmniej nukleotydów, często co najmniej 2 nukleotydów, a często co najmniej 0 nukleotydów. Ponieważ każdy z dwóch polinukleotydów może (1) zawierać sekwencję (tj. część pełnej sekwencji polinukleotydowej), która jest podobna między dwoma polinukleotydami i (2) może ponadto zawierać sekwencję, która jest rozbieżna pomiędzy dwiema sekwencjami polinukleotydowymi, porównania sekwencji między dwiema (lub więcej) polinukleotydami przeprowadza się zwykle poprzez 21
23 1 2 3 porównywanie sekwencji dwóch polinukleotydów w oknie porównywania w celu zidentyfikowania i porównania lokalnych regionów podobieństwa sekwencji. [011] Wskaźnik powtarzalności (RI, ang. Repetitive Index), stosowany w niniejszym opisie, jest średnią liczbą kopii jednostek quasi-powtórzonych zawartych w wektorze do klonowania. [0116] Termin saturacja odnosi się do techniki ewolucji, gdzie wszystkie możliwe zmiany są dokonane w każdej pozycji polinukleotydu matrycowego lub polipeptydu matrycowego; jednakże zmiana w każdej pozycji nie jest potwierdzana przez testowanie, a jedynie zakładana statystycznie, gdzie szacuje się, że większość możliwych zmian lub prawie każda możliwa zmiana pojawi się w każdej pozycji matrycy. [0117] Termin identyczność sekwencji znaczy, że dwie sekwencje polinukleotydowe są takie same (tj., na zasadzie nukleotyd na - nukleotyd) w oknie porównywania. Termin procent identyczności sekwencji oblicza się przez porównanie dwóch optymalnie przyrównanych sekwencji w oknie porównywania, określając liczbę pozycji, w których identyczna zasada kwasu nukleinowego (na przykład, A, T, C, G, U lub I) występuje w obu sekwencjach, co daje liczbę dopasowanych pozycji, dzieląc liczbę pasujących pozycji przez całkowitą liczbę pozycji w oknie porównywania (tj., wielkość okna) i mnożąc wynik przez 0 w celu uzyskania procentu identyczności sekwencji. Ta zasadnicza identyczność, stosowana w niniejszym opisie, oznacza cechę sekwencji polinukleotydowej, gdzie polinukleotyd zawiera sekwencję wykazującą co najmniej 80 procent identyczności sekwencji, korzystnie co najmniej 8 procent identyczności, często od 90 do 9 procent identyczności sekwencji, a najczęściej co najmniej 99 procent identyczności sekwencji przy porównywaniu do sekwencji odnośnikowej w oknie porównywania obejmującym co najmniej 2-0 nukleotydów, gdzie procent identyczności sekwencji oblicza się przez porównanie sekwencji odnośnikowej z sekwencją polinukleotydową, która może obejmować delecje lub addycje, stanowiące łącznie procent lub mniej sekwencji odnośnikowej w oknie porównywania. [0118] Termin mutacja cicha odnosi się do zmiany kodonu, która nie powoduje zmiany aminokwasu w wyrażanym polipeptydzie i opiera się na nadmiarowości stosowania kodonów do wprowadzania aminokwasów. [0119] Jak wiadomo w stanie techniki, podobieństwo między dwoma enzymami określa się przez porównanie sekwencji aminokwasowej i ich konserwowanych podstawników aminokwasowych jednego białka z sekwencją drugiego białka. Podobieństwo może zostać określone przy zastosowaniu procedur, które są dobrze znane w stanie techniki, na przykład, program BLAST (Basic Local Alignment Search Tool w National Center for Biological Information). [01] Stosowany tu termin przeciwciało jednołańcuchowe odnosi się do polipeptydu zawierającego domenę VH i domenę VL w powiązaniu polipeptydowym, połączone generalnie poprzez peptyd łącznikowy (np. [Gly-Gly-Gly-Gly-Ser] x ), i który może zawierać dodatkowe sekwencje aminokwasowe na końcach aminowych i/lub karboksylowych. Przykładowo, jednołańcuchowe przeciwciało może zawierać odcinek wiążący do łączenia z polinukleotydem kodującym. Przykładem scfv jest przeciwciało jednołańcuchowe. Przeciwciała jednołańcuchowe są generalnie białkami składającymi z jednego lub większej liczby odcinków polipeptydowych z co najmniej sąsiadujących aminokwasów zasadniczo kodowanych przez geny z nadrodziny immunoglobulin (np. patrz Williams i Barclay, 1989, 22
24 1 2 3 str ), najczęściej kodowane są przez sekwencję genu łańcucha ciężkiego lub lekkiego gryzonia, naczelnego innego niż człowiek, ptaka, świni, bydlęcia, owcy, kozy lub człowieka. Funkcjonalne przeciwciało jednołańcuchowe zawiera generalnie wystarczającą część produktu genu z nadrodziny immunoglobulin, tak, aby zachować właściwości wiązania z określoną docelową cząsteczką, typowo receptorem lub antygenem (epitopem). [0121] Uważa się, że przedstawiciele pary cząsteczek (np., pary przeciwciało-antygen lub pary kwasów nukleinowych) wiążą się swoiście ze sobą, jeżeli wiążą się one ze sobą z większym powinowactwem niż z innymi, nieswoistymi cząsteczkami. Na przykład, przeciwciało uzyskane przeciwko antygenowi, z którym wiąże się bardziej skutecznie niż z białkiem nieswoistym, można opisać jako wiążące się swoiście z antygenem. [0122] Hybrydyzacja specyficzna jest tu zdefiniowana jako tworzenie hybrydy pomiędzy pierwszym polinukleotydem i drugim polinukleotydem (np. polinukleotyd mając różną, ale zasadniczo identyczną sekwencję co pierwszy polinukleotyd), gdzie zasadniczo niespokrewnione sekwencje polinukleotydowe nie tworzą hybryd w mieszaninie. [0123] Termin polinukleotyd specyficzny oznacza polinukleotyd mający określone punkty końcowe i mający określoną sekwencję kwasu nukleinowego. Dwa polinukleotydy, gdzie jeden polinukleotyd ma identyczną sekwencję co część drugiego polinukleotydu, ale różne końce, obejmują dwa różne swoiste polinukleotydy. [0124] Ostre warunki hybrydyzacji oznaczają hybrydyzację, która nastąpi tylko wówczas, gdy jest co najmniej 90% identyczności, korzystnie co najmniej 9% identyczności, a najkorzystniej co najmniej 97% identyczności pomiędzy sekwencjami. Patrz Sambrook i wsp., [012] Wynalazkiem objęte są również polipeptydy mające sekwencje, które są zasadniczo identyczne do sekwencji polipeptydu, takiej jak dowolna z ujawnionych tu SEK ID NR. Sekwencja aminokwasowa zasadniczo identyczna jest to sekwencja, która różni się od sekwencji odnośnikowej tylko konserwatywnymi podstawieniami aminokwasowymi, na przykład, podstawieniami jednego aminokwasu innym aminokwasem z tej samej klasy (np., podstawienie jednego aminokwasu hydrofobowego, takiego jak izoleucyna, walina, leucyna lub metionina, innym, lub podstawienie jednego aminokwasu polarnego innym, tak jak podstawienie argininy lizyną, kwasu glutaminowego kwasem asparaginowym lub glutaminy asparaginą). [0126] Dodatkowo, sekwencja aminokwasowa zasadniczo identyczna, jest to sekwencja, która różni się od sekwencji odnośnikowej tylko jednym lub większą liczbą niekonserwatywnych podstawień, delecji lub insercji, zwłaszcza, kiedy takie podstawienie następuje w miejscu, które nie jest aktywnym miejscem cząsteczki oraz pod warunkiem, że polipeptyd zasadniczo zachowuje swoje właściwości behawioralne. Na przykład, jeden lub większą liczbę aminokwasów można usunąć z polipeptydu fitazy, powodując zmiany struktury polipeptydu, bez znaczącej zmiany jej aktywności biologicznej. Na przykład, można usunąć aminokwasy amino- lub karboksykońcowe, które nie są wymagane do aktywności biologicznej. Takie zmiany mogą prowadzić do uzyskania mniejszych aktywnych polipeptydów fitazy. [0127] Stosowany w niniejszym opisie termin zasadniczo czysty oznacza, że dany rodzaj obiektu jest dominującym rodzajem, który jest obecny (tj. molowo jest go więcej niż dowolnych innych 23
25 1 2 poszczególnych rodzajów makrocząsteczek w kompozycji), a korzystnie zasadniczo oczyszczona frakcja jest kompozycją, w której dany rodzaj obiektu zawiera co najmniej 0 procent (molowo) wszystkich obecnych rodzajów makrocząsteczek. Ogólnie, kompozycja zasadniczo czysta będzie zawierać więcej niż 80 do 90 procent wszystkich rodzajów makrocząsteczek obecnych w kompozycji. Najkorzystniej, jeżeli dany rodzaj obiektu oczyszcza się do zasadniczej jednorodności (rodzajów zanieczyszczających nie można wykryć w kompozycji konwencjonalnymi sposobami wykrywania), gdzie kompozycja składa się zasadniczo z jednego rodzaju makrocząsteczek. Rozpuszczalniki, małe cząsteczki (<00 daltonów) i jony pierwiastków nie są uważane za rodzaje makrocząsteczek. [0128] Stosowany w niniejszym opisie oligopeptyd matrycowy oznacza przeciwciało matrycowe, dla którego pożądana jest drugorzędowa biblioteka wariantów. Co będzie wiadome specjalistom w tej dziedzinie, w niniejszym wynalazku może znaleźć zastosowanie dowolna liczba matryc. Konkretnie objęte definicją przeciwciała i oligopeptydy są fragmenty domen znanych przeciwciał, w tym domen funkcjonalnych, takich domeny wiążące itd., oraz mniejsze fragmenty, takie jak skręty, pętle, itd. Oznacza to, że mogą być również stosowane fragmenty przeciwciała. Ponadto, mogą być stosowane warianty przeciwciał, tj., struktury analogów przeciwciał niewystępujące w przyrodzie. [0129] Odpowiednie szkielety przeciwciał obejmują, lecz nie są do tego ograniczone, wszystkie te znajdujące się w bazie danych zebrane i obsługiwane przez Research Collaboratory for Structural Bioinformatics (RCSB, dawniej Brookhaven National Lab). [01] Stosowany w niniejszym opisie określenie odcinek zmienny odnosi się do części nowopowstającego peptydu, który zawiera losową, pseudolosową lub określoną sekwencję kernelową. Odcinek zmienny odnosi się do części nowopowstającego peptydu, który zawiera losową, pseudolosową lub określoną sekwencję kernelową. Odcinek zmienny może zawierać zarówno zmienne, jak i niezmienne pozycje reszt, a stopień zmienności reszt w pozycji reszty zmiennej może być ograniczony: obie opinie są w gestii specjalisty. Typowo, odcinki zmienne mają długość do reszt aminokwasowych (np., 8 do ), jakkolwiek odcinki zmienne mogą być dłuższe i mogą zawierać części przeciwciał, takie jak fragment przeciwciała. [0131] Termin typu dzikiego lub typ dziki oznacza, że polinukleotyd ten nie zawiera żadnych mutacji. Przeciwciało typu dzikiego oznacza, że przeciwciało będzie aktywne na poziomie aktywności występującej w naturze i będzie zawierać sekwencję aminokwasową występującą w naturze. SZCZEGÓŁOWY OPIS WYNALAZKU 3 [0132] Ujawnienie dostarcza sposobu zintegrowanego wytwarzania i/lub selekcji, ewolucji i wyrażania przeciwciała terapeutycznego u gospodarza ssaka, do wytwarzania w jednym systemie. W jednym wykonaniu sposobu CIAO, przeciwciała terapeutyczne są uzyskiwane, optymalizowane i wytwarzane w tym samym systemie gospodarza-ssaka. W innym wykonaniu, lecznicze białko terapeutyczne jest znajdywane i wytwarzane w tym samym gospodarzu. To optymalizuje wytwarzanie od samego początku, oszczędzając koszty (czas i zasoby). 24
26 1 2 3 [0133] Historycznie, uzyskiwanie przeciwciał przeprowadzano w gospodarzach eukariotycznych (euk) i prokariotycznych (prok). Typowo w bakterii (E. coli), uzyskuje się przeciwciała o częściowej długości; na przykład, w technologii prezentacji na fagach Fab są odzyskiwane i czasem przekształcane później w przeciwciała pełnej długości. Istnieje kilka potencjalnych wad tych podejść. [0134] W jednym przykładzie, istnieją dowody, że regiony Fc i Fv komunikują się dla uzyskania właściwości przeciwciała, takich jak wiązanie i wyrażanie. Dlatego też, gdy fragment przeciwciała jest zoptymalizowany dla właściwości, takich jak wyrażanie, polepszenie nie zawsze przekłada się na lepsze wyrażanie złożonego przeciwciała pełnej długości. Na przykład, wytworzono bibliotekę Fc, aby próbować znaleźć Fc świętego Graala, który mógłby być dołączony do dowolnego Fv dla polepszenia wyrażania w dowolnym gospodarzu. [013] W jednym aspekcie, przeprowadzono mutagenezę kodonów w regionie stałym w celu optymalizacji wyrażania w komórkach ssaków. Konkretnie, wytworzono 326 mutantów w regionie stałym i wyrażono w komórkach HEK 293 i komórkach CHO-S. Przeszukiwanie przeprowadzono przy zastosowaniu testu ELISA. Kilka Fc spełniało kryteria polepszonego wyrażania i zostały nawet zidentyfikowane pewne zoptymalizowane Fc, które przenoszą pozytywne efekty w wielu liniach komórkowych; jednak, gdy inny Fv został dołączony do Fc, nie było przełożenia na polepszenie wyrażania. To pokazuje, że Fc i Fv komunikują się. [0136] W celu uniknięcia nieoczekiwanych wyników po rekombinowaniu fragmentów przeciwciał, w jednym korzystnym aspekcie, sposób CIAO jest stosowany do wykrywania cząsteczek przeciwciał pełnej długości. W innym korzystnym aspekcie, sposób CIAO wykorzystuje gospodarzy euk. [0137] W jednym wykonaniu, systemem eukariotycznym jest system ssaczy wybrany spośród jednej z grup składających się z linii komórkowych CHO, HEK293, IM9, DS-1, THP-1, Hep G2, COS, NIH 3T3, C33a, A49, A37, SK-MEL-28, DU 14, PC-3, HCT 116, Mia PACA-2, ACHN, Jurkat, MM1, Ovcar 3, HT 80, Panc-1, U266, 769P, BT-474, Caco-2, HCC 194, MDA-MB-468, LnCAP, NRK-49F i SP2/0; oraz splenocytów myszy i PBMC królika. W jednym z aspektów, system ssaczy jest wybrany z linii komórkowej CHO lub HEK293. W jednym konkretnym aspekcie, systemem ssaczym jest linia komórkowa CHO-S. W innym konkretnym aspekcie, systemem ssaczym jest linia komórkowa HEK293. W innym wykonaniu, systemem eukariotycznym jest system komórek drożdży. W jednym z aspektów, system eukariotyczny jest wybrany z komórek drożdży S. cerevisiae lub komórek drożdży Pichia. [0138] W innym wykonaniu, wytworzanie ssaczej linii komórkowej może być przeprowadzone przez zakontraktowane badania naukowe przez usługową organizację produkcyjną. Przykładowo, dla rekombinowanych przeciwciał lub innych białek, Lonza (Lonza Group Ltd., Bazylea, Szwajcaria) może wytworzyć wektory do wyrażania produktu przy zastosowaniu technologii GS Gene Expression System z CHOK1SV lub NSO jako liniami komórkowymi gospodarza. [0139] W innym wykonaniu, ewolucję można przeprowadzić w gospodarzu prok (takim jak E. coli), a przeszukiwanie może nastąpić w gospodarzu euk (na przykład, CHO), o ile przeszukiwanie następuje w tym samym gospodarzu jakim jest gospodarz wytwarzający. Selekcja wiodącego przeciwciała (przeciwciał) - kandydata(ów) 2
27 1 2 3 [01] Można zastosować rozmaite sposoby do znajdywania, wytwarzania i/lub selekcji jednego lub więcej przeciwciał terapeutycznych-kandydatów do ewolucji. Wyselekcjonowanymi cząsteczkami mogą być istniejące przeciwciała rekombinowane lub mogą one pochodzić ze kolekcji rekombinowanych przeciwciał. Rekombinowane przeciwciała można znajdywać przy zastosowaniu dowolnej liczby dostępnych platform do wytwarzania i przeszukiwania. Przeciwciała mogą występować w dowolnej postaci, w tym przeciwciał lub ich fragmentów jednołańcuchowych, w pełni ludzkich, Fab, Fv, Fc, dwufunkcyjnych, chimeryczne, humanizowanych lub w pełni ludzkich. W jednym korzystnym wykonaniu sposób CIAO jest stosowany do znajdywania cząsteczek przeciwciał pełnej długości. Można wytwarzać biblioteki rekombinowanych przeciwciał i przeszukiwać je przy zastosowaniu systemów selekcji lub przeszukiwania w zoptymalizowanym lub nieoptymalizowanym gospodarzu-ssaku aby uzyskać kandydatów do ewolucji. Opublikowano kilka eukariotycznych systemów ekspresyjnych, na przykład, systemy komórek ssaczych lub drożdżowych. [0141] W sposobie według niniejszego wynalazku, takie ssacze systemy ekspresyjne, w szczególności systemy z zastosowaniem prezentacji cząsteczek do przeszukiwania i selekcji na powierzchni komórek, stosuje się do identyfikacji i selekcji kandydatów do wytwarzania lub ewolucji, po której następuje wytwarzanie. Korzystnie, takimi ssaczymi gospodarzami są komórki fibroblastów (3T3, mysz; BHK21, chomik syryjski), komórki nabłonkowe (MDCK, pies; Hela, człowiek; PtK1, kanguroszczur), komórki plazmatyczne (SP2/0 i NS0, mysz), komórki nerkowe (293 człowiek; COS, małpa), komórki jajnika (CHO, chomik chiński), komórki embrionalne (R1 i E14.1, mysz; H1 i H9, człowiek; PER C.6 człowiek). Technologia prezentacji na powierzchni komórki jest wykorzystywana do prezentacji przeciwciał na powierzchni komórek ssaków do przeszukiwania. Sklonowano białka jako fuzje z cząsteczkami błonowymi, które, po wyrażeniu, prezentują białka na powierzchni komórek, na przykład do szybkiego i wysokoprzepustowego przeszukiwania. [0142] W niektórych wykonaniach, ujawnienie dostarcza sposobu dostarczania zoptymalizowanego przeciwciała. W jednym wykonaniu, wybiera się antygen i wytwarza i wyraża bibliotekę ludzkich przeciwciał w systemie ssaczym, na przykład komórkach CHO-S. Bibliotekę przeszukuje się w celu zidentyfikowania w pełni ludzkich przeciwciał, na przykład, poprzez sortowanie komórek aktywowane fluorescencją (FACS). Znalezione w pełni ludzkie przeciwciała przeszukuje się/charakteryzuje następnie przy zastosowaniu odpowiedniego testu pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności. W jednym aspekcie, poddane ewolucji cząsteczki bada się pod kątem różnych właściwości jednocześnie, na przykład, polepszonej funkcji i ekspresji. Odpowiedni test może obejmować, na przykład test ELISA lub technologię macierzy. Przeciwciało matrycowe wybiera się ze znalezionych przeciwciał ludzkich. Dowolną metodę ewolucji przeprowadza się na przeciwciele matrycowym albo jego fragmencie polipeptydowym w celu przygotowania zbioru zmutowanych przeciwciał. W jednym z aspektów, ewolucję przeprowadza się przy zastosowaniu metody kompleksowej inżynierii białek w celu wytworzenia zbioru zmutowanych przeciwciał. Metodę kompleksowej inżynierii białek można wybrać, na przykład, z jednego lub kombinacji kompleksowej ewolucji pozycyjnej (CPE ), kompleksowej syntezy białka (ang. Comprehensive Protein Synthesis, CPS ), ewolucji podatnych miejsc (ang. Flex Evolution), ewolucji synergicznej (ang. Synergy 26
28 1 2 3 Evolution), kompleksowej insercyjnej ewolucji pozycyjnej (CPI ) lub kompleksowej delecyjnej ewolucji pozycyjnej (CPD ). W innym aspekcie, zmutowane przeciwciała są wyrażone w tym samym systemie ssaczym, co zastosowany do wytworzenia biblioteki ludzkich przeciwciał. [0143] Zbiór zmutowanych przeciwciał charakteryzuje się/przeszukuje pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności. W jednym aspekcie, zbiór mutantów przeciwciał przeszukuje się jednocześnie, na przykład, pod kątem polepszonej funkcji i wyrażania. W jednym aspekcie, w baza danych swoista dla cząsteczki w postaci funkcyjnej mapy pozycyjnej (EvoMap ) służy do dodatkowej optymalizacji przy zastosowaniu jednej lub więcej technik ewolucji białek znanych w tej dziedzinie; po której następuje identyfikacja i charakteryzowanie polepszonych mutantów. Zoptymalizowane przeciwciało wybiera się poprzez porównanie z przeciwciałem matrycowym pod względem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności. [0144] W jednym aspekcie, przeprowadza się wyrażanie wybranego zoptymalizowanego przeciwciała na skalę gramową; po czym następuje toksykologia nie-glp. Przeprowadza się stabilną transfekcję linii komórkowej, opracowanie procesu i wytworzenie macierzystego banku komórek (ang. Master Cell Bank). Wytwarzanie GMP odbywa się w tym samym systemie ssaczym co zastosowany do wytworzenia biblioteki ludzkich przeciwciał, na przykład, w komórkach CHO-S, aby uzyskać w rezultacie zoptymalizowane terapeutyczne rekombinowane mab. [014] W innym wykonaniu, metoda CIAO rozpoczyna się od wyboru matrycowej hybrydomy lub rekombinowanego przeciwciała; ewolucji przeciwciała dla dostarczenia zbioru zmutowanych przeciwciał, które są przeszukiwane przez zastosowanie prezentacji przeciwciał na powierzchni komórek w systemie komórek ssaczych; a wytwarzanie jest przeprowadzane w tym samym systemie komórek ssaczych co stosowane do przeszukiwania. [0146] W innym wykonaniu, wybrana matrycowa hybrydoma/rekombinowane przeciwciało jest humanizowane i przeszukiwane w gospodarzu do wytwarzania, po czym następuje wytwarzanie w gospodarzu do wytwarzania, gdzie etap optymalizacji (ewolucji) jest w ogóle pominięty. [0147] W innych aspektach niniejszego wynalazku, dalszą optymalizację wyrażania w gospodarzu do wytwarzania prowadzi się przez poddanie ewolucji regionu Fc przeciwciała, zastosowanie cichych kodonów w przeciwciele i/lub wektorze i/lub genach gospodarza stosowanych do wyrażania białka. W jednym aspekcie, wytwarzana jest biblioteka Fc przy zastosowaniu dowolnej techniki ewolucji. W jednym konkretnym aspekcie optymalizacji wyrażania, CPE przeprowadza się wobec domeny Fc przeciwciała w celu wytworzenia biblioteki mutantów Fc, które mogą być zastosowane do wyboru optymalnego partnera dla każdego Fv. Optymalizacja jest przeznaczona do szybkiego przyłączania wszystkich wariantów CPE dla Fc do każdego nowego regionu Fv. Alternatywnie, podzbiór tych Fc można stosować do przyłączania różnych Fv. Każdy z tych wariantów CPE dla Fc/kombinacje Fv przeszukuje się jako przeciwciała o pełnej długości wyrażane w komórkach ssaków (np. CHO, tania pożywka) dla optymalnego wyrażania. Ponadto, CPS można przeprowadzić w celu zbadania wszystkich teoretycznych permutacji do 12 lub więcej z tych trafień CPE w komórkach ssaków, do poprawy wyrażania. Specyficzne pożądane zmiany kodonów można również wybrać w celu zidentyfikowania klonów o zwiększonym wyrażaniu. Identyfikowane są ciche kodony i w tych 27
29 pozycjach przeprowadza się CPE. Tę bibliotekę CPE przeszukuje się w celu zidentyfikowania optymalnych wartości wyrażania. Ponadto, wszystkie teoretyczne permutacje do 12 lub więcej wyników CPE można zastosować w procesie CPS w celu wytworzenia nowej biblioteki, która może być przeszukiwana w komórkach ssaków, pod kątem polepszenia wyrażania. Najlepsze ciche mutacje po CPS są wykorzystywane do uzyskiwania białka dla optymalnego wyrażania w konkretnej linii komórkowej i pożywce. Daje to szansę na kontrolę precyzyjnej biopodobnej struktury. [0148] Inne obszary wzmacniania wyrażania obejmują: optymalizację wektora, w tym promotora, miejsc składania RNA, końców i 3, sekwencji flankujących, zmniejszenie delecji i rearanżacji genu, polepszenie działania genów komórki gospodarza, optymalizację enzymów glikozylujących gospodarza oraz mutagenezę i selekcję obejmującą cały chromosom komórki gospodarza. Pokazano, że sekwencje aminokwasowe po stronie ' są istotne dla wzmocnienia wyrażania. Ewolucja wiodących kandydatów [0149] Można zastosować dowolny sposób ewolucji białka do jednoczesnej ewolucji jakości przeciwciał i optymalizacji wyrażania. Optymalizacja jakości przeciwciała może obejmować polepszenie różnych cech, takich jak powinowactwo, właściwości farmakokinetyczne, kierowanie do tkanek, agregacja przeciwciało-białko, problemy wysokiej zmienności testu i modyfikowanie innych cech in vivo. [0] Sposoby przeprowadzania ewolucji cząsteczek, w tym wyselekcjonowanych kandydatów według niniejszego wynalazku, obejmują metody stochastyczne i niestochastyczne. Opublikowane metody obejmują podejścia z losową i nielosową mutagenezą. Można zastosować dowolne z tych podejść, aby przeprowadzić ewolucję właściwości przeciwciał terapeutycznych według wynalazku w kierunku pożądanej cechy, takiej jak lepsza stabilność w różnych warunkach temperatury i ph lub lepsze wyrażanie w komórce gospodarza. Do doświadczeń związanych z ewolucją można wybrać inne potencjalnie pożądane właściwości, takie jak polepszenie aktywności katalitycznej, zwiększona stabilność białka w różnych warunkach, polepszona wybiórczość i/lub rozpuszczalność i polepszone wyniki wyrażania będące wynikiem polepszenia cech, takich jak zmniejszenie agregacji. [011] Ewolucję przeprowadza się bezpośrednio w gospodarzu eukariotycznym, takim gospodarzkomórka ssacza lub gospodarz-komórka drożdży, które mają być stosowane później wytwarzania przeciwciała terapeutycznego. Kandydatów można poddawać ewolucji pod kątem optymalnego wyrażania w tym samym gospodarzu, co stosowany do przeszukiwania i/lub ewolucji i wytwarzania. Optymalizację wyrażania można osiągnąć przez optymalizację stosowanych wektorów (składników wektorów, takich jak promotory, miejsca składania RNA, końce ' i 3' oraz sekwencje flankujące), modyfikację genetyczną komórek gospodarza w celu zmniejszenia poziomu delecji i rearanżacji genowych, ewolucję aktywności genów komórek gospodarza poprzez metody ewolucji odpowiednich genów in vivo lub in vitro, optymalizację enzymów glikozylujących gospodarza poprzez ewolucję odpowiednich genów i/lub poprzez zakrojoną na skalę chromosomową mutagenezę komórki gospodarza i wybór strategii w celu wyselekcjonowania komórek z wzmocnionymi możliwościami wyrażania. Komórki gospodarza są tu opisane bardziej szczegółowo. 28
30 1 2 3 [012] Technologia wyrażania i przeszukiwania poprzez prezentację na powierzchni komórek (na przykład, jak zdefiniowano powyżej) mogą być wykorzystywane do przeszukiwania bibliotek poddanego ewolucji przeciwciała dla kandydatów do wytwarzania. [013] Obecnymi sposobami szeroko stosowanymi do wytwarzania mutantów z cząsteczki wyjściowej są technologie mutagenezy kierowanej oligonukleotydami, reakcja łańcuchowa polimerazy podatna na błędy i mutageneza kasetowa, w której określony region, który ma być optymalizowany jest zastępowany przez syntetycznie mutagenizowany oligonukleotyd. W takich przypadkach, wytwarza się pewną liczbę miejsc mutacji wokół określonych miejsc w wyjściowej sekwencji. [014] W mutagenezie kierowanej oligonukleotydami, krótka sekwencja jest zastępowana przez syntetycznie mutagenizowany oligonukleotyd. Podatna na błędy reakcja PCR wykorzystuje warunki polimeryzacji o niskiej wierności do wprowadzania niskiego poziomu mutacji punktowych losowo wzdłuż sekwencji. W mieszaninie fragmentów o nieznanej sekwencji podatną na błędy reakcję PCR można stosować do mutagenizowania mieszaniny. W mutagenezie kasetowej, blok sekwencji pojedynczej matrycy jest zwykle zastępowany przez sekwencję (częściowo) randomizowaną. [01] Chimeryczne geny wytworzono przez łączenie 2 fragmentów polinukleotydowych wykorzystując kompatybilne lepkie końce wytworzone przez enzym(y) restrykcyjny(e), gdzie każdy fragment pochodzi od odrębnej cząsteczki progenitorowej (lub rodzicielskiej). Innym przykładem jest mutageneza pozycji pojedynczego kodonu (tj. aby uzyskać podstawienie, addycję lub delecję kodonu) w polinukleotydzie rodzicielskim w celu wytworzenia pojedynczego polinukleotydu potomnego kodującego pojedynczy miejscowo zmutowany polipeptyd. [016] Ponadto, zastosowano systemy specyficznej miejscowo rekombinacji in vivo do wytworzenia hybryd genów, jak również losowe metody rekombinacji in vivo oraz rekombinację pomiędzy homologicznymi, ale skróconymi genami w plazmidzie. Opisano również mutagenezę poprzez nakładające się na siebie przedłużenia i reakcję PCR. [017] Metody nielosowe zastosowano do uzyskania większej liczby mutacji punktowych i/lub chimeryzacji, na przykład zastosowano kompleksowe lub wyczerpujące podejścia do wytworzenia wszystkich rodzajów cząsteczek w obrębie konkretnego zgrupowania mutacji w celu przypisania funkcji określonym grupom strukturalnym w przeciwciele matrycowym (np. konkretnej pozycji pojedynczego aminokwasu lub sekwencji składającej się z dwóch lub więcej pozycji aminokwasowych) i kategoryzacji i porównania poszczególnych grup mutacji. Patent USA numer zatytułowany Whole cell engineering my mutagenizing a substantial portion of a starting genome, combining mutations, and optionally repeating opisuje sposób przeprowadzania ewolucji organizmu w kierunku pożądanych właściwości. Patent USA numer zatytułowany Saturation mutagenesis in directed evolution i Patent USA numer zatytułowany Synthetic ligation reassembly in directed evolution opisuje sposoby wyczerpującej ewolucji i przeszukiwania pod kątem pożądanych cech cząsteczek. Dowolną taką metodę można zastosować w sposobie według niniejszego wynalazku. [018] Istnieje różnica pomiędzy wcześniej znanymi sposobami mutagenezy saturacyjnej i technik kompleksowej ewolucji tu preferowanych. Mutageneza saturacyjna odnosi się do techniki ewolucji, gdzie każda ewentualna zmiana jest dokonywana w każdej pozycji polinukleotydu matrycowego lub 29
31 1 polipeptydu matrycowego; jednak zmiana w każdej pozycji nie jest potwierdzana poprzez testowanie, a jedynie zakładana statystycznie. Ewolucja kompleksowa odnosi się do techniki ewolucji, gdzie każda ewentualna zmiana jest dokonywana w każdej pozycji polinukleotydu matrycowego lub polipeptydu matrycowego i polinukleotyd lub polipeptyd jest testowany, aby potwierdzić, że zamierzone zmiany zostały dokonane. [019] Sposoby saturacyjne są właściwiemetodami statystycznymi, niewyczerpującymi, a także nie były nigdy naprawdę kompleksowe we wszystkich etapach (na przykład, przy wytwarzaniu mutanta, identyfikowaniu mutanta, wyrażaniu zmutowanego białka, przeszukiwaniu zmutowanego białka i wytwarzaniu, identyfikacji, wyrażania i przeszukiwania rekombinowanego ulepszonego mutanta). W technikach kompleksowej ewolucji każda cząsteczka jest poddana badaniu i potwierdzona zarówno w pierwszym etapie mutagenezy, jak i dalej w drugim etapie rekombinowania ulepszonych mutantów lub trafień. [0160] O ile mutageneza saturacyjna nie jest potwierdzona przez sekwencjonowanie lub inne metody, technika ta nie może być uważana za kompleksową z kilku możliwych powodów. Na przykład, 1) system do klonowania nie jest w 0% wydajny ze względu na błędy klonowania lub syntezy albo trudności w klonowaniu cząsteczek, lub 2) niektóre białka są toksyczne po wyrażeniu, a zatem nie mogą być wydajnie wytwarzane. Dlatego ważne jest, aby potwierdzać przez sekwencjonowanie lub inną technikę, na każdym etapie. Przydatne jest sprawdzenie każdego etapu, aby przeprowadzić badanie pod kątem wyrażania, tak, że klony niewyrażające nie są oznaczane jako negatywne, jak w poprzedniej pracy, ale po prostu są klasyfikowane jako niezdolne do wyrażania. Kompleksowe techniki są więc uważane za bardziej czysty system niestochastyczny niż techniki saturacyjne, co zostało potwierdzone przez etap potwierdzenia. 2 3 Kompleksowa ewolucja pozycyjna [0161] Odnosząc się do Fig. 1, stosując liniowy peptyd jako prosty przykład, w pierwszym etapie, zbiór naturalnie występujących wariantów aminokwasowych (lub jego podzbiór lub pochodne aminokwasów) dla każdego kodonu od pozycji 1 do n (n odpowiadającej liczbie reszt w łańcuchu polipeptydowym) jest wytwarzany w procesie, określanym tu jako kompleksowa ewolucja pozycyjna (CPE ). Procedura ta jest powtarzana dla każdego łańcucha polipeptydowego cząsteczki docelowej. Minimalny zbiór mutacji aminokwasowych zawiera tylko jeden kodon dla każdego z 19 naturalnych aminokwasów. Jednak uważa się, że każdy system ekspresyjny może cierpieć z powodu preferencji kodonów, gdzie niewystarczające pule trna mogą prowadzić do zatrzymania translacji, przedwczesnego zakończenia translacji, zmiany ramki odczytu przy translacji i błędnego włączania aminokwasów. Dlatego też, w celu optymalizacji wyrażania każdy zbiór zawiera aż 63 różnych kodonów, obejmujących kodon stop. W następnym etapie, mutacje potwierdza się przez sekwencjonowanie każdej nowej cząsteczki. Można również zastosować inne metody potwierdzania. [0162] Każdy zbiór aminokwasów przeszukuje się następnie pod kątem co najmniej jednego spośród: - Polepszonej funkcji
32 Mutacji neutralnych - Mutacji hamujących - Wyrażania - Zgodności klonu z systemem gospodarza. [0163] Korzystnie, jeżeli wiele cech bada się pod kątem występowania jednocześnie, takich jak, na przykład, polepszenie funkcji i wyrażania. [0164] Dane z każdego zbioru łączy się dla całego łańcucha(ów) polipeptydowego(ych) i wytwarza się szczegółową mapą funkcyjną (określaną tu jako EvoMap ) cząsteczki docelowej. Ta mapa zawiera szczegółowe informacje o tym, jak każda mutacja wpływa na jakość/wyrażanie i/lub możliwości klonowania cząsteczki docelowej. Pozwala to na identyfikację wszystkich miejsc, gdzie nie można dokonać zmiany bez utraty funkcji białka (np., wiązania antygen/receptor w przypadku przeciwciała). Pokazuje to również, gdzie można dokonać zmiany być bez wpływu na funkcję. Ponadto identyfikuje to zmiany, których rezultatem są cząsteczki, które nie ulegają wyrażaniu w systemie gospodarza, a zatem nie można ocenić wpływu mutacji. [016] Schematyczna hipotetyczna mapa EvoMap jest pokazana na Fig. 1. Każda pozycja na matrycy jest identyfikowana jako miejsce restrykcyjne (niepodlegające mutacji), miejsce w pełni mutowalne, miejsce częściowo mutowane lub ulepszony mutant dla konkretnego podstawienia aminokwasowego. Każde miejsce częściowo podlegające mutacji może być dalej określone jako podatne na podstawienie, na przykład, resztą naładowaną lub podstawienie resztą niepolarną, i klon i/lub cząsteczka nieulegająca wyrażaniu, która nie może być klonowana w systemie gospodarza. [0166] Możliwe jest wykorzystanie EvoMap w celu rozpoznania i włączenia korzystnych pojedynczych podstawień aminokwasowych, i przebadania w celu dalszej optymalizacji cechy w cząsteczce docelowej. Jednak ewolucja pewnych cech może wymagać dwóch lub więcej jednoczesnych mutacji, by stać się widoczną. EvoMap może być wykorzystana do efektywnego, opłacalnego wytwarzania zbioru polipeptydów zmutowanych w wielu miejscach w sposób nielosowy. Zbiór polipeptydów zmutowanych w wielu miejscach można następnie przeszukiwać pod kątem polepszonych wielomiejscowych mutantów. [0167] CPE umożliwia uzyskanie pełnej potwierdzonej in vivo mapy mutacji przeciwciała. Identyfikacja całego zbioru polepszonych mutantów umożliwia dalszy etap(y) kombinatorycznej ewolucji. CPE można wykorzystywać w celu zmniejszenia ryzyka immunogenności uzyskanych poprzez ewolucję przeciwciał poprzez selekcję mutacji niepowierzchniowych; eliminację epitopów komórek T; i naśladowanie mutacji somatycznych. [0168] W jednym z aspektów, CPE można zastosować do wytworzenia biblioteki z, lub 1 aminokwasów lub aż do wszystkich 19 aminokwasów. Zmiany są dokonywane w każdej pozycji w przeciwciele i przeszukiwane pod kątem pożądanej cechy, takiej jak powinowactwo wiązania lub wyrażanie, i wytwarzana jest mapa Evomap. Późniejsze rundy mutacji i przeszukiwania można zastosować do wytworzenia danych dla wszystkich 19 aminokwasów. Z mapy identyfikowane są miejsca w pełni podlegające mutacji. Miejsca te są przydatne do identyfikacji pozycji, które mogą być modyfikowane, aby utworzyć nowy zbiór cząsteczek, które mogą być wytwarzane i testowane pod 31
33 kątem nowych cech. Na przykład, można wykorzystać informatykę do identyfikacji haplotypów w sekwencji i można dokonać pożądanych zmian aby uniknąć tych haplotypów poprzez dokonanie konkretnych ukierunkowanych zmian w miejscach neutralnych ( w pełni mutowalnych ) zidentyfikowanych z mapy, gdzie nie będzie wpływu na wyjściową cechę. Może to potencjalnie zmniejszyć ryzyko immunogenności (można wybrać mutacje niepowierzchniowe, wyeliminować epitopy komórek T, naśladować mutacje hipersomatyczne). Ponadto, mapa może pokazać miejsca dla konkretnych modyfikacji (glikozylacji i sprzęgania chemicznego), aby poprawić różne cechy. Ponadto optymalizacja cichych mutacji może poprawić wyrażanie przeciwciał w różnych gospodarzach. Ewolucja synergiczna [0169] W jednym wykonaniu niniejszego wynalazku, wytwarzana jest mapa EvoMap i wykorzystywana do ewolucji synergicznej, jak pokazano na Fig. 2. W ewolucji synergicznej jednoczesna mutacja w 2- wybranych miejscach może być łączona w celu uzyskania efektu kombinatorycznego. EvoMap polipeptydu docelowego jest stosowana do wyboru konkretnych pojedynczych mutacji punktowych aminokwasów do połączenia ich razem w wielomiejscowe mutacje polipeptydowe. [0170] W ewolucji synergicznej, nieinaktywujące punktowe mutacje aminokwasowe są wybierane z miejsc częściowo podlegających mutacji, które są blisko miejsc niepodlegających mutacji na EvoMap. W jednym z aspektów, wybrane nieinaktywujące punktowe mutacje aminokwasowe sąsiadują z miejscami niepodlegającymi mutacji. W ewolucji synergicznej przeprowadza się jednoczesną mutację aminokwasów w dwóch do wybranych miejscach dla uzyskania efektów kombinatorycznych. W jednym aspekcie, rekombinację dwóch do dwudziestu mutacji stosuje się do wytworzenia biblioteki z wariantami kodonów kodujących populację polipeptydów zmutowanych w wielu miejscach. Po klonowaniu i wyrażaniu, populację wytwarzanych polipeptydów zmutowanych w wielu miejscach przeszukuje się następnie pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności w porównaniu do polipeptydu matrycowego. W ten sposób, można zidentyfikować ulepszone polipeptydy zmutowane w wielu miejscach. W jednym aspekcie, ulepszone polipeptydy zmutowane w wielu miejscach są wytwarzane poprzez kombinatoryczną syntezę białka. Jedną z zalet ewolucji synergicznej jest to, że nie wymaga rentgenowskiej struktury krystalicznej przeciwciała, aby kierować ewolucją matrycowego polipeptydu. Technika ta jest szczególnie przydatna dla przeciwciał z dużą zmiennością w testach i innymi wielomiejscowymi efektami. [0171] Zgodnie z niniejszym wynalazkiem, zastosowanie ewolucji synergicznej obejmuje, ale nie ogranicza się do tego, ewolucję złożonych molekularnych zmian mechanicznych, ewolucję przeciwciał z dużą zmiennością w testach, ewolucję swoistości przeciwciała, polepszenie wyrażania w różnych gospodarzach ekspresyjnych, stabilność i optymalizację ph. Ewolucja synergiczna jest stosowalna wobec przeciwciał terapeutycznych. [0172] W jednym aspekcie niniejszego wynalazku, ewolucja synergiczna może być wykorzystywana do optymalizacji jednego lub więcej aspektów polipeptydu, który jest częścią cząsteczki przeciwciała. 32
34 1 2 Cząsteczka przeciwciała może być składana przez poddawanie ligacji jednego lub więcej zmutowanych kwasów nukleinowych kodujących polipeptydy z zero, jednym lub większą liczbą kwasów nukleinowych kodujących polipeptydy zrębowe z wytworzeniem wariantu przeciwciała poprzez techniki klonowania, translacji i wyrażania znane w tej dziedzinie. W jednym aspekcie, polipeptyd zrębowy pochodzi z cząsteczki przeciwciała typu dzikiego. W tym aspekcie, ewolucję synergiczną można zastosować w połączeniu z technikami humanizacji przeciwciała. Przykładowo, można wyselekcjonować mysie przeciwciało monoklonalne w celu ewolucji i humanizacji. Regiony CDR przeciwciała klonuje się i sekwencjonuje, i poszczególne regiony CDR (CDR1, CDR2, CDR3) można zsyntetyzować i poddać ligacji z innymi nukleotydami kodującymi polipeptydy zrębowe przeciwciała ludzkiego, a następnie wytworzyć bibliotekę wariantów ludzkiej IgG. Bibliotekę wariantów ludzkiej IgG przeszukuje się następnie pod kątem co najmniej jednej właściwości, na przykład dwóch lub więcej właściwości, w tym lepszej funkcji i wyrażania w porównaniu do mysich mab. W innym aspekcie, polipeptydem zrębowym jest sztuczny polipeptyd szkieletowy. Konkretne techniki wytwarzania fragmentu ds DNA, ligacji i składania kwasów nukleinowych, klonowania, transfekcji, wyrażania, syntezy bibliotek w fazie stałej, syntezy bibliotek w fazie roztworu, kompleksowa ewolucja pozycyjna, kombinatoryczna synteza białka, ocena ilościowa ekspresji przy zastosowaniu oceny ilościowej w teście ELISA i teście β-galaktozydazy oraz funkcjonalny test ELISA są przedstawione w części Przykłady. [0173] W innym wykonaniu wynalazku, ewolucja synergiczna może być stosowana w celu zwiększenia powinowactwa wiązania przeciwciała. W tym wykonaniu, można dokonać optymalizacji regionu zmiennego przeciwciała. Na przykład, aby wytworzyć mutanty przeciwciał, CPE przeprowadza się dla regionów zmiennych łańcucha lekkiego i łańcucha ciężkiego wybranego przeciwciała i wytwarza się EvoMap. Selekcjonuje się mutanty do ponownego składania; na przykład, wybiera się warianty łańcucha lekkiego i wybiera się warianty łańcucha ciężkiego do składania. Nieinaktywujące aminokwasowe mutacje punktowe są wybierane spośród miejsc częściowo podlegających mutacji, które są blisko miejsc niepodlegającymi mutacji. Technologię ponownego łączenia wykorzystującą CPS można zastosować do wytworzenia biblioteki łańcuchów ciężkich. Warianty łańcucha lekkiego mogą być łączone z wariantami łańcucha ciężkiego, klonowane, poddawane wyrażaniu i warianty przeszukuje się jako pełne IgG z nadsączy eukariotycznych linii komórkowych. Powinowactwo wiązania dla niektórych wariantów ocenia się przy zastosowaniu, na przykład, ELISA, testów z oprzyrządowaniem BIAcore i/lub Sapidyne lub inne techniki znane w tej dziedzinie. Ewolucja miejsc podatnych 3 [0174] W innym wykonaniu, CPE/EvoMap można zastosować do identyfikacji i wykorzystania miejsc w pełni podlegających mutacji. W jednym z aspektów, wykorzystanie wielu miejsc w pełni podlegających mutacji określa się jako ewolucję miejsc podatnych i jest ona stosowana do dokonywania ukierunkowanych zmian, takich jak wprowadzanie miejsc glikozylacji (np. kodonów dla aminokwasów dla N- lub O-glikozylacji; Asn w obrębie sekwencji konsensusowej Asn-Aa-Ser-Thr lub Ser/Thr) i sprzęgania chemicznego. Ewolucja miejsc podatnych może być również stosowana do 33
35 1 2 3 projektowania miejsc cięcia dla proteaz, wprowadzania znaczników do oczyszczania i/lub wykrywania, znakowania swoistego miejscowo i tym podobnych. Ponadto, optymalizacja kodonów dla mutacji cichych może być wykorzystana do polepszenia wyrażania przeciwciał. W tym wykonaniu, nazywanym ewolucją miejsc podatnych, po wyrażeniu przeciwciała, wytwarzane biblioteki zmutowanych polipeptydów przeszukuje się ponownie pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności w porównaniu do polipeptydu matrycowego. W jednym aspekcie, określona uprzednio właściwość obejmuje zmniejszenie agregacji przeciwciało-białko, wzmocnienie stabilności przeciwciała lub zwiększoną rozpuszczalność przeciwciała. W jednym aspekcie, biblioteki zmutowanych polipeptydów przeszukuje się pod kątem dwóch lub więcej właściwości jednocześnie. W innym aspekcie, można do wprowadzenia miejsc glikozylacji zastosować dowolny eukariotyczny system ekspresyjny, który glikozyluje, taki jak, na przykład, u ssacze, roślinne, drożdżowe i owadzie linie komórkowe. [017] W ewolucji miejsc podatnych, ocena bioinformatyczna i rentgenowskich struktur krystalicznych przeciwciał spokrewnionych lub przeciwciała lub polipeptydu matrycowego, jest przydatna do optymalizacji matrycy. W jednym z aspektów, wybrane miejsca nie są w pozycjach reszt kontaktowych. W innym aspekcie, selekcja mutacji niepowierzchniowych przeciwciała pozwala na zmniejszenie ryzyka immunogenności. [0176] Zastosowania ewolucji miejsc podatnych obejmują, ale nie są to tego ograniczone, zmniejszenie agregacji przeciwciało-białko, polepszenie rozpuszczalności przeciwciała, optymalizację farmakokinetyki z wykorzystaniem bibliotek glikozylacji, optymalizację struktury drugorzędowej i trzeciorzędowej przeciwciała i deimmunizację miejsc antygenowych bądź bezpośrednio poprzez zbiory mutacji, bądź pośrednio poprzez maskowanie glikozylacji. [0177] W jednym z aspektów ewolucji miejsc podatnych mapa EvoMap jest wykorzystywana do identyfikacji miejsc w pełni podlegających mutacji, wytwarzanie CPS przeprowadza się z wprowadzeniem reszt do glikozylacji do miejsc w pełni podlegających mutacji (lub mutacji cichych dla efektów translacji), a przeszukiwanie kombinatorycznej glikozylowanej biblioteki przeprowadza się przy zastosowaniu analizy analitycznej (np. analizy poprzez spektroskopię mas, dynamiczne rozpraszanie światła), zmniejszenie immunogenności (przy zastosowaniu bioinformatyki lub testu) i/lub analizy farmakokinetycznej (np. w myszach Foxn1nu). [0178] W jednym z aspektów, ewolucję miejsc podatnych można zastosować do deimmunizacji aby wyeliminować immunogenność utrzymując funkcję. Deimmunizację poprzez ewolucję miejsc podatnych można przeprowadzić maskując immunogenność przy zastosowaniu glikozylacji, identyfikowanie podstawień aminokwasowych stanowiących spectra ludzkich mutacji hipersomatycznych, które mogą wyeliminować immunogenność przy utrzymaniu funkcji, zmniejszenie dawki dla obniżenia potencjału immunogenności i minimalizację zmian niepowierzchniowych reszt aminokwasowych. Ponadto, można zastosować bazy danych i algorytmy dla immunogenności w celu zidentyfikowania i zastąpienia potencjalnych epitopów wiążących MHC. W jednym aspekcie, aby wytworzyć warianty przewidywanie modyfikowania in silico jest łączone z danymi CPE/CPS. [0179] Obniżoną tendencję do wytwarzania epitopów dla komórek T i/lub deimmunizacji można mierzyć technikami znanymi w tej dziedzinie. Korzystnie, deimmunizację białek można testować in 34
36 1 2 3 vitro, stosując test proliferacji komórek T. W tym teście PBMC od dawcy reprezentujące > 80% alleli HLA-DR na świecie, przeszukuje się pod kątem proliferacji w odpowiedzi bądź na peptydy typu dzikiego, bądź deimmunizowane. Idealnie, jeżeli proliferacja komórek jest wykrywana tylko po załadowaniu komórek prezentujących antygen peptydami typu dzikiego. Dodatkowe testy dla deimmunizacji obejmują testy ponownej stymulacji ludzkich PBMC in vitro (na przykład test ELISA dla interferonu gamma (TH1) lub IL4 (TH2). Alternatywnie, można zbadać deimmunizację poprzez wyrażanie tetramerów HLA-DR reprezentujących wszystkie haplotypy. W celu zbadania, czy deimmunizowane peptydy są prezentowane na haplotypach HLA-DR, można zmierzyć wiązanie np. peptydów wyznakowanych fluorescencyjnie na PBMC. Jest to pomiar HLA klasy I i klasy II myszy transgenicznych pod kątem odpowiedzi na antygen docelowy (np. interferon gamma lub IL4). Alternatywnie, to przeszukiwanie biblioteki epitopów przy zastosowaniu nauczonych komórek T (MHCI 9mer; MHC klasy II mer) z PBMC i/lub testy z zastosowaniem myszy transgenicznych. Ponadto deimmunizację można udowodnić przez określenie, czy przeciwciała przeciw deimmunizowanym cząsteczkom zostały wytworzone po podaniu u pacjentów. [0180] W innym przykładzie wykonania, techniki ewolucji miejsc podatnych według niniejszego wynalazku można zastosować do optymalizacji wyrażania. W jednym z aspektów, niniejszy wynalazek ujawnia zastosowanie metod inżynierii białek do uzyskania wariantów Fc zoptymalizowanych pod względem kodonów dla mutacji cichej z polepszonym wyrażaniem w komórkach eukariotycznych. Mutacja cicha to taka, w której zmiana sekwencji DNA nie powoduje zmian w sekwencji aminokwasowej przeciwciała. W jednym aspekcie, mutagenezę kodonów przeprowadza się w regionie stałym w celu optymalizacji wyrażania w komórkach eukariotycznych. Wariant Fc zoptymalizowany pod względem kodonów o polepszonych właściwościach wyrażania przy zachowaniu zdolności do pośredniczenia w funkcjach efektorowych polepsza wytwarzanie przeciwciał terapeutycznych. W tym aspekcie, na przykład, region stały cząsteczki przeciwciała może zostać poddany ewolucji dla przeszukiwania w różnych gospodarzach do wyrażania, na przykład, do przeszukiwania pod kątem wyrażania w ssaczych liniach komórkowych, stosując CHO, HEK293 i COS-7. Przykład optymalizacji wyrażania poprzez mutagenezę kodonów w regionie stałym do wyrażania w komórkach ssaków jest przedstawiony na Fig. 3 i opisany w Przykładzie 19. Każdy z pokazanych poziomów wyrażania jest średnią z 4 punktów danych i jest potwierdzony na wielu doświadczeniach. Zwielokrotnioną zdolność linii komórkowej wykazano dla pierwszego badanego mutanta w systemach ekspresyjnych dla linii komórkowych HEK293 i CHO. [0181] Ponadto, EvoMap można zastosować do wytworzenia 3-wymiarowych obliczeniowych modeli cząsteczkowych oligopeptydu lub jego konkretnych regionów, w celu zbadania mechanizmów strukturalnych uczestniczących w, np., swoistości i stabilności przeciwciało-epitop. Hipotetyczna trójwymiarowa mapa EvoMap jest pokazana na Fig. 13. [0182] Informacje z EvoMap mogą być również łączone z informacją strukturalną (jeśli jest dostępna), aby wybrać, np., tylko reszty powierzchniowe dla mutacji w celu zwiększenia rozpuszczalności/zmniejszenia agregacji. Kompleksowa insercyjna ewolucja pozycyjna 3
37 1 2 3 [0183] W jednym wykonaniu, wynalazek dostarcza sposobów identyfikowania i mapowania zmutowanych polipeptydów utworzonych z lub w oparciu o polipeptyd matrycowy. Odnosząc się do Fig. 8, stosując liniowy peptyd jako prosty przykład, w pierwszym etapie, zbiór naturalnie występujących wariantów aminokwasowych (lub jego podzbiór lub pochodne aminokwasów) dla każdego kodonu od pozycji 2 do n (gdzie n odpowiada liczbie reszt w łańcuchu polipeptydowym) jest wytwarzany w procesie określanym tu jako kompleksowa insercyjna ewolucja pozycyjna (CPI ). [0184] W CPI aminokwas jest wstawiany po każdym aminokwasie na przestrzeni polipeptydu matrycowego po jednym na raz aby wytworzyć zbiór wydłużonych polipeptydów. CPI można stosować do wprowadzania 1, 2, 3, 4 lub do nowych miejsc w tym samym czasie. Każdy z aminokwasów jest dodawany w każdej nowej pozycji, po jednym na raz, tworząc zbiór różnych cząsteczek dla każdej nowej pozycji dodanej do matrycy. W tym przypadku, pozycja 1, którą jest metionina i jest niezmienna, jest pomijana. Procedura ta jest powtarzana dla każdego łańcucha polipeptydowego cząsteczki docelowej. Minimalny zbiór mutacji aminokwasowych zawiera tylko jeden kodon dla każdego z naturalnych aminokwasów. [018] Niniejszy wynalazek dotyczy sposobów identyfikowania i mapowania zmutowanych polipeptydów utworzonych z, lub w oparciu o, polipeptyd matrycowy. Zwykle, polipeptyd będzie zawierać n reszt aminokwasowych, gdzie sposób obejmuje: (a) wytwarzanie n+[ x (n-1)] odrębnych polipeptydów, gdzie każdy polipeptyd różni się od polipeptydu matrycowego tym, że ma wstawiony po każdej pozycji w matrycy każdy z aminokwasów, po jednym na raz (jak pokazano na Fig. 1); badanie każdego polipeptydu pod kątem co najmniej jednej określonej właściwości, cechy lub aktywności; i (b) dla każdego przedstawiciela zidentyfikowanie jakichkolwiek zmiany w tej właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego. [0186] W jednym przykładzie wykonania, jeden lub więcej regionów wybiera się do mutagenezy, aby dodać jedną pozycję na raz, jak opisano powyżej. W takim przypadku, n reprezentuje podzbiór lub region polipeptydu matrycowego. Na przykład, gdy polipeptydem jest przeciwciało, całe przeciwciało lub jeden albo więcej regionów determinujących dopasowanie (CDR) przeciwciała są poddawane mutagenezie, aby dodać jedną pozycję na raz w polipeptydzie matrycowym po każdej pozycji. [0187] Wynalazek obejmuje zatem sposoby mapowania zbioru zmutowanych przeciwciał, utworzonych z przeciwciała matrycowego mającego co najmniej jeden, a korzystnie sześć regionów determinujących dopasowanie (CDR), gdzie CDR razem zawierają n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n+[ x (n-1)] oddzielnych przeciwciał, gdzie każde przeciwciało różni się od przeciwciała matrycowego tym, że ma wstawioną pojedynczą określoną uprzednio pozycję, po jednej na raz, po każdej pozycji w sekwencji matrycowej; (b) badanie każdego zbioru pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; oraz (c) dla każdego członka zidentyfikowanie jakichkolwiek zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego. W przypadku przeciwciał, określoną uprzednio właściwością, cechą lub własnością może być, na przykład, powinowactwo wiązania i/ lub immunogenność. [0188] Ponadto, dostarczone są sposoby wytwarzania zbioru zmutowanych przeciwciał, wytworzonych z przeciwciała matrycowego, zawierających co najmniej jeden region determinujący 36
38 1 2 3 dopasowanie (CDR), gdzie CDR zawierają n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n+[ x (n-1)] oddzielnych przeciwciał, gdzie każde przeciwciało różni się od przeciwciała matrycowego tym, że ma ekstra aminokwas dodany w jednej określonej uprzednio pozycji w CDR. W innym przykładzie wykonania, przeciwciało zawiera sześć CDR i razem CDR zawierają n reszt aminokwasowych. [0189] W innym przykładzie wykonania, nowe wydłużone polipeptydy opisane powyżej są dalej mutowane i mapowane po przeszukiwaniu w celu zidentyfikowania zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu skróconego. Zazwyczaj wydłużony polipeptyd będzie zawierać n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n (n-1 w przypadku, w którym początkową resztą jest metionina) oddzielnych zbiorów polipeptydów, gdzie każdy zbiór zawiera elementy-polipeptydy mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji polipeptydu; gdzie każdy polipeptyd różni się jedną pojedynczą określoną uprzednio pozycją; badanie każdego zbioru pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; (b) dla każdego członka zidentyfikowanie jakichkolwiek zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego; i ewentualnie (c) wytworzenie mapy funkcyjnej odzwierciedlającej takie zmiany. Korzystnie, jeżeli liczba różnych wytwarzanych elementów-polipeptydów jest równa n x X (lub [n-1] x X, w zależności od przypadku). [0190] Jako alternatywa, sposób obejmuje wytwarzanie pojedynczej populacji zawierającej zbiór zmutowanych polipeptydów z wydłużonych polipeptydów. W tym wykonaniu całą nową populację przeszukuje się, identyfikuje się poszczególnych przedstawicieli i wytwarza mapę funkcyjną. [0191] Typowo, jeśli stosowany jest każdy naturalnie występujący aminokwas, X będzie wynosić 19 (co odpowiada naturalnie występującym resztom aminokwasowym, wyłączając konkretną resztę obecną w danym położeniu polipeptydu matrycowego). Jednakże, może być stosowany dowolny zbiór aminokwasów, a każdy zbiór polipeptydów mogą być podstawiony wszystkimi lub podzbiorem ogółu X stosowanego w całej populacji. [0192] Jednakże uważa się, że każdy system ekspresyjny może cierpieć z powodu preferencji stosowania kodonów, gdzie niewystarczające pule trna mogą prowadzić do zatrzymania translacji, przedwczesnego zakończenia translacji, przesunięcia ramki odczytu przy tłumaczenia i błędnego wprowadzania aminokwasów. Dlatego też, w celu optymalizacji wyrażania każdy zestaw zawiera aż do 63 różnych kodonów. [0193] Każdy zbiór aminokwasów przeszukuje się pod kątem jednej, a korzystnie dwóch lub więcej, pożądanych cech, takich jak polepszona funkcja; mutacje neutralne, hamujące mutacje i wyrażanie. [0194] W jednym aspekcie, wydłużone polipeptydy można zmapować aby zidentyfikować zmianę we właściwości, cesze lub aktywności pojawiającej się w rezultacie w skróconych polipeptydach w stosunku do typu dzikiego. Dane z każdego zestawu łączy się dla całego polipeptydu lub cząsteczki docelowej. Wyniki dodatnie z przeszukiwania wydłużonych polipeptydów (cząsteczek docelowych) można następnie wykorzystać do dalszej kompleksowej mutagenezy łańcucha(ów) i przeszukiwania, jak tu opisano. Dane z mutagenezy dostarczają szczegółowej mapy funkcyjnej (określanej tu jako EvoMap ) wytworzonej cząsteczki docelowej. Ta mapa zawiera szczegółowe informacje, jak każda mutacja wpływa na wydajność/wyrażanie cząsteczki docelowej. Pozwala to na identyfikację 37
39 wszystkich miejsc, gdzie żadne zmiany nie mogą być dokonywane bez utraty funkcji przeciwciała (lub wiązania antygenu/receptora przeciwciał). Pokazuje to również, gdzie zmiany mogą być dokonywane bez wpływu na funkcję. [019] W innym aspekcie, CPE można zastosować, aby wytworzyć bibliotekę,, aż do 1 lub do wszystkich 19 aminokwasów w każdej pozycji będącej przedmiotem zainteresowania. Kompleksowa delecyjna ewolucja pozycyjna [0196] Kompleksowa delecyjna ewolucja pozycyjna (CPD ) odnosi się do sposobów identyfikowania i mapowania zmutowanych polipeptydów utworzonych z lub w oparciu o polipeptyd matrycowy. Ewolucja CPD usuwa każdy aminokwas na przestrzeni przeciwciała, jedną pozycję na raz. Zazwyczaj polipeptyd będzie zawierać n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n-1 (n-2 w przypadku, gdzie początkową resztą jest metionina) oddzielnych zbiorów polipeptydów, gdzie każdy polipeptyd różni się od polipeptydu matrycowego tym, że nie ma jednej określonej uprzednio pozycji; badanie każdego polipeptydu pod kątem co najmniej jednej określonej właściwości, cechy lub aktywności; i (b) dla każdego przedstawiciela zidentyfikowanie jakichkolwiek zmiany w tej właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego. [0197] W jednym przykładzie wykonania ewolucji CPD, jeden lub więcej regionów wybiera się do mutagenezy, aby usunąć jedną pozycję na raz, jak opisano powyżej. W takim przypadku, n reprezentuje podzbiór lub region polipeptydu matrycowego. Na przykład, gdy polipeptydem jest przeciwciało, całe przeciwciało lub jeden albo więcej regionów determinujących dopasowanie (CDR) przeciwciała są poddawane mutagenezie, aby usunąć jedną pozycję na raz w polipeptydzie matrycowym po każdej pozycji. [0198] W jednym przykładzie wykonania, CPD obejmuje zatem sposoby mapowania zbioru zmutowanych przeciwciał, utworzonych z przeciwciała matrycowego mającego co najmniej jeden, a korzystnie sześć regionów determinujących dopasowanie (CDR), gdzie CDR razem zawierają n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie (n-1) oddzielnych przeciwciał, gdzie każde przeciwciało różni się od przeciwciała matrycowego tym, że nie ma pojedynczej określonej uprzednio pozycji; (b) badanie każdego zbioru pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; i (c) dla każdego członka zidentyfikowanie jakichkolwiek zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego. W przypadku przeciwciał, określoną uprzednio właściwością, cechą lub własnością może być, na przykład, powinowactwo wiązania i/ lub immunogenność. [0199] Jeden z aspektów ewolucji CPD obejmuje sposoby wytwarzania zbioru zmutowanych przeciwciał, wytworzonych z przeciwciała matrycowego, zawierających co najmniej jeden region determinujący dopasowanie (CDR), gdzie CDR zawierają n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie (n-1) oddzielnych przeciwciał, gdzie każde przeciwciało różni się od przeciwciała matrycowego tym, że ma nie ma jednej określonej uprzednio pozycji w CDR. W innym przykładzie wykonania, przeciwciało zawiera sześć CDR i razem CDR zawierają n reszt aminokwasowych. 38
40 1 2 3 [00] W innym przykładzie wykonania ewolucji CPD, nowe skrócone polipeptydy opisane powyżej są dalej mutowane i mapowane po przeszukiwaniu w celu zidentyfikowania zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu skróconego. Zazwyczaj skrócony polipeptyd będzie zawierać n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n (n-1 w przypadku, w którym początkową resztą jest metionina) oddzielnych zbiorów polipeptydów, gdzie każdy zbiór zawiera elementy-polipeptydy mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji polipeptydu; gdzie każdy polipeptyd różni się jedną pojedynczą określoną uprzednio pozycją; badanie każdego zbioru pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; (b) dla każdego członka zidentyfikowanie jakichkolwiek zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego; i ewentualnie (c) wytworzenie mapy funkcyjnej odzwierciedlającej takie zmiany. Korzystnie, jeżeli liczba różnych wytwarzanych elementów-polipeptydów jest równa n x X (lub [n-1] x X, w zależności od przypadku). [01] Jako alternatywę, sposób obejmuje wytwarzanie pojedynczej populacji zawierającej zbiór zmutowanych polipeptydów ze skróconych polipeptydów. W tym wykonaniu całą nową populację przeszukuje się, poszczególnych przedstawicieli identyfikuje i wytwarza mapę funkcyjną. Typowo, jeśli stosowany jest każdy naturalnie występujący aminokwas, X będzie wynosić 19 (co odpowiada naturalnie występującym resztom aminokwasowym, wyłączając konkretną resztę obecną w danym położeniu polipeptydu matrycowego). Jednakże, może być stosowany dowolny zbiór aminokwasów, a każdy zbiór polipeptydów mogą być podstawiony wszystkimi lub podzbiorem ogółu X stosowanego w całej populacji. [02] Dowolne sposoby mutacji lub syntezy można zastosować do wytworzenia zbioru mutantów w ewolucji CPD. W jednym wykonaniu, wytwarzanie polipeptydów obejmuje (i) poddawanie polinukleotydu zawierającego kodony kodujące polipeptyd matrycowy amplifikacji opartej na polimerazie przy użyciu 64-krotnie zdegenerowanego oligonukleotydu dla każdego kodonu, który ma być mutagenizowany, gdzie każdy z tych 64-krotnie zdegenerowanych oligonukleotydów składa z pierwszej sekwencji homologicznej i zdegenerowanej sekwencji tripletu N, N, N, tak aby wytworzyć zbiór polinukleotydów potomnych; i (ii) poddanie tego zbioru polinukleotydów potomnych amplifikacji klonalnej tak, że polipeptydy kodowane przez polinukleotydy potomne są wyrażane. [03] W jednym wykonaniu ewolucji CPD cały skrócony polipeptyd jest poddawany mutagenezie saturacyjnej. W innym wykonaniu, jeden lub więcej regionów wybiera się do mutagenezy saturacyjnej. W takim przypadku, N oznacza podzbiór lub region polipeptydu matrycowego. Na przykład, w przypadku, gdy polipeptydem jest przeciwciało, całe przeciwciało lub jeden albo więcej regionów determinujących dopasowanie (CDR) przeciwciała jest poddawanych mutagenezie saturacyjnej. [04] Ujawnienie ewolucji CPD obejmuje zatem sposoby mapowania zbioru zmutowanych przeciwciał, utworzonych ze skróconego przeciwciała matrycowego mającego co najmniej jeden, a korzystnie sześć, regionów determinujących dopasowanie (CDR), gdzie CDR zawierają n reszt aminokwasowych, gdzie sposób obejmuje (a) wytwarzanie n oddzielnych zbiorów polipeptydów, gdzie każdy zbiór zawiera elementy-polipeptydy mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji polipeptydu; gdzie każdy polipeptyd różni się jedną pojedynczą określoną uprzednio pozycją; i liczba różnych wytworzonych elementów-przeciwciał 39
41 1 2 jest równa n x X; (b) badanie każdego zbioru pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; (b) dla każdego elementu zidentyfikowanie jakichkolwiek zmiany we właściwości, cesze lub aktywności w stosunku do polipeptydu matrycowego; i ewentualnie (c) wytworzenie mapy funkcyjnej odzwierciedlającej takie zmiany; i (d) wytworzenie strukturalnej mapy pozycyjnej takich zmian. W przypadku przeciwciał, określoną uprzednio właściwością, cechą lub własnością może być, na przykład, powinowactwo wiązania i/ lub immunogenność. Jak przedstawiono powyżej, jako alternatywę, można wytworzyć jedną populację zawierającą wszystkie zbiory zmutowanych przeciwciał. [0] Ponadto, dostarczone są sposoby wytwarzania zbioru zmutowanych przeciwciał, utworzonych ze skróconego przeciwciała matrycowego zawierającego co najmniej jeden region determinujący dopasowanie (CDR), gdzie CDR zawierają n reszt aminokwasowych, gdzie sposób obejmuje: (a) wytwarzanie n oddzielnych zbiorów polipeptydów, gdzie każdy zbiór zawiera elementy-polipeptydy mające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji polipeptydu; gdzie każdy polipeptyd różni się jedną pojedynczą określoną uprzednio pozycją; i liczba różnych wytworzonych elementów-przeciwciał jest równa n x X. W innym przykładzie wykonania przeciwciało zawiera sześć regionów CDR i razem, CDR zawierają n reszt aminokwasowych. [06] Metoda ewolucji CPD obejmuje funkcyjną mapę pozycyjną (EvoMap ) wykonaną przy zastosowaniu opisanych tu sposobów. W dodatkowym wykonaniu, na EvoMap mogą być wskazane niektóre reszty szczególnie wrażliwe na zmiany. Dalsza optymalizacja może być realizowana poprzez dodatkowe zmiany mutacyjne w pozycjach poza tymi wrażliwymi miejscami. Jest również możliwe zastosowanie EvoMap do rozpoznania i rekombinowania korzystnych pojedynczych podstawień aminokwasowych i przeszukiwania, aby dalej optymalizować pożądane cechy w cząsteczce docelowej w procesie zwanym kombinatoryczną syntezą białek (CPS ). Kombinatoryczna synteza białek 3 [07] Kombinatoryczna synteza białek (CPS ) obejmuje łączenie poszczególnych wyników dodatnich z CPE, CPI, CPD lub jakiejkolwiek innej techniki ewolucji dla łączenia dwóch lub więcej mutacji. CPS jest stosowana do syntetyzowania przeciwciał z łączonymi mutacjami, które następnie są przeszukiwane pod kątem zoptymalizowanej cechy genu i przeciwciał. Schemat CPS jest pokazany na Fig. 3. W jednym aspekcie, dwie lub więcej mutacji punktowych, których rezultatem są ulepszone mutanty lub mutacje neutralne, są łączone w CPS. [08] W jednym przykładzie wykonania CPE jest łączona z CPS w celu wytworzenia mutantów, które są przeszukiwane pod kątem pożądanej właściwości. W jednym aspekcie, czas i zasoby mogą być oszczędzane w procesie CPE poprzez zmianę 2 aa lub 3 aa lub 4 aa naraz w przeciwieństwie do jednego naraz; zatem jeśli liczba aa w przeciwciele wynosi N, całkowita liczba wytwarzanych i przeszukiwanych pod kątem 2 aa naraz wynosiłaby ( 2 ) x ½N; 3 naraz wynosiłaby ( 3 ) x ⅓N, itd. Przykładowo, w jednym konkretnym aspekcie, (w przykładzie z 2 aa): 1. aa w 1. pozycji jest łączony z wszystkimi w 2. pozycji aa, a wszystkie inne aa pozostają takie same, a następnie 2. aa w 1.
42 1 2 3 pozycji aa jest łączony z wszystkimi w 2. pozycji aa, a wszystkie inne aa pozostają takie same. Cała populacja jest przeszukiwana pod kątem mutantów polepszonych, a następnie dokonywana jest mutacja w drugim zbiorze następnych kolejnych dwóch aa. W podobnym aspekcie, może to być wykonane dla 3 aa naraz lub 4 aa naraz. W innym aspekcie, ewentualnie następuje proces CPE dla ulepszonych mutantów po CPS (w tym dowolnych ich podgrup). [09] W jednym aspekcie, do procesu mogą być włączone aminokwasy nienaturalne (a więc wszystkich 19 innych aminokwasów lub ich podzbiór plus aminokwasy nienaturalne) poprzez zastosowanie nowych technologii, takich jak kodon kwadrupletowy opisany w załączonych i pokrewnych publikacjach. Neumann i wsp. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, (14 lutego ). W tym aspekcie CPE/CPS przeprowadza się do wprowadzania aminokwasów nienaturalnych. [02] W dalszym aspekcie, do cała biblioteka CPE jest wytwarzana syntetycznie (syntetyzowanie wszystkich cząsteczek na komercyjnie dostępnych maszynach). W przypadku, gdy synteza maszynowa nie może wytworzyć dostatecznie dużych nici, syntetyzowane są fragmenty, a następnie poddawane ligacji z wytworzeniem cząsteczek pełnej długości. Ta biblioteka jest przeszukiwana, a następnie przeprowadza się CPS, aby połączyć pożądane mutacje. Jest to proces dwuetapowy, w którym po CPE następuje CPS, a nie tylko jeden etap CPE. [0211] W innym aspekcie, wytwarzana jest i przeszukiwana biblioteka CPE, po czym następuje CPS łącząca ulepszone mutanty, jak następuje: jeżeli jest ulepszonych mutantów, testuje się jedną cząsteczkę ze wszystkimi zmianami, następnie testuje się wszystkie wersje 9 mutacji, następnie 8, 7, 6, itd., aż w jednej z grup nie ma ulepszonej cząsteczki względem dowolnej z poprzednich grup. Po zidentyfikowaniu ulepszonej cząsteczki proces można zakończyć. [0212] W kolejnym aspekcie, CPE przeprowadza się w celu zidentyfikowania ulepszonych mutantów i mutacji neutralnych pod względem powinowactwa i wyrażania, a następnie przeprowadza się CPS z łączeniem ulepszonych mutantów i mutacji neutralnych i biblioteki przeszukuje się ponownie pod kątem dalszego polepszenia cech, takich jak funkcja, powinowactwo i/lub wyrażanie. [0213] W kolejnym aspekcie, CPE przeprowadza się na kodonach domeny Fc lub innej domeny dla zmian glikozylacji. [0214] W innym aspekcie, można przeprowadzić CPE lub CPE w połączeniu z CPS wobec mikrorna lub intronów. [021] W kolejnym aspekcie przeprowadza się CPE lub CPE w połączeniu z CPS wobec CDR przeciwciała gryzonia, a następnie przeszukuje ulepszone mutanty, po czym następuje humanizacja. [0216] W jednym z aspektów, CPE lub CPE w połączeniu z CPS przeprowadza się w celu wytworzenia alternatywnych pośrednich nukleotydów, które prowadzą do pożądanej mutacji w końcowej reakcji, na przykład, metylowanej cytozyny, który przekształca się w uracyl. [0217] W jednym aspekcie, CPE lub CPE w połączeniu z CPS wraz z informatyką stosuje się do przekształcania CDR myszy w ludzki CDR i vice versa. [0218] W jednym z aspektów, CPE lub CPE w połączeniu z CPS stosuje się z 2 i 3 mutacjami rozmieszczonymi na całej długości przeciwciała. 41
43 1 2 3 [0219] W innym aspekcie, CPE lub CPE w połączeniu z CPS stosuje się w dwułańcuchowym wektorze do oceny przeszukiwania pod kątem zwiększonej czułości. [02] W kolejnym aspekcie, CPE lub CPE w połączeniu z CPS stosuje się do selekcji pod kątem zmian allosterycznych. [0221] Do oceny mutantów CPE/CPS można zastosować dowolną z kilku technik przeszukiwania. W jednym z aspektów mutanty po CPE lub CPE w połączeniu z CPS mogą być wydzielane i prezentowane w gospodarzach eukariotycznych. Alternatywnie, mutanty po CPE lub CPE w połączeniu z CPS mogą być wytwarzane w E. coli i przeszukiwane w gospodarzach eukariotycznych. W innym aspekcie, CPE przeprowadza się zaczynając od 1 aa i aa, a następnie przeprowadza się CPS; następnie robi się to z pozostałymi 19 aa. W innym aspekcie, CPE lub CPE w połączeniu z CPS stosuje się do ewolucji przeciwciał specyficznie ze zmianami aminokwasów niepowierzchniowych. W jednym aspekcie, CPE można stosować do wielowymiarowego mapowania epitopów. W innym aspekcie, CPE lub CPE w połączeniu z przeszukiwaniem CPS można przeprowadzać przejściowo w komórkach eukariotycznych. W dalszym aspekcie, przeprowadza się CPE lub CPE w połączeniu z CPS, a następnie przeprowadza się sekwencjonowanie i analizę macierzy dla wszystkich klonów, na przykład w formacie w oparciu o czipy lub w oparciu o studzienki dla wyrażania i przeszukiwania. W innym aspekcie, CPE lub CPE w połączeniu z CPS stosuje się uzyskania zmian koordynacji jonów metalu poprzez dokonywanie selekcji w różnych stężeniach jonów. W kolejnym aspekcie, przeprowadza się CPE lub CPE w połączeniu z CPS i przeciwciała są wyrażane i przeszukiwane w warunkach wolnych od komórek i w żywych organizmach innych niż człowiek. W jednym z aspektów, CPE lub CPE w połączeniu z przeszukiwaniem poprzez CPS komórek macierzystych przeprowadza się pod kątem różnych efektów na różnicowanie i wyrażanie przeciwciała oraz RNA i mrna. W kolejnym aspekcie, CPE lub CPE w połączeniu z multipleksowym przeszukiwaniem poprzez CPS przeprowadza się pod kątem wielu cech przeciwciała, takich jak wyrażanie i wiązanie. W innym aspekcie, CPE lub CPE w połączeniu z CPS przeprowadza się na cząsteczkach matrycowych uczestniczących w transporcie mózgowym i przechodzeniu przez błony; i mutanty przeszukuje się pod kątem polepszonych cech. W jednym z aspektów, mutanty po CPE lub CPE w połączeniu z CPS przeszukuje się pod kątem przeciwciał higroskopijnych. W innym aspekcie, mutanty po CPE lub CPE w połączeniu z CPS bada się pod kątem selekcji przeciwciał dynamicznych. W jednym z aspektów, CPE lub CPE w połączeniu z przeszukiwaniem CPS przeprowadza się poza stanem docelowym w celu zidentyfikowania mutantów w obrębie stanu docelowego i odwrotnie. [0222] W jednym przykładzie wykonania, dowolny z powyższych aspektów CPE lub CPE połączonych CPS stosuje się w połączeniu ze sposobem wybranym z CPT, CPD i CPD w kombinacji z CPI. [0223] W innym przykładzie wykonania, dowolny z powyższych aspektów CPE lub CPE w połączeniu z CPS stosuje się w połączeniu ze sposobem wybranym z ewolucji miejsc podatnych i ewolucji synergicznej przeprowadzanej na matrycy. [0224] Termin matryca może odnosić się do polipeptydu bazowego lub polinukleotydu kodującego taki polipeptyd. Co będzie wiadome specjaliście w tej dziedzinie, w sposobach i kompozycjach według niniejszego wynalazku można zastosować dowolną matrycę. Matryca, która może być mutowana, a tym samym podlegać ewolucji, może być stosowana do kierowania syntezą innego polipeptydu lub 42
44 1 2 3 biblioteki polipeptydów, jak to opisano w niniejszym wynalazku. Jak tu opisano bardziej szczegółowo, podlegająca ewolucji matryca koduje syntezę polipeptydu i może być stosowana następnie do dekodowania historii syntezy polipeptydu, do pośredniej amplifikacji polipeptydu i/lub do przeprowadzenia ewolucji (tj. dywersyfikacji, selekcji i amplifikacji) polipeptydu. Podlegającą ewolucji matrycą jest, w niektórych wykonaniach, kwas nukleinowy. W pewnych wykonaniach niniejszego wynalazku, matryca jest oparta na kwasie nukleinowym. W innych wykonaniach wynalazku, matrycą jest polipeptyd. [022] Matryce-kwasy nukleinowe stosowane w niniejszym wynalazku stanowią DNA i RNA, hybryda DNA i RNA lub pochodna DNA i RNA i mogą być jedno- lub dwuniciowe. Sekwencja matrycy jest stosowana do zakodowania syntezy polipeptydu, korzystnie związku, które nie jest lub nie przypomina kwasu nukleinowego lub analogu kwasu nukleinowego (np., polimeru nienaturalnego lub małej cząsteczki). W przypadku niektórych polimerów nienaturalnych, matrycę-kwas nukleiny, stosuje się w celu uszeregowania jednostek monomeru w kolejności pojawiania się w polimerze i zbliżania ich do sąsiednich jednostek monomeru wzdłuż matrycy tak, że będą one reagować i zostaną połączone wiązaniem kowalencyjnym. W niektórych innych wykonaniach, matrycę można zastosować do wytworzenia polimerów nienaturalnych poprzez amplifikację w reakcji PCR syntetycznej biblioteki dla matrycy DNA składającej się z losowego regionu nukleotydów. [0226] Należy zauważyć, że matryca może się znacznie różnić liczbą zasad. Na przykład, w niektórych wykonaniach, matryca może mieć długość od do 000 zasad, korzystnie od do 00 zasad. Długość matrycy będzie oczywiście zależeć od długości kodonów, złożoności biblioteki, długości nienaturalnego polimeru, który ma być zsyntetyzowany złożoności małych cząsteczek, które mają być zsyntetyzowane, stosowania sekwencji odstępnikowych, itp. Taką sekwencję kwasu nukleinowego można wytwarzać przy zastosowaniu dowolnej metody znanej w tej dziedzinie do przygotowywania sekwencji kwasu nukleinowego. Metody te obejmują metody in vivo i in vitro, w tym, PCR, wytworzenie plazmidu, trawienie endonukleazami, syntezę w fazie stałej, transkrypcję in vitro, rozdzielanie nici, itd. W niektórych wykonaniach, matrycę kwasu nukleinowego syntetyzuje się przy zastosowaniu zautomatyzowanego syntetyzatora DNA. [0227] Jak omówiono powyżej, w niektórych wykonaniach niniejszego wynalazku, sposób stosuje się do syntezowania polipeptydów, które nie są lub nie przypominają kwasów nukleinowych lub analogów kwasów nukleinowych. Zatem, w pewnych wykonaniach niniejszego wynalazku, matryca-kwas nukleinowy składa się z sekwencji zasad, które kodują syntezę nienaturalnego polimeru lub małej cząsteczki. mrna zakodowany w matrycy-kwasie nukleinowym korzystnie zaczyna się od konkretnego kodonu, który umiejscawia chemicznie reaktywne miejsce, od którego może odbywać się polimeryzacja albo w przypadku syntezy małej cząsteczki kodon startowy może kodować antykodon związany ze szkieletem małej cząsteczki lub pierwszym uczestnikiem reakcji. Kodon startowy według niniejszego wynalazku jest analogiczny do kodonu startowego ATG, który koduje aminokwas-metioninę. [0228] W jeszcze innych wykonaniach wynalazku, matryca-kwas nukleinowy sama może być modyfikowana, aby zawierać miejsce inicjacji syntezy polimeru (np. nukleofilu) lub szkieletu małej cząsteczki. W niektórych wykonaniach, matryca-kwas nukleinowy zawiera pętlę spinki do włosów na 43
45 1 2 3 jednym ze swoich końców kończącą się grupą reaktywną wykorzystywaną do inicjowania polimeryzacji jednostek monomeru. Na przykład, matryca-dna może zawierać pętlę spinki do włosów kończącą się grupą '-aminową, która może być zabezpieczona lub nie. Z grupy aminowej może rozpocząć się polimeryzacja nienaturalnego polimeru. Reaktywna grupa aminowa może być również wykorzystywana do łączenia szkieletu małej cząsteczki z matrycą-kwasem nukleinowym w celu syntezy biblioteki małych cząsteczek. [0229] Aby zakończyć syntezę nienaturalnego polimeru, w matrycy-kwasu nukleinowym powinien być obecny kodon stop, korzystnie na końcu sekwencji kodującej. Kodon stop według niniejszego wynalazku jest analogiczny do kodonów stop (tj. TAA, TAG, TGA) znajdujących się w transkryptach mrna. Te kodony prowadzą do zakończenia syntezy przeciwciała. W pewnych wykonaniach, wybiera się kodon stop, który jest kompatybilny ze sztucznym kodem genetycznym zastosowanym do zakodowania nienaturalnego polimeru. Na przykład, kodon stop nie powinien kolidować z jakimikolwiek innymi kodonami wykorzystywanymi do kodowania syntezy i powinien być z tego samego ogólnego formatu, jak inne kodony stosowane w matrycy. Kodon stop może kodować jednostkę monomeru, który kończy polimeryzację nie dostarczając grupy reaktywnej do dalszego przyłączania. Na przykład, jednostka monomeru stop może zawierać zablokowaną grupę reaktywną, taką jak acetamid zamiast pierwszorzędowej aminy. W jeszcze innych wykonaniach, jednostka monomeru stop zawiera koniec biotynylowany, dostarczając dogodnego sposobu zakończenia etapu polimeryzacji i oczyszczania uzyskanego polimeru. [02] W jednym wykonaniu, mutagenizowane produkty DNA stosuje się bezpośrednio jako matrycę do syntezy odpowiednich zmutowanych przeciwciał in vitro. Ze względu na dużą skuteczność, z którą można wytworzyć wszystkich 19 podstawień aminokwasowych jako pojedynczą resztę, można przeprowadzić mutagenezę saturacyjną wobec licznych reszt będących przedmiotem zainteresowania, bądź niezależnie, bądź łącznie z innymi mutacjami w obrębie przeciwciała. Stosowany tu termin mutageneza z pełną saturacją jest zdefiniowany jako zastąpienie danego aminokwasu w obrębie przeciwciała innymi 19 naturalnie występującymi aminokwasami. Na przykład, mutageneza saturacyjna miejsc genu, która systematycznie bada minimalnie wszystkie możliwe pojedyncze podstawienia aminokwasowe wzdłuż sekwencji przeciwciała, jest ujawniona w Kretza i wsp., Methods in Enzymology, 04, 388: 3-11; Short, patent USA nr 6,171,8; i Short, patent USA nr 6,62,94. [0231] W jednym z aspektów, wynalazek dostarcza zastosowania starterów dla kodonów (zawierających zdegenerowaną sekwencję N, N, G/T) dla wprowadzania mutacji punktowych do polinukleotydu, tak, aby wytworzyć zestaw polipeptydów potomnych, w których w każdej pozycji aminokwasowej reprezentowany jest pełen zakres pojedynczych podstawień aminokwasowych (patrz patent USA nr 6,171,8; patrz też, patent USA nr,677,149). Stosowane oligo składają się z sąsiadujących ze sobą: pierwszej sekwencji homologicznej, zdegenerowanej sekwencji N, N, G/T i, korzystnie, ale niekoniecznie, drugiej sekwencji homologicznej. Dalsze potomne produkty translacji po zastosowaniu takich oligo obejmują wszystkie możliwe zmiany aminokwasowe w każdym miejscu aminokwasu wzdłuż polipeptydu, ponieważ degeneracja sekwencji N, N G/T obejmuje kodony dla wszystkich aminokwasów. 44
46 1 2 3 [0232] Wykorzystywanie kodonów jest jednym z ważnych czynników w ekspresji genów eukariotycznych. Częstotliwość, z jaką różne kodony są wykorzystywane różni się znacznie pomiędzy różnymi gospodarzami i między przeciwciałami wyrażanymi na wysokich lub niskich poziomach w obrębie tego samego organizmu. Najbardziej prawdopodobnym powodem tego zróżnicowania jest to, że korzystne kodony korelują z ilością pokrewnych trna dostępnych w komórce. Jest możliwe, że wykorzystywanie kodonów i stężenie akceptorowych trna podlegały koewolucji i że presja selekcyjna dla tej koewolucji jest bardziej widoczna dla genów z wysoką ekspresją niż genów z ekspresją na niskich poziomach. [0233] W jednym aspekcie, jeden taki zdegenerowany oligo (składający się z jednej zdegenerowanej kasety N, N, G/T) jest stosowany do poddawania każdego wyjściowego kodonu w matrycypolinukleotydzie rodzicielskim pełnemu zakresowi podstawień kodonów. W innym aspekcie stosowane są co najmniej dwie zdegenerowane kasety N, N, G/T - w tym samym oligo lub nie, aby poddać każdy wyjściowy kodon w matrycy-polinukleotydzie rodzicielskim pełnemu zakresowi podstawień kodonów. A zatem więcej niż jedna sekwencja N, N, G/T może być zawarta w jednym oligo, aby wprowadzić mutacje aminokwasowe w więcej niż jednym miejscu. Tych wiele sekwencji N, N, G/T może bezpośrednio sąsiadować ze sobą lub być rozdzielonych jedną lub więcej dodatkowych sekwencji nukleotydowych. W innym aspekcie, oligo, które mogą służyć do wprowadzania addycji i delecji można zastosowane bądź same, bądź łączenie z kodonami zawierającymi sekwencję N, N, G/T, aby wprowadzić dowolną kombinację lub permutację addycji, delecji i/lub podstawień aminokwasowych. [0234] W innym aspekcie, niniejszy wynalazek dostarcza zastosowania zdegenerowanych kaset o mniejszej degeneracji niż sekwencja N, N, G/T. Na przykład, może być pożądane w niektórych przypadkach zastosowanie (np. w oligo) zdegenerowanej sekwencji trypletu składającej się tylko z jednego N, gdzie N może być w pierwszej, drugiej lub trzeciej pozycji trypletu. Dowolne inne zasady, w tym wszelkie ich kombinacje i permutacje mogą być stosowane w dwóch pozostałych pozycjach trypletu. Alternatywnie, może być pożądane w niektórych przypadkach zastosowanie (np., w oligo) zdegenerowanej sekwencji trypletu N, N, N. [023] Jednak należy zauważyć, że użycie zdegenerowanego trypletu N, N, G/T, jak tu ujawniono, jest korzystne z kilku powodów. W jednym aspekcie, ten wynalazek dostarcza sposobów do systematycznego i dość łatwego wytwarzania podstawień pełnego zakresu możliwych aminokwasów (dla ogółem aminokwasów) w każdej ze wszystkich pozycji aminokwasowych w polipeptydzie. A zatem, dla polipeptydu 0-aminokwasowego, niniejszy wynalazek dostarcza sposobu systematycznego i dość łatwego wytwarzania 00 różnych rodzajów (tj. możliwych aminokwasów na pozycję x 0 pozycji aminokwasowych). Należy zauważyć, że poprzez zastosowanie zdegenerowanego oligonukleotydu zawierającego tryplet N, N, G/T, dostarczono 32 indywidualne sekwencje, które kodują możliwych aminokwasów. A zatem w naczyniu reakcyjnym, w którym rodzicielska sekwencja polinukleotydowa jest poddawana mutagenezie saturacyjnej przy zastosowaniu jednego takiego oligo, wytworzonych jest 32 odrębnych polinukleotydów potomnych kodujących różnych polipeptydów. W przeciwieństwie do tego, zastosowanie oligo niezdegenerowanych w mutagenezie ukierunkowanej, prowadzi tylko do jednego produktu potomnego-polipeptydu w naczyniu reakcyjnym. 4
47 1 2 3 [0236] A zatem, w korzystnym przykładzie wykonania, każde naczynie reakcyjne dla mutagenezy saturacyjnej zawiera polinukleotydy kodujące co najmniej cząsteczek polipeptydu potomnego tak, że wszystkie z aminokwasów są reprezentowane w jednej konkretnej pozycji aminokwasowej odpowiadającej pozycji kodonu mutagenizowanego w polinukleotydzie rodzicielskim. 32-krotnie zdegenerowane polipeptydy potomne wytwarzane w każdym naczyniu reakcyjnym dla mutagenezy saturacyjnej można poddawać amplifikacji klonalnej (np. klonować w odpowiednim gospodarzu E. coli stosując wektor ekspresyjny) i poddawać przeszukiwaniu pod kątem wyrażania. Gdy indywidualny polipeptyd potomny zostanie zidentyfikowany przez przeszukiwanie pod kątem wykazywania zmiany we właściwości (w porównaniu do polipeptydu matrycowego), można go zsekwencjonować w celu zidentyfikowania podstawienia aminokwasowego odpowiedzialnego za taką zmianę w nim zawartą. [0237] Do wytworzenia populacji zmutowanych polipeptydów można zastosować dowolną metodę syntezy chemicznej lub mutagenezy i rekombinowania. Przy wykonywaniu niniejszego wynalazku można zastosować, jeśli nie wskazano inaczej, konwencjonalne techniki biologii komórki, hodowli komórkowych, biologii molekularnej, mikrobiologii, biologii transgenicznej, rekombinowania DNA i immunologii, które są w zakresie specjalisty w tej dziedzinie. Takie techniki są w pełni wyjaśnione w piśmiennictwie. Patrz, na przykład, Molecular Cloning A Laboratory Manual, wyd. 2., red. Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press: 1989); DNA Cloning, tomy I i II (red. D. N. Glover, 198); Oligonucleotide Synthesis (red. M. J. Gait, 1984); Mullis i wsp., patent USA nr: 4,683,19; Nucleic Acid Hybridization (red. B. D. Hames & S. J. Higgins 1984); Transcription And Translation (red. B. D. Hames & S. J. Higgins 1984); Culture Of Animal Cells (R. I. Freshney, Alan R. Liss, Inc., 1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical Guide To Molecular Cloning (1984); rozprawa, Methods In Enzymology (Academic Press, Inc., N.Y.); Gene Transfer Vectors For Mammalian Cells (red. J. H. Miller i M. P. Cabs, 1987, Cold Spring Harbor Laboratory); Methods In Enzymnology, tomy 14 i 1 (Wu i wsp. red.), Immunochemical Mehods In Cell And Molecular Biology (red. Mayer and Walker, Academic Press, London, 1987); Handbook Of Experimental Immunology, tomy I-IV (red. D. M. Weir i C. C. Blackwell, 1986); Manipulating the Mouse Embiyo, (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986). [0238] W jednym przykładzie wykonania, polipeptydem matrycowym jest przeciwciało. Przeciwciało poddaje się opisanym tu sposobom, na przykład, mapuje się, żeby zrozumieć, które pozycje w obrębie CDR wpływają na powinowactwo wiązania. Techniki wytwarzania i stosowania różnych konstruktów opartych o przeciwciała i ich fragmenty są dobrze znane w tej dziedzinie. Ważnym aspektem niniejszego wynalazku jest identyfikacja reszt, które odgrywają lub mogą odgrywać rolę w oddziaływaniu będącym przedmiotem zainteresowania (na przykład oddziaływaniu antygenprzeciwciało, chelatowaniu metalu, wiązaniu receptora, wiązaniu substratu, itd). Zgodnie z niniejszym wynalazkiem może być stosowane dowolne przeciwciało lub fragment przeciwciała. [0239] W jednym wykonaniu, można zastosować dowolną platformę ewolucji: CPE, CPI, CPD i CPS, do wytwarzania przeciwciał agonistycznych, tj. przeciwciał aktywujących. Te technologie ewolucji umożliwiają wytwarzanie przeciwciał agonistycznych poza wiązaniem krzyżowym prostszego białka, a w szczególności pozwalają na aktywację receptorów, takich jak GPL-1 lub 2, które są tradycyjnie aktywowane przez peptydy. 46
48 1 2 3 [02] W jednym aspekcie przeciwciała są wybierane przy zastosowaniu FACS lub mikroskopii lub ekwiwalentnej metody pod względem przeciwciał słabo aktywujących, stosując komórki z sygnałami fluorescencyjnymi, które fluoryzują, kiedy receptor na powierzchni komórki jest aktywowany. Następnie narzędzia do ewolucji są stosowane w celu zwiększenia tej aktywacji. Technologia CPS jest następnie wykorzystywana do łączenia polepszonych mutantów. [0241] W innym aspekcie wybierane jest przeciwciało, które wiąże się z miejscem aktywacji receptora, co określa się poprzez mapowanie epitopów. Techniki CPE, CPI i/lub CPD są stosowane do selekcji mutantów, które powodują stymulację receptora, co określa się poprzez wewnątrzkomórkowy odczyt, na przykład fluorescencji w odpowiedzi na uwalnianie jonów wapnia, lub innych testów, które są dobrze znane w tej dziedzinie. Technologię CPS stosuje się następnie do łączenia ulepszonych mutantów. [0242] W konkretnym aspekcie, niektórymi z kluczowych zalet CPI z pojedynczymi, podwójnymi lub potrójnymi insercjami aminokwasowymi jest to, że te wstawione aminokwasy mogą rozciągać się na kieszeń wiążącą receptora dla aktywacji receptora. W innym konkretnym aspekcie, CPD może remodelować i i/lub zmieniać położenie aminokwasów oddziałujących z receptorami w celu polepszenia lub aktywacji, a na koniec CPE może przeprowadzić stosunkowo niewielkie zmiany w celu uzyskania aktywacji receptora. [0243] Swoistość przeciwciała jest określona przez regiony determinujące dopasowanie (CDR) w obrębie regionów zmiennych łańcucha lekkiego (VL) i regionów zmiennych łańcucha ciężkiego (VH). Fragment Fab przeciwciała, który stanowi około jedną trzecią wielkości pełnego przeciwciała, zawiera regiony zmienne łańcucha ciężkiego i lekkiego, cały region stały łańcucha lekkiego i część regionu stałego łańcucha ciężkiego. Cząsteczki Fab są stabilne i dobrze się łączą dzięki udziałowi sekwencji regionu stałego. Jednak wydajność funkcjonalnego Fab wyrażanego w systemach bakteryjnych jest niższa niż w przypadku mniejszego fragmentu Fv zawierającego jedynie regiony zmienne łańcuchów ciężkich i lekkich. Fragment Fv jest najmniejszą częścią przeciwciała, która zachowuje nadal funkcjonalne miejsce wiązania antygenu. Fragment Fv ma takie same właściwości wiązania jak Fab, jednakże bez stabilności nadawanej przez regiony stałe: dwa łańcuchy Fv stosunkowo łatwo dysocjują w warunkach rozcieńczenia. [0244] W jednym aspekcie, regiony VH i VL mogą być połączone przez linker polipeptydowy (Huston i wsp., 1991) w celu stabilizacji miejsca wiązania antygenu. Ten pojedynczy polipeptyd Fv jest znany jako przeciwciało jednołańcuchowe (scfv). VH i VL mogą być zorganizowane z każdą z domen jako pierwszą. Łącznik łączy koniec karboksylowy pierwszego łańcucha z końcem aminowym drugiego łańcucha. [024] Specjalista w tej dziedzinie będzie wiedział, że można zastosować w tym systemie pojedynczy łańcuch Fv, łańcuch ciężki lub lekki fragmentów Fv lub Fab. Łańcuch ciężki lub lekki może być mutagenizowany, a następnie do roztworu dodaje się łańcuch komplementarny. Umożliwia się następnie łączenie tych dwóch łańcuchów i utworzenie funkcjonalnego fragmentu przeciwciała. Dodanie losowych nieswoistych sekwencji łańcucha lekkiego lub ciężkiego umożliwia wytwarzanie układu kombinatorycznego do wytwarzania biblioteki różnych elementów składowych. 47
49 1 2 3 [0246] Generalnie, wytwarzany jest jednołańcuchowy polinukleotyd do wyrażania. Ten polinukleotyd do wyrażania obejmuje: (1) kasety dla przeciwciała jednołańcuchowego składającej się z domeny V H, peptydu łącznikowego i domeny V L, połączonych funkcjonalnie tak, aby kodować przeciwciało jednołańcuchowe, (2) promotora odpowiedniego do transkrypcji in vitro (np., promotora T7, promotora SP6 i tym podobnych), przyłączonych funkcjonalnie dla zapewnienia transkrypcji in vitro kasety dla przeciwciała jednołańcuchowego, z wytworzeniem mrna kodującego przeciwciało jednołańcuchowe i (3) sekwencję terminacji transkrypcji, odpowiednią do funkcjonowania w reakcji transkrypcji in vitro. Ewentualnie, polinukleotyd do wyrażania może ponadto zawierać miejsce startu replikacji i/lub marker selekcyjny. Przykładem odpowiedniego polinukleotydu do wyrażania jest plm166. [0247] Sekwencje V H i V L można dogodnie uzyskać z biblioteki sekwencji V H i V L wytworzonych przez amplifikację PCR z użyciem starterów specyficznych dla rodziny genów V lub starterów specyficznych dla genu V (Nicholls i wsp., J. Immunol. Meth 1993, 16: 81; WO93/12227) lub zaprojektowanych zgodnie ze standardowymi sposobami znanymi w tej dziedzinie na podstawie dostępnych informacji o sekwencji. Typowo, izoluje się sekwencje V H i V L myszy lub człowieka. Sekwencje V H i V L poddaje się następnie ligacji, zazwyczaj z wtrąconą sekwencją łącznikową (np., kodującą w ramce elastyczny łącznik peptydowy), tworząc kasetę kodującą przeciwciało jednołańcuchowe. Typowo, stosuje się bibliotekę zawierającą wiele sekwencji V H i V L (czasami także z wielu rodzajami przedstawionych łączników), gdzie bibliotekę konstruuje się z zastosowaniem jednego lub więcej zmutowanych sekwencji V H i V L w celu zwiększenia różnorodności sekwencji, zwłaszcza reszt CDR, czasami reszt zrębowych. Sekwencje regionu V mogą być dogodnie klonowane jako cdna lub produkty amplifikacji PCR dla komórek wyrażających immunoglobuliny. Na przykład, komórki ludzkiej hybrydomy, chłoniaka lub innej linii komórkowej, która syntetyzuje immunoglobulinę bądź na powierzchni komórki, bądź jako wydzielaną, można zastosować do izolacji RNA polia+. RNA jest następnie stosowany do syntezy cdna przy użyciu starterów oligo dt z zastosowaniem enzymu odwrotnej transkryptazy (dla ogólnych metod patrz Goodspeed i wsp., Gene 1989, 76: 1; Dunn i wsp., J. Biol. Chem, 1989, 264: 17). Po wizolowaniu cdna lub produktu reakcji PCR dla regionu V, klonuje się go do wektora, z wytworzeniem kasety dla przeciwciała jednołańcuchowego. [0248] Aby dokonać konstrukcji przeciwciał i fragmentów przeciwciał, izoluje się i identyfikuje geny kodujące. Geny mogą być modyfikowane w celu umożliwienia klonowania do wektora ekspresyjnego i transkrypcji/translacji in vitro. Jakkolwiek mogą być wykorzystane metody, takie jak wykrywanie przy zastosowaniu sond DNA dla VH i VL z cdna hybrydomy (Maniatis i wsp., 1982) lub konstruowanie syntetycznego genu VH i VL (Barbas i wsp., 1992), dogodnym sposobem jest użycie sposobów amplifikacji sekwencji przeciwciał kierowanej matrycą. Zróżnicowana populacja genów przeciwciał może być powielana z próbki matrycy poprzez zaprojektowanie starterów do konserwatywnych sekwencji na końcach 3' i ' regionów zmiennych, znanych jako regiony zrębowe, lub regionów stałych przeciwciała (Iverson i wsp., 1989). W obrębie starterów mogą być umieszczone miejsca restrykcyjne w celu ułatwienia klonowania do wektora ekspresyjnego. Poprzez skierowanie tych starterów do tych konserwowanych regionów, utrzymywane jest zróżnicowanie populacji przeciwciał, aby umożliwić konstrukcję zróżnicowanych bibliotek. Konkretny rodzaj i klasę przeciwciała można zdefiniować 48
50 1 2 3 poprzez wybór sekwencji starterów, co jest zilustrowane przez dużą liczbę sekwencji dla wszystkich typów przeciwciał podaną w Kabat i wsp., [0249] Informacyjny RNA wyizolowany ze śledziony lub krwi obwodowej zwierzęcia można zastosować jako matrycę do amplifikacji biblioteki przeciwciał. W pewnych okolicznościach, gdzie pożądane jest, aby prezentować jednorodną populację fragmentów przeciwciał na powierzchni komórki, mrna można wyizolować z populacji przeciwciał monoklonalnych. Informacyjny RNA z obu źródeł można przygotować przy zastosowaniu standardowych metod i stosować bezpośrednio lub do wytwarzania matrycy cdna. Wytwarzanie mrna dla celów klonowania przeciwciał osiąga się łatwo, postępując zgodnie z dobrze znanymi sposobami wytwarzania i charakteryzowania przeciwciał (patrz, np., Antibodies: A Laboratory Manual, 1988). [0] Przy wytwarzaniu przeciwciał monoklonalnych (mab) postępuje się generalnie zgodnie z takimi samymi procedurami, jak te do wytwarzania przeciwciał poliklonalnych. Pokrótce, przeciwciało poliklonalne wytwarza się zgonie przez immunizację zwierzęcia kompozycją immunogenną i z tego immunizowanego zwierzęcia pobiera się antysurowicę. Do wytwarzania antysurowic można stosować szeroki wachlarz gatunków zwierzęcych. Zazwyczaj zwierzęciem wykorzystywanym do wytwarzania antysurowic odpornościowych jest królik, mysz, szczur, chomik, świnka morska lub koza. Ze względu na stosunkowo dużą objętość krwi u królika, króliki są zwykle preferowane do wytwarzania przeciwciał poliklonalnych. [021] Kompozycje immunogenne często różnią się immunogennością. Często konieczne jest zatem, wzmocnienie układu odpornościowego gospodarza, co można osiągnąć przez sprzężenie immunogenu peptydowego lub polipeptydowego z nośnikiem. Przykładowymi i korzystnymi nośnikami są hemocyjanina ze skałoczepa (KLH, ang. keyhole limpet hemocyanin) i albumina surowicy bydlęcej (BSA, ang. bovine serum albumin). Jako nośniki można zastosować inne albuminy, takie jak albumina jaja kurzego, albumina surowicy mysiej lub albumina surowicy królika. Uznane sposoby sprzęgania polipeptydu z białkiem nośnikowym są dobrze znane i obejmują glutaraldehyd, ester m- maleimidobenzoilo-n-hydroksysukcynimidu, karbodiimidy i bis-diazowaną benzydynę. [022] Immunogenność konkretnej kompozycji immunogenu może być wzmocniona przez zastosowanie nieswoistych stymulatorów odpowiedzi immunologicznej, znanych jako adiuwanty. Przykładowe i korzystne adiuwanty obejmują kompletny adiuwant Freunda (nieswoisty stymulator odpowiedzi immunologicznej, zawierający zabite Mycobacterium tuberculosis), niekompletny adiuwant Freunda oraz adiuwant-wodorotlenek glinu. [023] Ilość kompozycji immunogenu stosowana do wytwarzania przeciwciał poliklonalnych zmienia się w zależności od natury immunogenu, jak również zwierzęcia użytego do immunizacji. Do podawania immunogenu można stosować różnorodne drogi (podskórnią, domięśniową, śródskórną, dożylną i dootrzewnową). Wytwarzanie przeciwciał poliklonalnych można monitorować przez pobieranie próbek krwi immunizowanego zwierzęcia w różnych punktach po immunizacji. Można również podać drugie wstrzyknięcie wzmacniające. Sposób wzmacniania i miareczkowania powtarza się aż do uzyskania odpowiedniego miana. Gdy uzyskany zostanie pożądany poziom immunogenności, immunizowane zwierzę można skrwawić i surowicę wyizolować, przechowywać, a 49
51 1 2 3 śledziony zebrane do izolacji mrna z odpowiedzi poliklonalnej lub zwierzęcia można użyć do wytworzenia mab poprzez izolację mrna z jednorodnej populacji przeciwciał. [024] mab można łatwo wytworzyć przez stosowanie dobrze znanych technik, takich jak te podane jako przykłady w pat. USA nr 4,196,26. Typowo, technika ta obejmuje immunizację odpowiedniego zwierzęcia wybraną kompozycją immunogenu, np. małą cząsteczką haptenu sprzężoną z nośnikiem, oczyszczonym lub częściowo oczyszczonym białkiem, polipeptydem lub peptydem. Kompozycję immunizującą podaje się w sposób skuteczny do stymulowania komórek wytwarzających przeciwciała. Często stosowanymi zwierzętami są gryzonie, takie jak myszy i szczury; jednakże możliwe jest również stosowanie królika, owcy, komórek żaby. Pewnych korzyści może dostarczyć użycie szczurów (Goding, str , 1986), lecz korzystne są myszy, szczególnie myszy BALB/c, ponieważ są one stosowane najbardziej rutynowo i generalnie dają wyższy procent stabilnych fuzji. [02] Po immunizacji, komórki somatyczne z potencjałem do wytwarzania przeciwciał, szczególnie limfocyty B (komórki B), wybiera się do stosowania w protokole wytwarzania mab. Komórki te można otrzymać z biopsji śledziony, migdałków lub węzłów chłonnych albo z próbki krwi. Komórki śledziony i komórki krwi są korzystne, te pierwsze, ponieważ są bogatym źródłem komórek wytwarzających przeciwciała, które są w fazie dzielących się plasmablastów, a te drugie, ponieważ krew jest łatwo dostępna. Często, immunizowany będzie panel zwierząt i pobierana będzie śledziona zwierzęcia o najwyższym mianie przeciwciał, a limfocyty śledziony będą uzyskiwane poprzez homogenizację śledziony strzykawką. Zazwyczaj śledziona z immunizowanej myszy zawiera około x 7 do 2 x 8 limfocytów. [026] Limfocyty B wytwarzające przeciwciała z immunizowanego zwierzęcia poddaje się następnie fuzji z komórkami nieśmiertelnej komórki szpiczaka, generalnie jednego z tych samych gatunków zwierzęcia, które było immunizowane. Linie komórkowe szpiczaka odpowiednie do stosowania w procedurach fuzji hybrydom wytwarzających są, korzystnie, niewytwarzającymi przeciwciał, mają wysoką wydajność fuzji i niedobory enzymów, które czynią je następnie niezdolnymi do wzrostu w pewnych selektywnych pożywkach, które wspomagają wzrost tylko pożądanych komórek po fuzji (hybrydom). [027] Można stosować dowolną z licznych komórek szpiczaka, jak to jest znane specjalistom w tej dziedzinie (Goding, str. 6-66, 1986; Campbell, 1984). Na przykład, gdy immunizowanym zwierzęciem jest mysz, można użyć P3-X63/Ag8, X63-Ag8.63, NS1/1.Ag 4 1, SP2-Ag14, FO, NSO/U, MPC-11, MPC11-X4-GTG 1.7 i S194/XX0 Bul; dla szczurów można użyć R2.RCY3, Y3-Ag 1.2.3, IR983F i 4B2; a U-266, GM0-GRG2, LICR-LON-HMy2 i UC729-6 są przydatne w powiązaniu z fuzjami komórek ludzkich. [028] Jedną z korzystnych komórek mysiego szpiczaka jest linia komórkowa szpiczaka NS-1 (określana także jako P3-NS-1- Ag4-1), która jest łatwo dostępna w NIGMS Human Genetic Mutant Cell Repository, prosząc o numer depozytowy linii komórkowej GM373. Inną linią komórkową szpiczaka myszy, którą można stosować, jest odporna na 8-azaguaninę, niewytwarzająca linia komórek mysiego szpiczaka SP2/0. [029] Sposoby wytwarzania hybryd z komórek śledziony lub węzłów chłonnych wytwarzających przeciwciała oraz komórek szpiczaka, zwykle obejmują mieszanie komórek somatycznych z 0
52 1 2 3 komórkami szpiczaka w proporcji 2:1, choć proporcja może się zmieniać, odpowiednio od :1 do 1: 1, w obecności czynnika lub czynników (chemicznych lub elektrycznych), które promują fuzję błon komórkowych. Metody fuzji przy użyciu wirusa Sendai zostały opisane przez Kohler i Milstein (197; 1976), a te z użyciem glikolu polietylenowego (PEG), na przykład 37% (obj./obj.) PEG, przez Gefter i wsp., 1977). Odpowiednie jest także stosowanie metod fuzji indukowanych elektrycznie (Goding, str , 1986). [0260] Procedury fuzji zazwyczaj dają żywotne hybrydy z niskimi częstościami, od 1 x -6 do 1 x -8. Jednakże, nie stanowi to problemu, jako że żywe fuzje - hybrydy są odróżniane od komórek rodzicielskich, które nie uległy fuzji (szczególnie komórki szpiczaka, które nie uległy fuzji,a normalnie kontynuują podział w nieskończoność), przez hodowlę w pożywce selektywnej. Pożywka selektywna jest zwykle taką, która zawiera czynnik, który hamuje syntezę de novo nukleotydów w pożywce do hodowli tkankowej. Przykładowymi i korzystnymi czynnikami są aminopteryna, metotreksat i azaseryna. Aminopteryna i metotreksat blokują syntezę de novo zarówno puryn, jak i pirymidyn, podczas gdy azaseryna blokuje tylko syntezę puryny. W przypadku, gdy stosuje się aminopterynę lub metotreksat, pożywka jest uzupełniana hipoksantyną i tymidyną jako źródłem nukleotydów (pożywka HAT). W przypadku, gdy stosuje się azaserynę, pożywka jest uzupełniana hipoksantyną. [0261] Korzystną pożywką selekcyjną jest HAT. Tylko komórki zdolne do wykorzystywania szlaków ratunkowych dla nukleotydów są zdolne do przeżycia w pożywce HAT. Komórki szpiczaka są wadliwe pod względem kluczowych enzymów szlaku ratunkowego, np. fosforybozylotransferazy hipoksantyny (HPRT) i nie mogą przeżyć. Komórki B mogą wykorzystywać ten szlak, lecz mają ograniczoną długość życia w hodowli i generalnie umierają w ciągu około dwóch tygodni. A zatem, tylko komórki, które mogą przeżyć w pożywce selektywnej są tymi hybrydami utworzonymi z komórek szpiczaka i komórek B. [0262] To hodowanie dostarcza populacji hybrydoma, spośród których selekcjonuje się swoiste hybrydomy. Typowo, selekcję hybrydom przeprowadza się hodując komórki przez rozcieńczanie do pojedynczych klonów na płytkach do mikromiareczkowania, a następnie testowanie poszczególnych nadsączy klonalnych (po około dwóch do trzech tygodni) pod kątem pożądanej reaktywności. Proste i szybkie testy obejmują testy radioimmunologiczne, enzymatyczne testy immunologiczne, testy cytotoksyczności, testy łysinkowe, kroplowe testy immunowiązania i tym podobne. [0263] Wybrane hybrydomy rozcieńcza się seryjnie i klonuje do poszczególnych linii komórkowych wytwarzających przeciwciała, z których klony można następnie namnażać w nieskończoność w celu dostarczenia mab. Linie komórkowe można wykorzystywać do wytwarzania mab na dwa podstawowe sposoby. Próbkę hybrydomy można wstrzyknąć (często do jamy otrzewnowej) do histokompatybilnego zwierzęcia typu, który został użyty do dostarczenia komórek somatycznych i szpiczaka do wyjściowej fuzji. U zwierzęcia po wstrzyknięciu rozwijają się guzy nowotworowe wydzielające swoiste przeciwciało monoklonalne wytwarzane przez hybrydę komórek po fuzji. Następnie można pobrać płyny ciała zwierzęcia, takie jak surowica, płyn puchlinowy, aby dostarczyć mab w wysokim stężeniu. Poszczególne linie komórkowe można także hodować in vitro, gdzie mab są naturalnie wydzielane do pożywki hodowlanej, z której można je łatwo uzyskać w wysokim stężeniu. mab wytwarzane którymkolwiek ze sposobów można dalej oczyszczać, jeśli trzeba, przy 1
53 1 2 3 użyciu filtracji, wirowania i różnych metod chromatograficznych, takich jak HPLC lub chromatografia powinowactwa. [0264] Po izolacji i scharakteryzowaniu pożądanego przeciwciała monoklonalnego, mrna można izolować przy zastosowaniu technik dobrze znanych w tej dziedzinie i stosować jako matrycę do amplifikacji sekwencji docelowej. [026] Dostępnych jest szereg procesów zależnych od matrycy do amplifikacji sekwencji docelowej przed i po mutagenezie. Jednym z najbardziej znanych sposobów amplifikacji jest łańcuchowa reakcja polimerazy (określana dalej jako PCR, ang. polymerase chain reaction), która jest opisana szczegółowo Pat. USA nr 4,683,19, 4,683,2 i 4,800,19 oraz w Innis i wsp., Pokrótce, w reakcji PCR przygotowuje się dwie sekwencje starterów, które są komplementarne do regionów na przeciwnych komplementarnych niciach docelowej sekwencji. Do mieszaniny reakcyjnej dodaje się nadmiar trifosforanów deoksynukleozydów wraz z polimerazą DNA, np. polimerazą Taq. Jeżeli docelowa sekwencja jest obecna w próbce, startery będą się z nią wiązać i polimeraza spowoduje, że startery będą wydłużane wzdłuż sekwencji docelowej poprzez dodawanie nukleotydów. Poprzez podnoszenie i obniżanie temperatury mieszaniny reakcyjnej, wydłużone startery będą oddysocjowywać od celu z wytworzeniem produktów reakcji, nadmiar starterów wiąże się z sekwencją docelową i produktami reakcji i proces jest powtarzany. Korzystnie, jeżeli można przeprowadzić procedurę amplifikacji PCR z odwrotną transkrypcją w celu określenia ilości powielonego celu. Metody dla reakcji łańcuchowej polimerazy są dobrze znane w tej dziedzinie. Stosując techniki enzymatycznej amplifikacji, takie jak PCR, można zaprojektować pożądane elementy kontrolne w starterze, a więc będą one włączone do produktu DNA. [0266] Innym sposobem amplifikacji jest łańcuchowa reakcja ligazy ( LCR, ang. ligase chain reaction), ujawniona w EPA nr 3 8. W LCR, przygotowuje się dwie komplementarne pary sond i w obecności sekwencji docelowej każda para będzie wiązać się z przeciwległymi komplementarnymi nićmi docelowymi, tak, że stykają się one ze sobą. W obecności ligazy dwie pary sond zostaną połączone, tworząc pojedynczą jednostkę. Poprzez cykliczne zmiany temperatury, jak w reakcji PCR, związane, połączone poprzez ligację jednostki oddysocjowują od celu, a następnie służą jako sekwencje docelowe do ligacji nadmiaru par sond. Pat. USA nr 4,883,70 opisuje metodę podobną do LCR do wiązania pary sond z sekwencją docelową. [0267] Jako metoda amplifikacji może być również stosowana replikaza Qbeta, opisana w zgłoszeniu PCT nr PCT/US87/ W tej metodzie, do próbki jest dodawana replikacyjna sekwencja RNA, która zawiera region komplementarny do tego w sekwencji docelowej, w obecności polimerazy RNA. Polimeraza będzie kopiować sekwencję replikacyjną, która następnie może być wykrywana. [0268] Do amplifikacji kwasów nukleinowych może być również przydatna metoda amplifikacji izotermicznej, w której endonukleazy restrykcyjne i ligazy są stosowane do uzyskania amplifikacji docelowych cząsteczek, które zawierają nukleotydo- -[alfa-tio]-trifosforany w jednej nici miejsca restrykcyjnego (Walker i wsp., 1992). [0269] Amplifikacja poprzez wymianę nici (SDA, ang. Strand Displacement Amplification) jest inną metodą przeprowadzania izotermicznej amplifikacji kwasów nukleinowych, który obejmuje wiele rund wypierania nici i syntezy, tj. przesunięcia nacięć. Podobna metoda, zwana reakcją naprawy łańcucha 2
54 1 2 3 (RCR, ang. Repair Chain Reaction) obejmuje przyłączanie kilku sond wzdłuż regionu będącego celem dla amplifikacji, a następnie przeprowadzenie reakcji naprawy, w której obecne są tylko dwie z czterech zasad. Dwie inne zasady mogą być dodawane jako biotynylowane pochodne dla łatwego wykrywania. Podobne podejście stosuje się w SDA. Specyficzne sekwencje docelowe mogą być wykrywane przy użyciu cyklicznej reakcji z sondą (CPR, ang. cyclic probe reaction). W CPR sondę mającą po stronie 3 i sekwencje DNA niespecyficznego i środkową sekwencję RNA specyficznego, poddaje się hybrydyzacji z DNA, który jest obecny w próbce. Po hybrydyzacji, mieszaninę reakcyjną traktuje się RNazą H i produkty sondy identyfikuje jako odrębne produkty, które są uwalniane po trawieniu. Wyjściowa matryca jest przyłączana po innej cyklicznej sondy i reakcję powtarza się. [0270] Inne metody amplifikacji są opisane w zgłoszeniu GB nr i zgłoszeniu PCT nr PCT/US89/02 i mogą być stosowane zgodnie z niniejszym wynalazkiem. W pierwszym zgłoszeniu, zmodyfikowane startery są stosowane w reakcji typu PCR - syntezie zależnej od matrycy i enzymu. Startery mogą być zmodyfikowane poprzez znakowanie ugrupowaniem wychwytującym (na przykład biotyną) i/lub ugrupowaniem do wykrywania (na przykład enzymem). W drugim zgłoszeniu, do próbki dodawany jest nadmiar znakowanych sond. W obecności docelowej sekwencji, sonda wiąże się i jest cięta katalitycznie. Po przecięciu, sekwencja docelowa jest uwalniana w stanie nienaruszonym do związania się z nadmiarem sondy. Cięcie wyznakowanej sondy sygnalizuje obecność sekwencji docelowej [0271] Inne procedury amplifikacji kwasu nukleinowego obejmują systemy amplifikacji oparte na transkrypcji (TAS, ang. transcription-based amplification system), w tym amplifikację opartą na sekwencji kwasu nukleinowego (NASBA, ang. nucleic acid sequence based amplification) i 3SR (Kwoh i wsp., 1989). W NASBA, kwasy nukleinowe mogą być przygotowane do amplifikacji przy zastosowaniu standardowej ekstrakcji fenol/chloroform, denaturacji cieplnej próbki klinicznej, traktowania buforem do lizy i zastosowania kolumny do wirowania MiniSpin do izolacji DNA i RNA lub ekstrakcji RNA chlorkiem guanidyny. Te techniki amplifikacji obejmują przyłączanie startera, który ma specyficzne sekwencje docelowe. Po polimeryzacji, hybrydy DNA/RNA są trawione RNazą H, podczas gdy dwuniciowe cząsteczki DNA są ponownie poddawane denaturacji cieplnej. W każdym przypadku jednoniciowy DNA jest przekształcany w całkowicie dwuniciowy przez dodanie drugiego startera specyficznego względem sekwencji docelowej, a następnie polimeryzację. Dwuniciowe cząsteczki DNA podlegają następnie wielokrotnej transkrypcji przez polimerazę, taką jak T7 lub SP6. W izotermicznej reakcji cyklicznej RNA poddaje się odwrotnej transkrypcji do dwuniciowego DNA i jeszcze raz transkrypcji przez polimerazę taką jak T7 lub SP6. Powstałe produkty, czy to skrócone, czy kompletne, wskazują na sekwencje specyficzne dla celu. [0272] Davey i wsp., EPA nr ujawniają sposób amplifikacji kwasu nukleinowego obejmujący cykliczne syntetyzowanie jednoniciowego RNA ( ssrna, ang. single-stranded RNA), ssdna i dwuniciowego DNA (dsdna, ang. double-stranded DNA), które mogą być stosowane zgodnie z niniejszym wynalazkiem. ssrna jest pierwszą matrycą dla pierwszego oligonukleotydu starterowego, który jest przedłużany przez odwrotną transkryptazę (polimerazę DNA zależną od RNA). RNA jest następnie usuwany z otrzymanego w rezultacie dupleksu DNA: RNA przez działanie rybonukleazy H (RNaza H, RNaza swoista dla RNA w dupleksie bądź z DNA, bądź z RNA). Otrzymany w rezultacie 3
55 1 2 3 ssdna jest drugą matrycą dla drugiego startera, który również zawiera sekwencje promotora dla polimerazy RNA (której przykładem jest polimeraza RNA T7) po stronie względem jego homologii do matrycy. Starter jest następnie wydłużany przez polimerazę DNA (której przykładem jest duży fragment Klenowa polimerazy DNA I E. coli), wynikiem czego jest cząsteczka dwuniciowego DNA ( dsdna ), mająca sekwencję identyczną do wyjściowego RNA pomiędzy starterami i mającą dodatkowo w jednym końcu sekwencję promotora. Ta sekwencja promotora może być wykorzystywana przez odpowiednią polimerazę RNA, tak, aby wytworzyć wiele kopii RNA z DNA. Kopie te mogą ponownie wejść w cykl prowadzący do bardzo szybkiej amplifikacji. Dzięki odpowiedniemu doborowi enzymów, ta amplifikacja może odbywać się w warunkach izotermicznych, bez dodawania enzymów w każdym cyklu. Ze względu na cykliczną naturę procesu sekwencję wyjściową można dobrać tak, aby była w postaci bądź DNA, bądź RNA. [0273] Miller i wsp., zgłoszenie PCT WO 89/06700 ujawnia system amplifikacji sekwencji kwasu nukleinowego w oparciu o hybrydyzację sekwencji promotora/startera z docelowym jednoniciowym DNA ( ssdna ), a następnie transkrypcję wielu kopii sekwencji RNA. Ten system nie jest cykliczny, tj. z powstających transkryptów RNA nie są wytwarzane nowe matryce. Inne metody amplifikacji obejmują RACE i jednostronną reakcję PCR (Frohman, 1990; O'Hara i wsp., 1989). [0274] W etapie amplifikacji można również zastosować metody oparte na ligacji dwóch (lub więcej) oligonukleotydów w obecności kwasów nukleinowych mających sekwencje z powstałego w rezultacie dioligonukleotydu, powielając w ten sposób di-oligonukleotyd (Wu i wsp. 1989). [027] Produkty amplifikacji można analizować poprzez elektroforezę w żelu agarozowym, agarozowo-akryloamidowym lub poliakryloamidowym stosując standardowe metody (patrz, na przykład, Maniatis i wsp., 1982). Na przykład, można zastosować 1% żel agarozowy wybarwiony bromkiem etydyny i wizualizować w świetle UV. Alternatywnie, produkty amplifikacji mogą być integralnie wyznakowane nukleotydami znakowanymi radio- lub fluorymetrycznie. Żele można następnie, odpowiednio, eksponować wobec błony rentgenowskiej lub uwidaczniać w odpowiednich widmach stymulujących. [0276] Procedury mutagenne według niniejszego wynalazku mogą obejmować dowolne podejście mutagenezy, które może być dostosowane do konkretnego miejsca w genie, tj. mutagenezy ukierunkowanej lub mutagenezy miejscowo-specyficznej. Ponieważ niniejszy wynalazek opiera się na mutagenezie kompleksowej, niniejszy wynalazek bierze pod uwagę, jako korzystne wykonania, te procedury mutagenezy, które są szybkie, skuteczne i opłacalne. [0277] W jednym przykładzie wykonania, procedura mutagenezy wykorzystuje techniki syntezy chemicznej. Czyniąc to, możliwe jest dokładne umieszczenie podstawienia w jednym lub więcej konkretnych położeniach w obrębie genu, a także konkretne zdefiniowanie charakteru zmian. Chemiczne sposoby syntezy dla DNA są dobrze znane w tej dziedzinie. Korzystne w tym względzie są techniki w fazie stałej. [0278] Jedną z zalet metody syntezy genu w fazie stałej jest możliwość mutagenezy z wykorzystaniem kombinatorycznych technik syntezy. Kombinatoryczne techniki syntezy są zdefiniowane jako te techniki, gdzie wytworzone zostają duże kolekcje lub biblioteki związków jednocześnie, łącząc kolejno różne bloki budulcowe. Biblioteki można skonstruować przy użyciu 4
56 1 2 3 wolnych związków w roztworze, ale korzystnie, jeżeli związek jest związany z podłożem stałym, takim jak kulki, cząstki stałe lub nawet jako prezentowany na powierzchni mikroorganizmu. [0279] Istnieje szereg sposobów syntezy kombinatorycznej (Holmes i wsp., 199; Burbaum i wsp., 199; Martin i wsp., 199; Freier i wsp., 199; Pei i wsp., 1991; Bruce i wsp., 199; Ohlmeyer i wsp., 1993), w tym synteza dzielona lub synteza równoległa. Synteza dzielona może być stosowana do wytwarzania małych ilości stosunkowo dużej liczby związków, podczas gdy synteza równoległa będzie dawać większe ilości stosunkowo małej liczby związków. Generalnie, stosując syntezę dzieloną, związki syntetyzuje się na powierzchni mikrocząstek. Na każdym etapie, cząstki te dzieli się na kilka grup aby dodać następny składnik. Następnie różne grupy ponownie łączy się i rozdziela w celu utworzenia nowych grup. Proces ten jest powtarzany, aż związek jest ukończony. Każda cząstka niesie kilka kopii tego samego związku, umożliwiając łatwe rozdzielanie i oczyszczanie. Synteza dzielona może być przeprowadzana wyłącznie przy użyciu nośnika stałego. [0280] Technikę alternatywną, znaną jako synteza równoległa, można prowadzić bądź w fazie stałej, bądź w roztworze. Przy stosowaniu syntezy równoległej, różne związki są syntetyzowane w oddzielnych pojemnikach, często z wykorzystaniem automatyzacji. Syntezę równoległą można prowadzić na płytce do mikromiareczkowania, gdzie różne odczynniki można dodawać do każdej studzienki w określony uprzednio sposób w celu wytworzenia biblioteki kombinatorycznej. Synteza równoległa jest korzystnym podejściem do stosowania z technikami enzymatycznymi. Jest dobrze zrozumiałe, że istnieje wiele modyfikacji tej techniki i można ją przystosować do stosowania w niniejszym wynalazku. Stosując metody kombinatotyczne, można zsyntetyzować wiele matryc dla zmutowanych genów. [0281] Mutanty genów można również wytworzyć metodami półsyntetycznymi znanymi w tej dziedzinie (Barbas i wsp., 1992). Stosując regiony konserwatywne fragmentu przeciwciała jako regiony zrębowe, regiony zmienne mogą być umieszczone w losowych kombinacjach jeden lub więcej na raz, aby zmienić swoistość fragmentu przeciwciała i wytworzyć nowe miejsca wiązania, zwłaszcza przy wytwarzaniu przeciwciał wobec antygenów problematycznych przy immunizacji, takich jak związki toksyczne lub nietrwałe. Postępując w taki sam sposób, znaną sekwencję przeciwciała można zmieniać poprzez wprowadzanie mutacji losowo. Można tego dokonać przy zastosowaniu metod dobrze znanych w tej dziedzinie, takich jak stosowanie reakcji PCR podatnej na błędy. [0282] Stosując odpowiednie startery oligonukleotydowe, reakcja PCR jest wykorzystywana do szybkiej syntezy matrycy DNA zawierającej jedną lub większą liczbę mutacji w genie białka wiążącego. Mutageneza miejscowo-specyficzna jest techniką przydatną przy wytwarzaniu pojedynczych peptydów lub biologicznie funkcjonalnych, równoważnych białek albo peptydów, przez specyficzną mutagenezę bazowego DNA. Technika zapewnia dodatkowo łatwą zdolność do przygotowania i testowania wariantów sekwencji, biorąc pod uwagę jedną lub więcej z powyższych uwag, przez wprowadzenie do DNA jednej lub więcej zmian w sekwencji nukleotydowej. Mutageneza miejscowo-specyficzna pozwala na wytwarzanie mutantów przez użycie specyficznych sekwencji oligonukleotydowych, które kodują sekwencje DNA o żądanej mutacji, jak również wystarczającą liczbę sąsiednich nukleotydów, w celu dostarczenia sekwencji startera o dostatecznej wielkości i złożoności sekwencji dla wytworzenia stabilnego dupleksu po obu stronach złącza delecji, które jest
57 1 2 3 przekraczane. Zazwyczaj korzystny jest starter o długości od 17 do 2 nukleotydów, z do reszt po obu stronach złącza sekwencji, która jest zmieniana. [0283] Technika typowo wykorzystuje wektor bakteriofagowy, który istnieje zarówno postaci jednoniciowej, jak i dwuniciowej. Typowe wektory przydatne w mutagenezie ukierunkowanej to wektory takie jak fag M13. Te wektory fagowe są dostępne komercyjnie i ich stosowanie jest generalnie dobrze znane specjalistom w tej dziedzinie. Dwuniciowe plazmidy są także rutynowo stosowane w ukierunkowanej mutagenezie, która eliminuje etap przenoszenia genu będącego przedmiotem zainteresowania z plazmidu do faga. [0284] Generalnie, mutagenezę ukierunkowaną przeprowadza się najpierw otrzymując jednoniciowy wektor lub stapiając dwie nici dwuniciowego wektora, który zawiera w obrębie swojej sekwencji sekwencję DNA kodującą pożądane białko. Starter oligonukleotydowy niosący pożądaną zmutowaną sekwencję jest wytwarzany syntetycznie. Ten starter jest następnie przyłączany do preparatu jednoniciowego DNA, biorąc pod uwagę stopień niedopasowania przy wyborze warunków hybrydyzacji, i poddawany działaniu enzymów polimeryzacji DNA, takich jak fragment Klenowa polimerazy I E. coli, w celu zakończenia syntezy nici niosącej mutację. A zatem, tworzy się heterodupleks, w którym jedna nić koduje wyjściową, niezmutowaną sekwencję, a druga nić niesie pożądaną mutację. Ten heterodupleksowy wektor stosuje się następnie do transformacji odpowiednich komórek, takich jak komórki E. coli i wybiera się klony, które zawierają rekombinowane wektory niosące zmutowany układ sekwencji. [028] Wytwarzanie wariantów sekwencji wybranego genu przy zastosowaniu ukierunkowanej mutagenezy jest dostarczone jako sposób wytwarzania potencjalnie przydatnych rodzajów i nie ma na celu być ograniczające, ponieważ istnieją inne drogi, którymi można otrzymać warianty sekwencji genów. Przykładowo, aby otrzymać warianty sekwencji, rekombinowane wektory, kodujące pożądany gen, można traktować środkami mutagennymi, takimi jak hydroksyloamina. [0286] Wiadomo, że w pewnych zastosowaniach, przy podstawieniach aminokwasowych poprzez mutagenezę ukierunkowaną, wymagane są warunki o niższej ostrości. W tych warunkach hybrydyzacja może nastąpić nawet wtedy, gdy sekwencje sondy i nici docelowej nie jest idealnie komplementarne, ale występują niedopasowania w jednej lub w większej liczbie pozycji. Warunki mogą być uczynione mniej ostrymi poprzez zwiększenie stężenia soli i obniżenie temperatury. Na przykład, warunki średniej ostrości mogłyby być dostarczone przez 0,1 do 0,2 M NaCl w temperaturach 37 C do C, podczas gdy warunki niskiej ostrości mogą być dostarczone przez sól 0,1 M do 0,9 M w temperaturach od C do C. A zatem, warunkami hybrydyzacji można łatwo manipulować, a więc będą generalnie metodą wyboru w zależności od pożądanych wyników. [0287] W innych przykładach wykonania, hybrydyzację można osiągnąć w warunkach, na przykład, 0 mm Tris-HCl (ph 8,3), 7 mm KCl, 3 mm MgCl 2, mm ditiotreitolu w temperaturach od około C do 37 C. Inne stosowane warunki hybrydyzacji mogą obejmować około mm Tris-HCl (ph 8,3), 0 mm KCl, 1, M MgCl 2, w temperaturach w zakresie od około C do 72 C. Do zmiany warunków hybrydyzacji można również użyć formamid i SDS. [0288] W konkretnym wykonaniu, można zastosować nakładającą się reakcję PCR. Pokrótce, można zastosować plazmid jako matrycę do pierwszej rundy reakcji PCR. Produkty reakcji PCR z pierwszej 6
58 1 2 3 rundy są oczyszczane i wykorzystywane wraz z zewnętrznymi starterami w reakcji PCR z nakładającymi się wydłużeniami. Produkty końcowe będą zawierały ukierunkowane zastąpienie danego aminokwasu wszystkimi innymi możliwymi resztami aminokwasowymi. [0289] Zmutowane polipeptydy wytwarzane według niniejszego wynalazku mogą być charakteryzowane przy zastosowaniu rozmaitych technik. Generalnie, produkty białkowe można analizować pod kątem prawidłowej widocznej masy cząsteczkowej metodą SDS-PAGE. Umożliwia to wstępne wskazanie, że polipeptyd został faktycznie zsyntetyzowany. Po porównaniu z cząsteczką naturalną, wskazuje to również, czy u mutanta nastąpiło normalne fałdowanie lub obróbka. Z tego względu, może okazać się przydatne wyznakowanie polipeptydu. Alternatywnie, polipeptyd może być zidentyfikowany poprzez barwienie żelu. [0290] Poza jedynie syntezą, przeciwciała mogą być charakteryzowane pod względem różnych właściwości i szerokiej gamy funkcji. Właściwości obejmują punkt izoelektryczny, stabilność termiczną, szybkość sedymentacji i fałdowanie. Jednym ze sposobów badania fałdowania jest zdolność do bycia rozpoznawanym przez pokrewnego partnera wiązania. Najlepszym przykładem tej funkcji jest oddziaływanie przeciwciało-antygen. Szeroka gama różnych formatów testów immunologicznych, jest dostępna dla tego celu i jest dobrze znana w tej dziedzinie. Zasadniczo, można określić zmiany bądź powinowactwa, bądź swoistości, gdy przeciwciało ma kontakt z określonym antygenem lub panelami spokrewnionych antygenów. [0291] Testy immunologiczne można generalnie podzielić na dwa rodzaje: testy heterogenne wymagające wielu etapów rozdziału i testy homogenne, które są wykonywane bezpośrednio. Heterogenne testy immunologiczne na ogół obejmują przeciwciało unieruchomione na podłożu stałym. Doprowadza się do kontaktu próbki zawierającej ligand z unieruchomionym przeciwciałem i ilość utworzonego kompleksu na podłożu stałym określa się na podstawie znacznika dołączonego bezpośrednio lub pośrednio do unieruchomionego kompleksu. Tak jak się stosuje w kontekście niniejszego wynalazku, ligand definiuje się jako rodzaj cząsteczki, która oddziałuje z nieidentyczną cząsteczką, z wytworzeniem ściśle związanego, stabilnego kompleksu. Dla celów praktycznych, powinowactwo wiązania jest zwykle większe niż 6 M -1, a korzystnie jest w zakresie od 9-1 M -1. Ligandem może być dowolna z kilku typów cząsteczek organicznych, w tym alicykliczne węglowodory wielopierścieniowe, związki aromatyczne, związki chlorowcowane, benzenoidy, węglowodory wielopierścieniowe, heterocykliczne związki azotowe, heterocykliczne związki siarkowe, heterocykliczne związki tlenowe i węglowodory alkanowe, alkenowe i alkinowe, itd. Cząsteczki biologiczne są przedmiotem szczególnego zainteresowania, w tym aminokwasy, peptydy, białka, lipidy, sacharydy, kwasy nukleinowe i ich kombinacje. Oczywiście należy rozumieć, że są to jedynie przykłady i że rozważane metody testów immunologicznych stosuje się do wykrywania niezwykle szerokiego zakresu związków, o ile można uzyskać przeciwciało, które wiąże się z ligandem będącym przedmiotem zainteresowania. [0292] Heterogeniczne testy immunologiczne można przeprowadzić jako testy kanapkowe, w których cząsteczkę będącą przedmiotem zainteresowania poddaje się reakcji z unieruchomionym przeciwciałem, które wiąże swoiście tę cząsteczkę z wysokim powinowactwem. W drugim etapie, poddaje się reakcji koniugat utworzony z tego samego lub innego przeciwciała z antygenem i 7
59 1 2 3 cząsteczką znacznika, z kompleksem antygen-przeciwciało na podłożu unieruchamiającym. Po usunięciu nadmiaru wolnego koniugatu mierzy się związany koniugat markerowy, który jest proporcjonalny do ilości ligandu w próbce. [0293] Wykrywanie tworzenia się immunokompleksu jest dobrze znane w tej dziedzinie i może być osiągnięte poprzez zastosowanie licznych podejść. Podejścia te są zwykle oparte na wykrywaniu znacznika lub markera, takiego jak dowolny ze znaczników (ang. tag, label) radioaktywnych, fluorescencyjnych, chemiluminescencyjnych, elektrochemiluminescencyjnych, biologicznych lub enzymatycznych znanych w tej dziedzinie. Patenty USA dotyczące stosowania takich znaczników obejmują pat. USA nr 3,817,837; 3,80,72; 3,939,; 3,996,34; 4,277,437; 4,27,149 i 4,366,241. Oczywiście, można znaleźć dodatkowe korzyści dzięki zastosowaniu drugorzędowego ligandu wiążącego, takiego jak drugie przeciwciało lub układ wiązania ligandu biotyna/awidyna, jak to jest znane w tej dziedzinie. [0294] Korzystne sposoby wykrywania obejmują test radioimmunologiczny (RIA, ang. radioimmunoassay) lub immunosorpcyjny test enzymatyczny (ELISA, ang. enzyme-linked immunosorbent assay), przy czym test ELISA jest najkorzystniejszy z uwagi na generalnie zwiększoną czułość. Testy ELISA są szeroko wykorzystywane w zastosowaniach biotechnologicznych, zwłaszcza w testach immunologicznych, w odniesieniu do szerokiej gamy substancji antygenowych. Czułość testu ELISA opiera się na enzymatycznej amplifikacji sygnału. [029] Stosowany tu termin doprowadzenie do kontaktu oznacza zbliżenie się składników reakcji do siebie na tyle, aby umożliwić zajście pożądanego oddziaływania. Doprowadzenie do kontaktu można uzyskać na przykład poprzez mieszanie składników w roztworze albo przez oddziaływanie heterogenne, takie jak w kontakcie w czasie przepływu przez kolumnę lub podłoże unieruchamiające, które wiąże się z jednym ze składników. [0296] Przeciwciała wytwarzane i izolowane przy zastosowaniu sposobu według wynalazku wybiera się tak, aby wiązały się z określonym uprzednio celem. Zwykle określony uprzednio cel będzie wybrany ze względu na możliwość jego stosowania jako celu diagnostycznego i/lub terapeutycznego. Określonym uprzednio celem może być znany lub nieznany epitop. Przeciwciała zazwyczaj wiążą się z określonym antygenem (np., immunogenem) z powinowactwem wynoszącym co najmniej 1 x 7 M - 1, korzystnie z powinowactwem wynoszącym co najmniej x 7 M -1, korzystniej z powinowactwem wynoszącym co najmniej od 1 x 8 M -1 do 1 x 9 M -1 lub większym, w niektórych przypadkach nawet do 1 x M -1 albo większym. Często określonym uprzednio antygenem jest białko ludzkie, takie jak na przykład antygen na powierzchni komórek ludzkich (na przykład, CD4, CD8, receptor IL-2, receptor EGF, receptor PDGF), inna ludzka makrocząsteczka biologiczna (na przykład trombomodulina, białko C, antygen węglowodanowy, sialyl-antygen Lewisa, L-selektyna) albo makrocząsteczka związana z chorobą nie człowieka (na przykład, bakteryjny LPS, białko kapsydu wirionu i glikoproteina otoczki) i tym podobne. [0297] W innym przykładzie, opublikowano kilka doniesień o możliwości stosowania diagnostycznego i terapeutycznego scfv (Gruber i wsp., 1994, tamże; Lilley i wsp., 1994, tamże.; Huston i wsp., Int. Rev. Immunol 1993, :a 19, Sandhu JS, Crit. Rev. Biotechnol., 1992, 12: 437). 8
60 1 2 3 [0298] Przeciwciała o wysokim powinowactwie z pożądaną swoistością można skonstruować i wyrazić w różnych systemach. Na przykład, scfv zostały wytworzone w roślinach (Firek i wsp. (1993) Plant Mol. Biol. 23: 861) i można je łatwo wytworzyć w systemach prokariotycznych (Owens RJ and Young RJ, J. Immunol. Meth., 1994,168: 149; Johnson S and Bird RE, Methods Enzymol., 1991, 3: 88). Ponadto, przeciwciała jednołańcuchowe można wykorzystać jako podstawę do skonstruowania całych przeciwciał lub różnych ich fragmentów (Kettleborough i wsp., Euro J. Immunol, 1994, 24: 92). Sekwencję kodującą region zmienny można wyizolować (na przykład, poprzez amplifikację PCR lub subklonowanie) i połączyć z sekwencją kodującą pożądanego ludzkiego regionu stałego dla zakodowania sekwencji przeciwciała ludzkiego bardziej odpowiedniego do zastosowań terapeutycznych u ludzi, gdzie immunogenność jest, korzystnie, zminimalizowana. Polinukleotyd(y) mający(e) powstałą w rezultacie w pełni ludzką sekwencję(e) kodującą(e) może być wyrażony w komórce gospodarza (np. z wektora ekspresyjnego w komórce eukariotycznej) i oczyszczony dla kompozycji farmaceutycznej. [0299] Konstrukty do ekspresji DNA będą zazwyczaj zawierać sekwencję DNA kontrolującą ekspresję, połączoną funkcjonalnie z sekwencjami kodującymi, w tym naturalnie związanymi lub heterologicznymi regionami promotorowymi. Korzystnie, jeżeli sekwencjami kontrolującymi ekspresję będą eukariotyczne układy promotorowe w wektorach zdolnych do transformowania albo transfekowania eukariotycznych komórek gospodarza. Po wprowadzeniu wektora do odpowiedniego gospodarza, gospodarza utrzymuje się w warunkach odpowiednich do wysokiego poziomu ekspresji sekwencji nukleotydowych oraz zbierania i oczyszczania zmutowanych poddanych manipulacjom przeciwciał. [00] Jak wspomniano poprzednio, sekwencje DNA będą ulegać ekspresji w gospodarzu po tym, jak sekwencje zostały połączone funkcjonalnie z sekwencją kontrolującą ekspresję (tj., umieszczone tak, aby zapewnić transkrypcję i translację genu strukturalnego). Te wektory ekspresyjne podlegają zazwyczaj replikacji w organizmach gospodarza jako episomy lub jako integralna część DNA chromosomalnego gospodarza. Zwykle, wektory ekspresyjne będą zawierać markery selekcyjne, np. tetracyklinę lub neomycynę, w celu umożliwienia wykrywania komórek transformowanych pożądanymi sekwencjami DNA (patrz, np., pat. USA nr 4,704,362). [01] Oprócz drobnoustrojów eukariotycznych, takich jak drożdże, do wytwarzania polipeptydów według niniejszego wynalazku można również zastosować hodowle komórkowe tkanek ssaków (patrz, Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y. (1987)). Korzystne są komórki eukariotyczne, ponieważ uzyskano w tej dziedzinie szereg odpowiednich linii komórek gospodarzy, zdolnych do wydzielania nienaruszonych immunoglobulin, i obejmują one linie komórkowe CHO, różne linie komórek COS, komórki HeLa, linie komórek szpiczaka, komórki B lub hybrydomy. Wektory ekspresyjne dla tych komórek mogą zawierać sekwencje kontrolujące ekspresję, takie jak początek replikacji, promotor, wzmacniacz (Queen i wsp., Immunol. Rev. 1986, 89: 49) i niezbędne miejsca dla obróbki informacji, takie jak miejsca wiązania rybosomu, miejsca składania RNA, miejsca poliadenylacji i sekwencje terminacji transkrypcji. Korzystnymi sekwencjami kontrolującymi ekspresję są promotory pochodzące z genów immunoglobulin, wirusa cytomegalii, SV, adenowirusa, bydlęcego wirusa brodawczaka, i tym podobne. 9
61 1 2 3 Sekwencję kodującą region zmienny można wyizolować (na przykład, poprzez amplifikację PCR lub subklonowanie) i połączyć z sekwencją kodującą pożądanego ludzkiego regionu stałego dla zakodowania sekwencji przeciwciała ludzkiego bardziej odpowiedniego do zastosowań terapeutycznych u ludzi, gdzie immunogenność jest, korzystnie, zminimalizowana. Polinukleotyd(y) mający(e) powstałą w rezultacie w pełni ludzką sekwencję(e) kodującą(e) może być wyrażony w komórce gospodarza (np. z wektora ekspresyjnego w komórce eukariotycznej) i oczyszczony dla kompozycji farmaceutycznej. [02] Konstrukty do ekspresji DNA będą zazwyczaj zawierać sekwencję DNA kontrolującą ekspresję, połączoną funkcjonalnie z sekwencjami kodującymi, w tym naturalnie związanymi lub heterologicznymi regionami promotorowymi. Korzystnie, jeżeli sekwencjami kontrolującymi ekspresję będą eukariotyczne układy promotorowe w wektorach zdolnych do transformowania albo transfekowania eukariotycznych komórek gospodarza. Po włączeniu wektora do odpowiedniego gospodarza, gospodarza utrzymuje się w warunkach odpowiednich do wysokiego poziomu ekspresji sekwencji nukleotydowych oraz zbierania i oczyszczania zmutowanych poddanych manipulacjom przeciwciał. [03] Jak wspomniano poprzednio, sekwencje DNA będą ulegać ekspresji w gospodarzu po tym, jak sekwencje zostały połączone funkcjonalnie z sekwencją kontrolującą ekspresję (tj., umieszczone tak, aby zapewnić transkrypcję i translację genu strukturalnego). Te wektory ekspresyjne podlegają zazwyczaj replikacji w organizmach gospodarza jako episomy lub jako integralna część DNA chromosomalnego gospodarza. Zwykle, wektory ekspresyjne będą zawierać markery selekcyjne, np. tetracyklinę lub neomycynę, w celu umożliwienia wykrywania komórek transformowanych pożądanymi sekwencjami DNA (patrz, np., pat. USA nr 4,704,362). [04] Oprócz drobnoustrojów eukariotycznych, takich jak drożdże, do wytwarzania polipeptydów według niniejszego wynalazku można również zastosować hodowle komórkowe tkanek ssaków (patrz, Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y. (1987)). Korzystne są komórki eukariotyczne, ponieważ uzyskano w tej dziedzinie szereg odpowiednich linii komórek gospodarzy zdolnych do wydzielania nienaruszonych immunoglobulin, i obejmują one linie komórkowe CHO, różne linie komórek COS, komórki HeLa, linie komórek szpiczaka, komórki B lub hybrydomy. Wektory ekspresyjne dla tych komórek mogą zawierać sekwencje kontrolujące ekspresję, takie jak początek replikacji, promotor, wzmacniacz (Queen i wsp., Immunol. Rev. 1986, 89: 49) i niezbędne miejsca dla obróbki informacji, takie jak miejsca wiązania rybosomu, miejsca składania RNA, miejsca poliadenylacji i sekwencje terminacji transkrypcji. Korzystnymi sekwencjami kontrolującymi ekspresję są promotory pochodzące z genów immunoglobulin, wirusa cytomegalii, SV, adenowirusa, bydlęcego wirusa brodawczaka, i tym podobne. [0] Eukariotyczna transkrypcja DNA może zostać zwiększona przez wstawienie do wektora sekwencji wzmacniacza. Wzmacniacze są to sekwencje działające w układzie cis z do par zasad zwiększające transkrypcję z promotora. Wzmacniacze mogą skutecznie zwiększać transkrypcję, gdy są po stronie lub 3 w stosunku do jednostki transkrypcyjnej. Są one równie skuteczne, jeśli znajdują się w obrębie intronu lub w samej sekwencji kodującej. Zazwyczaj stosowane są wzmacniacze wirusowe, w tym wzmacniacze SV, wzmacniacze wirusa cytomegalii, 60
62 1 2 3 wzmacniacze polioma oraz wzmacniacze adenowirusa. Powszechnie stosowane są również sekwencje wzmacniaczy z systemów ssaczych, takie jak wzmacniacz łańcucha ciężkiego mysiej immunoglobuliny. [06] Systemy ssaczych wektorów ekspresyjnych będą również zwykle zawierać podlegający selekcji gen markerowy. Przykłady odpowiednich markerów obejmują gen reduktazy dihydrofolianowej (DHFR), gen kinazy tymidynowej (TK) lub geny prokariotyczne, nadające oporność na leki. Pierwsze dwa geny markerowe preferują stosowanie zmutowanych linii komórkowych, które nie mają zdolności do wzrostu bez dodawania tymidyny do pożywki hodowlanej. Transformowane komórki można zidentyfikować na podstawie ich zdolności do wzrostu na podłożach nieuzupełnionych. Przykłady prokariotycznych genów oporności na lek przydatne jako markery obejmują geny nadające oporność na G418, kwas mykofenolowy i higromycynę. [07] Wektory zawierające odcinki DNA będące przedmiotem zainteresowania można przenieść do komórki gospodarza przy zastosowaniu dobrze znanych metod, zależnie od typu gospodarza komórkowego. Na przykład, transfekcję z użyciem chlorku wapnia stosuje się powszechnie wobec komórek prokariotycznych, natomiast traktowanie fosforanem wapnia, lipofekcję lub elektroporację można zastosować dla innych gospodarzy komórkowych. Inne metody stosowane do transformowania komórek ssaczych obejmują stosowanie polibrenu, fuzję protoplastów, liposomów, elektroporację i mikrowstrzyknięcie (patrz ogólnie, Sambrook i wsp., jak wyżej). [08] Po wyrażeniu, przeciwciała, poszczególne zmutowane łańcuchy immunoglobulinowe, zmutowane fragmenty przeciwciał i inne polipeptydy immunoglobulinowe według wynalazku można oczyszczać zgodnie ze standardowymi procedurami w tej dziedzinie, obejmującymi wytrącanie siarczanem amonowym, chromatografię kolumnową, elektroforezę żelową i tym podobne (patrz ogólnie, Scopes, R., Protein Purification, Springer-Verlag, N.Y. (1982)). Po oczyszczeniu, częściowym lub do homogenności, jak jest to pożądane, polipeptydy mogą być zastosowane terapeutycznie albo do opracowywania i wykonywania procedur testowych, barwienia immunofluorescencyjnego i tym podobnych (patrz, ogólnie, Immunological Methods, tomy I i II, red. Lefkovits and Pernis, Academic Press, N.Y. N.Y. (1979 i 1981)). [09] Oligopeptydy według niniejszego wynalazku można stosować w diagnostyce i terapii. Jako ilustracja, a nie ograniczenie, przeciwciała można stosować do leczenia nowotworu, chorób autoimmunizacyjnych lub infekcji wirusowych. Do leczenia nowotworu, przeciwciała zazwyczaj wiążą się z antygenem wyrażanym preferencyjnie na komórkach nowotworowych, takich jak ErbB-2, CEA, CD33 i wiele innych antygenów znanych specjalistom w tej dziedzinie. Do leczenia chorób autoimmunologicznych, przeciwciała zazwyczaj wiążą się z antygenem wyrażanym na komórkach T, takim jak CD4, receptor IL-2, różne receptory antygenu komórek T i wiele innych antygenów, znanych specjalistom w tej dziedzinie (np., patrz Fundamental Immunology, wyd. 2, W. E. Paul, red. Raven Press. Nowy Jork, N.Y.). Do leczenia infekcji wirusowych, przeciwciała zazwyczaj wiążą się z antygenem na komórkach zakażonych konkretnym wirusem, takim jak różne glikoproteiny (np., gb, gd, ge) wirusa opryszczki pospolitej i wirusa cytomegalii oraz wieloma innymi antygenami, znanymi specjalistom w tej dziedzinie (np., patrz Virology, wydanie 2, B. N. Fields i wsp., red. (1990), Raven Press: Nowy Jork, N.Y.). 61
63 1 [03] Kompozycje farmaceutyczne zawierające przeciwciała według niniejszego wynalazku są przydatne do podawania pozajelitowego, tj., podskórnie, domięśniowo lub dożylnie. Kompozycje do podawania pozajelitowego będą zazwyczaj zawierać roztwór przeciwciała lub ich mieszaninę rozpuszczoną w akceptowalnym nośniku, korzystnie nośniku wodnym. Można stosować różne nośniki wodne, na przykład, wodę, wodę buforowaną, 0,4% roztwór soli fizjologicznej, 0,3% glicynę i tym podobne. Roztwory te są jałowe i generalnie wolne od cząstek stałych. Kompozycje te można jałowić przy zastosowaniu konwencjonalnych, dobrze znanych technik jałowienia. Kompozycje mogą zawierać farmaceutycznie dopuszczalne substancje pomocnicze wymagane do przybliżania warunków fizjologicznych, takie jak środki regulujące ph i środki buforujące, czynniki regulujące toksyczność i tym podobne, na przykład octan sodu, chlorek sodu, chlorek potasu, chlorek wapnia, mleczan sodu, itd. Stężenie zmutowanych przeciwciał w tych kompozycjach może się zmieniać w szerokim zakresie, to jest od mniej niż 0,01%, zwykle co najmniej 0,1%, aż do % wagowych i będzie wybrane przede wszystkim w oparciu o objętości płynów, lepkości itp., zgodnie z konkretnym wybranym sposobem podawania. [0311] A zatem, typowa kompozycja farmaceutyczna do wstrzyknięć domięśniowych mogłaby być wykonana tak, aby zawierała 1 ml jałowej buforowanej wody i 1 mg zmutowanego przeciwciała. Typowa kompozycja do wlewu dożylnego może być zrobiona w objętości do ml jałowego roztworu Ringera i mg zmutowanego przeciwciała. Aktualne sposoby wytwarzania kompozycji do podawania pozajelitowego będą znane lub oczywiste dla specjalistów w tej dziedzinie i są opisane bardziej szczegółowo, na przykład w Remington s Pharmaceutical Science, wyd.., Mack Publishing Company, Easton, Pa. (00). Wytwarzanie kandydatów i/lub kandydatów poddanych ewolucji 2 3 [0312] W jednym wykonaniu, systemem eukariotycznym jest system ssaczy wybrany z jednej z grup składających się z linii komórkowych CHO, HEK293, IM9, DS-1, THP-1, Hep G2, COS, NIH 3T3, C33a, A49, A37, SK-MEL-28, DU 14, PC-3, HCT 116, Mia PACA-2, ACHN, Jurkat, MM1, Ovcar 3, HT 80, Panc-1, U266, 769P, BT-474, Caco-2, HCC 194, MDA-MB-468, LnCAP, NRK-49F i SP2/0; oraz splenocytów myszy i PBMC królika. [0313] W jednym wykonaniu, przy wytwarzaniu kandydata można zastosować wiele ssaczych komórek gospodarza, w tym komórki fibroblastów (3T3, mysz; BHK21, chomik syryjski), komórki nabłonkowe (MDCK, pies; Hela, człowiek; PtK1, kanguroszczur), komórki plazmatyczne (SP2/0 i NS0, mysz), komórki nerkowe (293 człowiek; COS, małpa), komórki jajnika (CHO, chomik chiński), komórki embrionalne (R1 i E14.1, mysz; H1 i H9, człowiek; PER C.6 człowiek). [0314] W pewnych aspektach, rekombinowane przeciwciała są wytwarzane w liniach komórkowych CHO i NS0 i SP2/0. W konkretnym aspekcie, system ssaczym jest linia komórkowa CHO-S. Najczęściej stosowanymi systemami wektora ekspresyjnego są systemy ekspresyjne syntetazy glutaminy i inne oparte na genach reduktazy dihydrofolianowej. [031] W innym wykonaniu, systemem eukariotycznym jest system komórek drożdży. W jednym aspekcie, system komórek drożdży jest wybrany z komórek S. cerevisiae lub Pichia. 62
64 [0316] W sposobie według wynalazku, gospodarze stosowani do przeszukiwania cząsteczek po ewolucji są tymi samymi, co gospodarze zastosowani do dalszego wytwarzania trafień. W innym aspekcie niniejszego wynalazku, system genetyczny zastosowany do znalezienia i ewolucji przeciwciał jest dokładnie tym samym co system genetyczny zastosowany do wytwarzania przeciwciała dla zastosowań komercyjnych. Środki biopodobne 1 2 [0317] Środki biopodobne są to terapeutyki oparte na białkach, które mają identyczną sekwencję aminokwasową (tj. skład chemiczny) co zatwierdzony etycznie lek, który nie jest już dłużej chroniony patentem. W jednym aspekcie, sposób CIAO jest szczególnie odpowiedni dla środków biopodobnych. Ponieważ konieczne jest wytwarzanie terapeutycznego przeciwciała w równoważnym preparacie i kompozycji, aby być konkurencyjnym na rynku, środek biopodobny powinien być wywarzany szybko i tak tanio, jak to tylko możliwe. Pożywki do hodowli komórkowych i proces opracowywania są jednymi z najbardziej kosztownych i czasochłonnych części przygotowywania i wytwarzania środków biopodobnych. [0318] Zmienianie kodonów z cichymi mutacjami w obrębie przeciwciała terapeutycznego zmienia kodon wykorzystywany do translacji białka, ale zachowuje sekwencję aminokwasową w obrębie przeciwciała. Te zmiany kodonów w różnych pozycjach w cząsteczce, w szczególności na końcu aminowym mogą mieć znaczący wpływ na ekspresję, a w niektórych przypadkach nawet glikozylację. W jednym aspekcie, sposób CIAO służy do selekcji i ewolucji kodonów z cichymi mutacjami przeciwciała w komórce gospodarza, podobnego do tego stosowanego ostatecznie do wytwarzania. Dlatego też czas obróbki może zostać zmniejszony dzięki wyższym wydajnościom przeciwciał i faktowi, że przeciwciało zostało wyselekcjonowanie do ekspresji w linii komórek gospodarza, tak, że większość tradycyjnych problemów z wytwarzaniem zostało z cząsteczki zostało odselekcjonowanych. Ponadto, selekcjonując cząsteczki w niedrogiej, wolnej od surowicy pożywce hodowlanej, mogą być wyselekcjonowane cząsteczki z kodonami, które pozwalają na niedrogie wytwarzanie i oczyszczanie. [0319] Bez dalszego omawiania, uważa się, że specjalista w tej dziedzinie może, stosując powyższy opis, zastosować niniejszy wynalazek w jego najpełniejszym zakresie. Poniższe przykłady mają być uważane jako ilustracyjne, a tym samym nieograniczające w jakikolwiek sposób pozostałej części ujawnienia. Przykłady 3 Przykład 1. Wytwarzanie i przeszukiwanie biblioteki przeciwciał [03] Przykład ten opisuje sposób wytwarzania i przeszukiwania biblioteki ludzkich przeciwciał prezentowanych na powierzchni komórek ssaczych w celu wyizolowania ludzkich przeciwciał o aktywności wiązania się z docelowym antygenem z wykorzystaniem kombinacji sortowania przez cytometrię przepływową i test ELISA. 63
65 Przeszukiwanie biblioteki poprzez analizę cytometrii przepływowej [0321] 1. Wytwórz bibliotekę ludzkich przeciwciał stabilnie zintegrowanych w komórkach ssaczych, takich jak opisane w Załączniku 1.2, 1.3 i 1.4, poniżej. 2. Namnóż stabilne klony z biblioteki w pełni ludzkich przeciwciał przed analizą przez cytometrię przepływową. 3. W dniu analizy przez cytometrię przepływową, przepłucz 1 x 7 komórek 1 x PBS. 4. Oderwij komórki przy zastosowaniu pożywki do odrywania komórek Detachin i zbierz komórki w 1 x PBS.. Zwiruj komórki przy 00 obrotach na minutę (rpm) przez minut. Usuń nadsącz. 6. Ponownie zawieś osad komórek w 1 ml zimnego 1 x PBS i zwiruj przy 00 rpm przez minut. 7. Usuń nadsącz i ponownie zawieś osad komórek w 00 l z 2 g/ml oczyszczonego ludzkiego białka 001 w zimnym 1 x PBS. 8. Inkubuj na lodzie przez 1 godzinę, od czasu do czasu mieszając ręcznie. 9. Zwiruj komórki przy 00 rpm przez minut. Usuń nadsącz.. Ponownie zawieś osad komórek w 1 ml zimnego 1 x PBS i wiruj przy 00 rpm przez minut. 11. Powtórz etapy 7 i Usuń nadsącz i ponownie zawieś osad komórek w 00 l 1 ng/ml króliczego przeciwciała poliklonalnego anty-ludzkie 001 w zimnym 1 x PBS z % kozią surowicą. 13. Inkubuj na lodzie przez minut od czasu do czasu mieszając ręcznie. 14. Zwiruj komórki przy 00 rpm przez minut. Usuń nadsącz. 1. Ponownie zawieś osad komórek w 1 ml zimnego 1 x PBS i zwiruj przy 00 rpm przez minut. 16. Powtórz etapy 7 i Usuń nadsącz i ponownie zawieś osad komórek w 00 l koniugatu koziego przeciwciała anty-króliczego z FITC oraz koniugatu koziego przeciwciała anty-ludzki Fc z fikoerytryną w zimnym 1 x PBS z % surowicą kozią. 18. Inkubuj na lodzie przez minut od czasu do czasu mieszając ręcznie. 19. Zwiruj komórki przy 00 rpm przez minut. Usuń nadsącz.. Ponownie zawieś osad komórek w 1 ml zimnego 1 x PBS i zwiruj przy 00 rpm przez minut. 21. Powtórz kroki 7 i Usuń nadsącz i ponownie zawieś osad komórek w 1 ml zimnego 1 x PBS z 2% surowicą kozią. 23. Kontynuuj cytometrię przepływową przy użyciu Dako MoFlo. 64
66 24. Wykreśl okno sortowania tak, aby objąć górną część 0,1% całkowitej ilości komórek pod względem stosunku fluorescencji PE/FITC. Zbierz komórki, które mieszczą się w oknie sortowania do 96-studzienkowych płytek z 0 l pożywki hodowlanej. Odzyskiwanie sekwencji regionu zmiennego łańcucha ciężkiego i łańcucha lekkiego [0322] Namnóż klony z 96-studzienkowych płytek w 6-studzienkowych płytkach. Gdy komórki osiągną 80% konfluencji w 6-studzienkowych płytkach, przejdź do izolacji genomowego DNA przy użyciu zestawu do izolacji Tissue Qiagen DNeasy. 2. Odessij pożywkę z komórek. Dodaj do każdej z 6 studzienek 00 ml 1 x PBS. Zeskrob komórki z płytki jałową końcówką do pipety. Przenieś zeskrobane komórki w PBS do jałowej probówki mikrowirówkowej. 3. Wiruj komórki przez minut przy 00 rpm. 4. Usuń nadsącz i ponowne zawieś osad komórek w 0 l 1 x PBS.. Dodaj l proteinazy K i 0 l buforu do próbek AL, zmieszaj dokładnie na worteksie i inkubuj w 6 C przez minut. 6. Dodaj do próbki 0 l etanolu i dokładnie wymieszaj na worteksie. 7. Przenieś pipetą mieszaninę z etapu 6 na kolumnę do wirowania. Odwiruj przy 8000 rpm. Wyrzuć wyciek. 8. Dodaj 00 l buforu AW1 i wiruj przez jedną minutę przy 8000 rpm. Usuń wypływ z kolumny. 9. Dodaj 00 l buforu AW2 i wiruj przez 2 min przy 100 rpm. Usuń wypływ z kolumny. Ponownie wiruj przez jedną minutę przy 100 rpm. Upewnij się, że membrana jest całkowicie sucha.. Umieść kolumnę do wirowania w jałowej probówce mikrowirówkowej i nanieś pipetą 0 l buforu AE bezpośrednio na membranę. 11. Inkubuj w temperaturze pokojowej przez jedną minutę i wiruj przez jedną minutę przy 8000 rpm w celu wymycia genomowego DNA. 12. Kontrola jakości (QC, ang. Quality Control) genomowego DNA poprzez wytworzenie następujących mieszanin reakcyjnych w 1, ml probówkach mikrowirówkowych: gdna x Bufor ładujący do próbek Maksymalna objętość l l l 3 Nanieś na 0,8% żel agarozowy w TAE 0, g/ml bromku etydyny. Użyj drabinkę DNA 1kB jako standard. Puszczaj żel przy 0 V przez - minut w buforze 1X TAE. 13. Skonfiguruj następujące reakcje PCR w jałowych probówkach do PCR: 6
67 gdna 2x HotStar Taq Master Mix Starter lewy dla domeny zmiennej * Starter prawy dla domeny zmiennej * H 2 O Maksymalna objętość 1 l 12, l 0, l 0, l, l 2 l *patrz załącznik Umieść probówki do PCR w termocyklerze i rozpocznij program przeprowadzania cykli. Początkowy etap aktywacji: 1 minut, 9 C 3-etapowe cykle Denaturacja: sekund, 94 C Przyłączanie: sekund, C Wydłużanie: 2 minuty, 72 C Liczba cykli: Etap końcowego wydłużania: minut, 72 C 1. QC reakcji PCR poprzez wytworzenie następujących mieszanin reakcyjnych w 1, ml probówkach mikrowirówkowych: Reakcja PCR x Bufor ładujący do próbek Maksymalna objętość l l l 1 Nanieś na 1% żel agarozowy w TAE 0, g/ml bromku etydyny. Użyj drabinkę DNA 1kB jako standard. Puszczaj żel przy 0 V przez - minut w buforze 1X TAE. 16. Skonfiguruj następujące reakcje do klonowania w 1, ml probówkach mikrowirówkowych przy zastosowaniu zestawu Invitrogen TOPO 2.1: Reakcja PCR Roztwór soli Wektor TOPO Maksymalna objętość 4 l 1 l 1 l 6 l 66
68 1 17. Delikatnie wymieszaj mieszaninę reakcyjną i inkubuj przez minut w temperaturze pokojowej. 18. Dodaj 2 l reakcji klonowania TOPO z etapu 17 do fiolki chemicznie kompetentnej E. coli One Shot i delikatnie wymieszaj. 19. Inkubuj w lodzie przez minut.. Poddaj komórki szokowi cieplnemu przez sekund w 42 C. 21. Przenieś probówki do lodu i inkubuj przez 2 minuty. 22. Dodaj l pożywki SOC o temperaturze pokojowej. 23. Wytrząsaj probówki poziomo w 37 C przez jedną godzinę przy 0 rpm. 24. Rozprowadź l transformacji na ponownie ogrzaną płytkę LB-karbenicylina. 2. Inkubuj płytkę przez noc w 37 C. 26. Pobierz 6 klonów z każdej transformacji do sekwencjonowania. 27. Analizuj sekwencje regionu zmiennego łańcucha ciężkiego i lekkiego łańcucha. Przejdź do drugiego cyklu przeszukiwania przy zastosowaniu metody ELISA. Trawienie wektora i klonów przeciwciała ludzkiego enzymami restrykcyjnymi [0323] Reakcje będą zależały od wybranego enzymu(ów) restrykcyjnego(ych) i będą zgodne z instrukcjami producenta; przykłady przedstawione są tutaj: Przygotuj następujące reakcje trawienia w probówce na lodzie: Wektor DNA (2 g) x Bufor Enz Rest x Woda wolna od nukleaz Enz Rest 1 ( U/ l) Enz Rest 2 ( U/ l) Całkowita objętość reakcji Klony ludzkiego przeciwciała ( g) x l l QS do 97 l 3 l 3 l 0 l x l x Bufor Enz Rest x l Woda wolna od nukleaz Enz Rest 1 ( U/ l) Enz Rest 2 ( U/ l) Całkowita objętość reakcji QS do 97 l 3 l 3 l 0 l 1. Wymieszaj delikatnie i krótko zwiruj ( sek.) w mikrowirówce. 67
69 2. Inkubuj mieszaninę reakcyjną w 37 C przez noc. Trawienie wektora CIP i oczyszczanie przy zastosowaniu zestawu QIAquick PCR Purification [0324] 1 3. Dodaj 2 l fosfatazy Apex do probówki zawierającej mieszaninę reakcyjną z trawienia wektora. 4. Inkubuj w temperaturze 37 C w ciągu minut.. Ogrzewaj w temperaturze 70 C przez minut w celu inaktywacji fosfatazy Apex 6. Dodaj 00 l buforu PBI do probówki do mikrowirówki 7. Wymieszaj na worteksie i szybko zwiruj. 8. Nanieś 70 l na raz na kolumnę. 9. Zwiruj przy x g przez 1 minutę i zdekantuj płyn z probówki do zbierania.. Powtarzaj, aż cała próbka zostanie przetworzona. 11. Przepłukuj przy użyciu 70 l buforu PE (z dodanym etanolem!). 12. Odwiruj przy x g przez 1 minutę, a następnie zdekantuj płyn z probówki do zbierania. 13. Umieść kolumnę z powrotem do probówki do zbierania i ponownie zwiruj. 14. Umieść kolumnę w nowych probówkach do mikrowirówki i poddaj elucji przy użyciu 0 l buforu EB. Oczyszczanie klonów przeciwciała ludzkiego strawionych enzymem restrykcyjnym przy zastosowaniu żelu 2 [032] 1. Skonfiguruj następujące reakcje w 1,-ml probówce do mikrowirówki: Klony w pełni ludzkiego przeciwciała strawione Enz Rest x Bufor ładujący do próbek Maksymalna objętość 0 l 3 l 3 l 3 2. Nanieś na 1% żel agarozowy w TAE z 0, g/ml bromku etydyny. Użyj drabinkę DNA 1kB jako standard. Puszczaj żel przy 0 V przez - minut w buforze 1X TAE. 4. Dodaj 3 objętości buforu QG na 1 objętość żelu.. Inkubuj w 0 C przez minut, aż skrawek żelu rozpuści się całkowicie. Dodaj do próbki izopropanol w objętość odpowiadającej objętości żelu i wymieszaj. 6. Umieść kolumnę do wirowania QIAquick w dostarczonej 2-ml probówce. 7. Nałóż próbkę do kolumnę QIAquick i zwiruj przez 1 minutę. 8. Usuń wypływ z kolumny i umieść kolumnę QIAquick z powrotem w tej samej probówce do 68
70 zbierania. 9. Dodaj 0,7 ml buforu PE do kolumny Qiaquick i zwiruj przez 1 minutę.. Usuń wypływ z kolumny i zwiruj kolumnę QIAquick przez dodatkową 1 minutę przy x g ( rpm). 11. Umieść kolumnę QIAquick w czystej 1,-ml probówce. 12. Dodaj 2 l buforu EB na środek membrany QIAquick i zwiruj kolumnę przez 1 minutę. Odstaw kolumnę na 1 minutę, a następnie zwiruj przez 1 minutę. Ligacja domeny zmiennej ludzkiego HC i LC ze strawionym DNA wektora [0326] Przygotuj następujące reakcje ligacji w probówce do mikrowirówki na lodzie: Strawiony wektorowy DNA (0 ng) domena zmienna ludzkiego HC i LC X bufor dla ligazy T4 Woda wolna od nukleaz Ligaza T4 (00 U/ l) Całkowita objętość reakcji x l y l 4 l QS do 19 l 1 l l Wymieszaj delikatnie i krótko zwiruj ( sek.) w mikrowirówce. 2. Inkubuj w temperaturze pokojowej przez 2 godziny lub w 16 C przez noc. 3. Transformuj superkompetentne komórki E. coli każdą z mieszanin reakcji ligacji. 4. Schłodź wstępnie 14 ml okrągłodenne probówki polipropylenowe BD Falcon na lodzie. Przygotuj pożywkę SOC o temperaturze 42 C.. Rozmroź superkompetentne komórki na lodzie. Po rozmrożeniu delikatnie wymieszaj i umieść porcję 0 l komórek w każdej z wstępnie schłodzonych probówek. 6. Do każdej porcji komórek dodaj 1,7 l -merkaptoetanolu. Inkubuj komórki na lodzie przez minut, mieszając delikatnie co 2 minuty. 7. Dodaj 2 l mieszaniny reakcyjnej z ligacji do jednej porcji komórek. Potrząśnij łagodnie probówkę. 8. Inkubuj probówki na lodzie przez minut. 9. Potraktuj probówki pulsowo ciepłem w 42 C w łaźni wodnej przez 4 sekund.. Inkubuj probówki na lodzie przez 2 minuty. 11. Dodaj 900 l ogrzanego podłoża SOC i inkubuj probówki w 37 C przez 1 godzinę z wytrząsaniem przy 22- rpm. 12. Wysiej l i 0 l mieszaniny transformacyjnej na płytki z agarem LB, zawierające karbenicylinę. 13. Inkubuj płytki w 37 C przez noc. 69
71 14. Zlicz kolonie na płytkach i pobierz 6 kolonii dla przeszukiwania poprzez PCR i sekwencjonowania. 1. Wybierz jeden klon z właściwą sekwencją, wypreparuj plazmidowy DNA, a następnie przejdź do transfekcji komórek 293F. Transfekcja komórek 293F [0327] Tydzień przed transfekcją, przenieś komórki 293F do jednowarstwowej hodowli w pożywce Dulbecco's Modified Eagle Medium (D-MEM) z dodatkiem surowicy. 2. Dzień przed transfekcją, wysiej 0,1 x komórek w 0 l D-MEM z dodatkiem surowicy, na próbkę do transfekcji, w formatach 96-studzienkowych. 3. Dla każdej próbki do transfekcji przygotuj kompleksy DNA-Lipofektamina. 4. Rozcieńcz 0,2 g DNA w 0 l pożywki ze zmniejszonym poziomem surowicy Opti-MEM Reduced Serum Medium. Delikatnie wymieszaj.. Rozpuść 0,12 l Lipofektaminy w 0 l pożywce Opti-MEM Reduced Serum Medium. Delikatnie wymieszaj i inkubuj przez min w temperaturze pokojowej. 6. Połącz rozcieńczony DNA z rozcieńczoną Lipofektaminą. Wymieszaj delikatnie i inkubuj przez min w temperaturze pokojowej. 7. Dodaj 0 l kompleksów DNA-Lipofektamina do każdej studzienki zawierającej komórki i pożywkę. Wymieszaj delikatnie, kołysząc płytkę w przód i w tył. 8. Inkubuj komórki w 37 C w inkubatorze z % CO Dodaj 0 l pożywki D-MEM z dodatkiem surowicy, do każdej studzienki po 6 godzinach. Inkubuj komórki w 37 C w inkubatorze z % CO 2 przez noc.. Odessij pożywkę z każdej studzienki. Przepłucz każdą studzienkę 0 l 293 II SFM z 4 mm L- glutaminy. Dodaj do każdej studzienki 0 l 293 SFM II z 4 mm L-glutaminy. 11. Zbierz nadsącz do testu ELISA po 96 godzinach od transfekcji. Załącznik 1.1: Przepisy na bufory [0328] 1x PBS z 2% kozią surowicą 3 2 ml koziej surowicy 98 ml 1 x PBS Bufor TAE 0x 242 g zasada Tris 70
72 7,1 ml lodowatego kwasu octowego 37,2 g Na 2 EDTA-2H 2 O Dodaj destylowanej H 2 O do końcowej objętości 1 litra Bufor TAE 1x ml buforu 0X TAE 800 ml destylowanej H 2 O 0,8% żel agarozowy z bromkiem etydyny 1 0,8 g agarozy LE 0 ml buforu 1X TAE Rozpuść agarozę w kuchence mikrofalowej i mieszaj obrotowo, aby zapewnić równomierne mieszanie. Schłodź agarozę do C. Dodaj do agarozy 2, l mg/ml bromku etydyny. Wlej do platformy dla żelu. 1% żel agarozowy z bromkiem etydyny 2 1 g agarozy LE 0 ml buforu 1X TAE Rozpuść agarozę w kuchence mikrofalowej i mieszaj obrotowo, aby zapewnić równomierne mieszanie. Schłodź agarozę do C. Dodaj do agarozy 2, l mg/ml bromku etydyny. Wlej do platformy dla żelu. LB 3 g NaCl g tryptonu g wyciągu z drożdży Dodaj destylowaną H 2 O do końcowej objętości 1 litra. Doprowadź ph do 7,0 przy użyciu N NaOH. Autoklawuj. Agar LB-karbenicylina 71
73 g NaCl g tryptonu g wyciągu z drożdży g agaru Dodaj destylowaną H 2 O do końcowej objętości 1 litra. Doprowadź ph do 7,0 przy użyciu N NaOH. Autoklawuj. Schłodź do C. Dodaj ml mg/ml karbenicyliny wyjałowionej poprzez filtrowanie. Wylej na płytki Petriego (2 ml/0-mm płytka). Pożywka SOC 1 0, g NaCl g tryptonu 0, g ekstraktu drożdżowego 2 ml wyjałowionego poprzez filtrację % glukozy Dodaj destylowaną H 2 O do końcowej objętości 1 litra. Autoklawuj. Dodaj przed użyciem ml wyjałowionego poprzez filtrację 1 M MgCl 2 i ml wyjałowionego poprzez filtrację 1 M MgSO 4. 2 Roztwór do płukania 0,0% Tween- w PBS Roztwór blokujący 2% odtłuszczonego mleka (Carnation) w PBS Inaktywowana termicznie płodowa surowica bydlęca 3 00 ml inaktywowanej termicznie płodowej surowicy bydlęcej w oryginalnej butelce od dostawcy. Ogrzewaj przez minut w 6 C z mieszaniem co minut. Przygotuj porcje 0 ml i przechowuj w - C. Pożywka Dulbecco's Modified Eagle Medium z dodatkiem surowicy 72
74 00 ml pożywki Dulbecco's Modified Eagle Medium 0 ml inaktywowanej termicznie płodowej surowicy bydlęcej ml mm nieistotne aminokwasy MEM Non-Essential Amino Acids 293 SFM II z 4 mm L-glutaminą 00 ml SFM II ml 0 mm L-glutaminy Roztwór DEAE-dekstranu 1 1 g dekstranu DEAE (dekstranu dietyloaminoetylowy) Rozpuść w 0 ml wody destylowanej. Poddaj jałowieniu przez filtr. Załącznik 1.2: Konstrukcja biblioteki dla w pełni ludzkiego przeciwciała [0329] Wszystkie funkcjonalne regiony V ludzkiego łańcucha lekkiego kappa (V k ) i łańcucha ciężkiego (V H ) linii zarodkowej można uzyskać z bazy V (http: //vbase.mrc-cpe.cam.ac.u/) i przyrównać. Przyrównania można następnie analizować w odniesieniu do różnorodności, zwłaszcza w trzech regionach zrębowych. Pożądaną liczbę genów V H i V k z otrzymanych rezultacie zgrupowań sekwencji można następnie wybrać do konstrukcji biblioteki. 2 3 Klonowanie regionu V [03] Geny regionu V łańcuchów ciężkich i lekkich (w tym ram regiony zrębowe 1, 2, i 3 oraz CDR1, CDR2 i CDR3) powiela się z ludzkiego genomowego DNA w dwóch kawałkach przy użyciu starterów specyficznych dla genu. Częściowe geny regionu V łączy się następnie przez nakładający się PCR. Do regionów V LC pełnej długości dodaje się łącznik przy zastosowaniu reakcji PCR, gniazdowe PCR (ang. nested), przed klonowaniem do ssaczego wektora ekspresyjnego. Dla sklonowanych domen zmiennych LC potwierdza się sekwencję (z uzyskaniem klonów V LC). Domeny zmienne łańcucha ciężkiego klonuje się przy zastosowaniu TOPO i potwierdza sekwencję (klony V HC TOPO). Regiony V HC z potwierdzoną sekwencją powiela się z odpowiednich plazmidów, do końca 3 dodaje się łącznik i powstałe w rezultacie produkty PCR klonuje się do klonów domen zmiennych V LC, z wytworzeniem kombinacji Vk/VH, i do wektora ssaczego. Wyrażanie IgG pełnej długości [0331] Wyrażanie łańcucha lekkiego kappa i łańcucha ciężkiego IgG1 pełnej długości w pożądanym wektorze ssaczym może być kierowana przez jeden promotor, na przykład, promotor CMV. Każdy 73
75 łańcuch jest poprzedzony przez sygnał sekrecyjny kierujący nowopowstający łańcuch polipeptydowy do retikulum endoplazmatycznego (ER). Do końca C łańcucha ciężkiego może być przyłączony sygnał kotwiący. Sygnał ten jest odcinany i zastępowany kotwicą, która wiąże się z IgG pełnej długości na zewnątrz błony komórkowej po wydzieleniu. Załącznik 1.3: Wytwarzanie biblioteki stabilnych, w pełni ludzkich przeciwciał w komórkach ssaków poprzez transfekcję [0332] 1. Tydzień przed transfekcją, przenieś komórki CHO-S do jednowarstwowej hodowli w pożywce Dulbecco's Modified Eagle Medium (D-MEM) z dodatkiem surowicy. 2. Dzień przed transfekcją, wysiej 6 x 6 komórek w 1 ml D-MEM z dodatkiem surowicy, na próbkę do transfekcji, w -cm płytce do hodowli tkankowej. 3. Dla każdej -cm płytki przygotuj kompleksy DNA-Lipofektamina zgodnie z etapami Rozcieńcz 2 g duże preparaty plazmidowego DNA dla biblioteki w pełni ludzkiego przeciwciała w 1, ml pożywki Opti-MEM ze zmniejszonym poziomem surowicy. Delikatnie wymieszaj.. Rozcieńcz 60 l Lipofektaminy w 1, l pożywki Opti-MEM Reduced Serum Medium. Delikatnie wymieszaj i inkubuj przez min w temperaturze pokojowej. 6. Połącz rozcieńczony DNA z rozcieńczoną Lipofektaminą. Wymieszaj delikatnie i inkubuj przez min w temperaturze pokojowej. 7. Dodaj 0 l kompleksów DNA-Lipofektamina do każdej studzienki zawierającej komórki i pożywkę. Wymieszaj delikatnie kołysząc płytkę w przód i w tył. 8. Inkubuj komórki w 37 C w inkubatorze z % CO Dodaj 0 l pożywki D-MEM z dodatkiem surowicy. Inkubuj komórki w 37 C w inkubatorze z % CO 2 przez 48 godziny.. Oderwij komórki przy zastosowaniu pożywki do odrywania Detachin i ponownie zawieś komórki w D-MEM z dodatkiem surowicy. 11. Wysiej 0,4 x 6 komórek w ml D-MEM z dodatkiem surowicy z 800 g/ml G418 w jednej -cm płytce do hodowli tkankowej. Przenieść wszystkie transfekowane komórki, co daje w rezultacie x -cm płytek. 12. Wysiej 0,4 x 6 nietransfekowanych komórek CHO-S w ml D-MEM z dodatkiem surowicy z 800 g/ml G418 w jednej -cm płytce do hodowli tkankowej. 13. Dokarmiaj komórki D-MEM z dodatkiem surowicy z 800 g/ml G418, co 4 dni. 14. Po upływie 14 dni, obejrzyj płytki nietransfekowanymi komórkami CHO-S. Nie powinno być żadnych żywych komórek na płytce. 1. Oderwij pozostałe transfekowane komórki przy zastosowaniu pożywki do odrywania Detachin i zamroź komórki w pożywce do zamrażania jako 1 x 7 komórek/ml. 74
76 Załącznik 1.4: Wytwarzanie biblioteki stabilnych w pełni ludzkich przeciwciał w komórkach ssaków poprzez infekcję retrowirusem [0333] 1. Jeden dzień przed transfekcją, wysiej 6 x 6 komórek EcoPack w 1 ml D-MEM z dodatkiem surowicy, na próbkę transfekcji, w -cm płytce do hodowli tkankowej. Przygotuj dziesięć płytek -cm. 2. Przygotuj podłoże zawierającego MBS. Robi się to bezpośrednio przed transfekcją. Dla każdej -cm płytki do hodowli tkankowych, trzeba przygotować 12 ml pożywki zawierającej MBS. 3. Dodaj 12 ml pożywki zawierającej MBS do każdej -cm płytki, umieść z powrotem, płytka po płytce, do inkubatora na 37 C. To musi być zrobione - minut przed dodaniem zawiesiny DNA. 4. Zawieś g dużego preparatu (ang. maxi-prep) osadu plazmidowego DNA dla biblioteki w pełni ludzkich przeciwciał w l jałowej H 2 O i przenieś DNA do oddzielnych -ml okrągłodennych probówek polistyrenowych BD Falcon.. Dodaj do DNA 0 l roztworu I i 00 μl roztworu II z zestawu do transfekcji Stratagene Transfection MBS Mammalian Transfection Kit. 6. Ostrożnie zawieś cały osad w zawiesinie DNA przez pipetowanie zawiesiny w górę i w dół pipetorem ustawionym na 00 l. 7. Inkubuj zawiesinę DNA w temperaturze pokojowej przez minut. 8. Wyjmij -cm płytki do transfekcji z inkubatora i dodaj do płytek zawiesinę DNA kroplami, mieszając delikatnie, aby zapobiec podnoszeniu komórek z płytki i rozprowadź równomiernie zawiesinę DNA. 9. Umieść z powrotem płytki do hodowli tkankowej w inkubatorze na 37 C.. Po inkubowaniu przez 3 godzin, usuń pożywkę z płytek i zastąp ją 4 ml D-MEM z dodatkiem surowicy uzupełnionej 2 M chlorochininy. Umieść płytki z powrotem w inkubatorze na 37 C. 11. Po inkubowaniu przez kolejne 6-7 godzin, usuń pożywkę zawierającą 2 M chlorochiny i zastąp 4 ml D-MEM z dodatkiem surowicy bez chlorochininy. 12. Inkubuj płytkę w inkubatorze na 37 C przez noc. 13. Usuń pożywkę z płytek i zastąp 3,0 ml świeżej pożywki D-MEM z dodatkiem surowicy. Umieść płytki z powrotem w inkubatorze na 37 C. 14. Wyjmij komórki do pakowania wytwarzające wirusa z inkubatora. 1. Zbierz nadsącz zawierający wirusa z pierwszej płytki i przefiltruj przez filtr 0,4 m do jałowej 0-ml probówki stożkowej. 16. Rozporcjuj nadsącz wirusowy do 1, ml kriofiolek i szybko zamroź w łaźni suchy lód/etanol. Przechowuj nadsącz wirusowy w -80 C. 17. Wysiej 0, x 6 komórek NIH-3T3 w ml D-MEM z dodatkiem surowicy w jednej -cm płytce do hodowli tkankowej. Wysiej 2 x -cm płytka do hodowli tkankowych. 18. Szybko rozmroź nadsącz przez gwałtowne mieszanie w 37 C w łaźni z H 2 O. 7
77 1 19. Rozcieńcz wirusa DMEM z dodatkiem surowicy bydlęcej z roztworem DEAE-dekstranu przy mianie 0,3 x cząstek wirusowych/ml. Przygotuj 3 ml rozcieńczonego wirusa na 0 mm-płytkę do infekcji (zakażonych zostanie % komórek). Przygotuj koktajl-ślepą próbę z pożywki wzrostowej plus DEAE-dekstran, do zastosowania jako kontrola ujemna.. Odessij pożywkę z komórek NIH-3T3. Dla każdej płytki, rozprowadź 3,0 ml rozcieńczonego wirusa równomiernie nad komórkami. Umieść płytki z powrotem w inkubatorze na 37 C na 3 godziny. 21. Po 3-godzinnej inkubacji dodaj do każdej płytki dodatkowe 7,0 ml D-MEM z dodatkiem surowicy i umieść płytki z powrotem w inkubatorze na 37 C. Inkubuj płytki przez 48 godzin. 22. Wymień pożywkę na ml D-MEM z dodatkiem surowicy cielęcej z 800 g/ml G418 w każdej z -cm płytce do hodowli tkankowej. 23. Dokarmiaj komórki D-MEM z dodatkiem surowicy cielęcej z 800 g/ml G418, co 4 dni. 24. Po 14 dniach obejrzyj płytki z komórkami NIH-3T3 infekowanymi ślepą próbą. Nie powinno być żadnych żywych komórek na płytce. 2. Oderwij pozostałe transfekowane komórki przy zastosowaniu pożywki do odrywania Detachin i zamroź komórki w pożywce do zamrażania przy 1 x 7 komórek/ml. Przykład 2. Reakcje dla kompleksowej ewolucji pozycyjnej (CPE) Reakcja mutagenezy 2 [0334] Zaprojektowano jedną parę starterów (Mieszanina starterów 1 i Mieszanina starterów 2) dla każdego kodonu, który ma być wstawiony. Projekt będzie zależeć od sekwencji genów i do zaprojektowania starterów można wykorzystać analizę sekwencji baz danych, taką jak Sequencher (Gene Codes Corporation) lub VectorNTI (Life Technologies). Dla CPE jedną parę starterów projektuje się dla każdego kodonu, który ma zostać zmutowany. Zdegenerowany kodon docelowy (NNK lub NNN) znajduje się w środku, otoczony przez zasad po każdej ze stron (całkowita długość startera: 43 zasad, 9f klonów do sekwencjonowania w celu zidentyfikowania unikatowych mutantów). Matrycowym DNA jest DNA wektora z genem(ami) docelowym(i). [033] Przygotuj następujące reakcje w 96-studzienkowych cienkościennych płytkach do PCR lub 0,2 ml cienkościennych probówkach do PCR na lodzie: Mieszanina starterów 1 (2, mm) Mieszanina starterów 2 (2, mm) x bufor dla polimerazy DNA Pfu turbo Matrycowy DNA (,, 2 ng) dntp l l 2, l x l 2 l 76
78 Woda wolna od nukleaz polimeraza DNA Pfu turbo (2, U/μl) Całkowita objętość reakcji QS do 24, l 0, l 2 l 1. Przygotuj jedną reakcję-kontrolę ujemną na jednej 96-studzienkowej płytce (zastąp startery buforem TE) 2. Wymieszaj delikatnie i krótko zwiruj ( sek.) w wirówce stołowej. 3. Przeprowadź cykl reakcji wykorzystując parametry cykli przedstawione poniżej: Odcinek Cykle Temperatura Czas C sekund C sekund C 1 minuta 68 C 16 minut Analiza kontroli jakości [0336] 1. Aby przeprowadzić QC dla reakcji amplifikacji, skonfiguruj następujące reakcje w 96- studzienkowych cienkościennych płytkach do PCR lub 0,2 ml cienkościennych probówkach do PCR: Reakcja mutagenezy Woda Bufor ładujący do próbek Objętość l 4 l 1 l l 1 2. Nanieś l na 1% żel agarozowy w TAE z 0, g/ml bromku etydyny. Użyj drabinkę DNA 1kB jako standard. Puszczaj żel przy 0 V przez - minut w buforze 1X TAE. Trawienia reakcji mutagenezy odpowiednimi enzymami restrykcyjnymi do klonowania do DNA wektora - Przykład dla enzymu restrykcyjnego DpnI [0337] 77
79 1 1. Dodaj 0, l enzymu restrykcyjnego DpnI ( U/ l) bezpośrednio do każdej reakcji. 2. Wymieszaj delikatnie i krótko zwiruj ( sek.) w wirówce stołowej. 3. Inkubuj w 37 C w urządzeniach do PCR przez 2 godziny. 4. Transformuj 6 mieszanin reakcyjnych z każdej z 96-studzienkowej płytki do superkompetentnych komórek XL1 Blue. Przechowuj resztę reakcji w - C.. Schłodź wstępnie 0,2-ml probówki do PCR na lodzie. Ogrzej pożywkę SOC do 42 C. 6. Rozmroź superkompetentne komórki XL1 Blue na lodzie. Po rozmrożeniu delikatnie wymieszaj i rozporcjuj 0 l komórek do każdej z wstępnie schłodzonych probówek. 7. Dodaj 0,8 l beta-merkaptoetanolu do każdej porcji komórek. Inkubuj komórki na lodzie przez minut, mieszając delikatnie co 2 minuty. 8. Dodaj 2 l mieszaniny reakcyjnej do jednej porcji komórek. Zamieszaj probówki łagodnie. 9. Inkubuj probówki w blokach chłodzących przez minut.. Poddaj probówki pulsowi cieplnemu w 42 C w łaźni wodnej przez 4 sekund. 11. Inkubuj probówki na lodzie przez 2 minuty. 12. Dodaj 0 l wstępnie ogrzanej pożywki SOC i probówki inkubuj w 37 C przez 1 godzinę z wytrząsaniem przy 22- rpm. 13. Wysiej całą mieszaninę transformacjyjną na płytki agarowe LB zawierające karbenicylinę. 14. Inkubuj płytki w temperaturze 37 C przez noc. 1. Zlicz kolonie na płytkach i pobierz 12 kolonii z każdej reakcji transformacji dla minipreparatu i sekwencjonowania. Transformacja na dużą skalę 2 3 [0338] 1. Rozmroź superkompetentne komórki XL1 Blue na lodzie. Rozmroź probówek komórek kompetentnych na 96 reakcji. Po rozmrożeniu do każdej probówki l komórek kompetentnych dodaj 4 l beta-merkaptoetanolu. Inkubuj komórki na lodzie przez minut, mieszając delikatnie co 2 minuty. 2. Schłodź wstępnie 0,2 ml probówki do PCR na lodzie. Ogrzej pożywkę SOC do 42 C. 3. Rozporcjuj 0 l komórek do każdej z wstępnie schłodzonych probówek. 4. Dodaj 2 l mieszaniny reakcyjnej do jednej porcji komórek. Zamieszaj probówki łagodnie.. Inkubuj probówki w blokach chłodzących przez minut. 6. Poddaj probówki pulsowi cieplnemu w 42 C w łaźni wodnej przez 4 sekund. 7. Inkubuj probówki na lodzie przez 2 minuty. 8. Dodaj 0 l wstępnie ogrzanej pożywki SOC i probówki inkubuj w 37 C przez 1 godzinę z wytrząsaniem przy 22- rpm. 9. Wysiej całą mieszaninę transformacjyjną na płytki agarowe LB zawierające karbenicylinę.. Inkubuj płytki w temperaturze 37 C przez noc. 11. Hoduj komórki do miniprepeparatów w 96-studzienkowych blokach 78
80 12. Przygotuj minipreparaty DNA przy użyciu zestawu 96 QIAVac postępując zgodnie z protokołem producenta. Przykład 3. Przeszukiwanie pod kątem polepszenia powinowactwa Transfekcja [0339] Tydzień przed transfekcją, przenieś komórki 293F do jednowarstwowej hodowli w pożywce Dulbecco's Modified Eagle Medium (D-MEM) z dodatkiem surowicy. Dzień przed transfekcją, wysiej 0,2 x i 0,4 x komórek w 0 l D-MEM z dodatkiem surowicy, na próbkę do transfekcji, w formatach 96-studzienkowych Dla każdej próbki do transfekcji przygotuj kompleksy DNA-Lipofektamina. 2. Rozcieńcz 0,2 g DNA w 0 l pożywki Opti-MEM Reduced Serum Medium. Delikatnie wymieszaj. 3. Rozcieńcz 0,12 l Lipofektaminy w 1, l pożywce Opti-MEM Reduced Serum Medium. Delikatnie wymieszaj i inkubuj przez min w temperaturze pokojowej. 4. Połącz rozcieńczony DNA z rozcieńczoną Lipofektaminą. Wymieszaj delikatnie i inkubuj przez min w temperaturze pokojowej.. Dodaj 0 l kompleksów DNA-Lipofektamina do każdej studzienki zawierającej komórki i pożywkę. Wymieszaj delikatnie kołysząc płytkę w przód i w tył. 6. Inkubuj komórki w 37 C w inkubatorze z % CO Dodaj 0 l pożywki D-MEM z dodatkiem surowicy do każdej studzienki po 6 godzinach. Inkubuj komórki w 37 C w inkubatorze z % CO 2 przez noc. 8. Odessij pożywkę z każdej studzienki. Przepłucz każdą studzienkę 0 l 293 II SFM z 4 mm L-glutaminy. Dodaj do każdej studzienki 0 l 293 SFM II z 4 mm L-glutaminy. 9. Zbierz nadsącz do testu ELISA po 96 godzinach po transfekcji. Funkcjonalny test ELISA [03] 3 1. Powlecz 96-studzienkowe płytki Nunc-Immuno Maxisorp 0 l 2 mg/ml antygenu w roztworze do powlekania. 2. Zakryj płytki uszczelniaczami i inkubuj przez noc w 4 C. 3. Zdekantuj płytki i wystukaj pozostałości płynu. 4. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej.. Zdekantuj płytki i wystukaj pozostałości płynu. 79
81 1 6. Dodaj 0 l roztworu blokującego. Wytrząsaj przy 0 rpm przez 1 godzinę w temperaturze pokojowej. 7. Zdekantuj płytki i wystukaj pozostałości płynu. 8. Dodaj jako duplikat 0 l/studzienkę przeciwciała kontrolnego (2 g/ml) w roztworze do blokowania płytek. 9. Dodaj jako duplikat 0 l nadsączu z transfekcji (SOP A) do płytek.. Wytrząsaj przy 0 rpm przez jedną godzinę w temperaturze pokojowej. 11. Zdekantuj płytki i wystukaj pozostałości płynu. 12. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej. 13. Powtórz etapy razy. 14. Dodaj do każdej studzienki 0 l rozcieńczenia 1:000 oczyszczonego przez powinowactwo koniugatu koziego anty-ludzkiego przeciwciała z HRP w roztworze blokującym. 1. Wytrząsaj przy 0 rpm przez jedną godzinę w temperaturze pokojowej. 16. Zdekantuj płytki i wystukaj pozostałości płynu. 17. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej. 18. Powtórz etapy razy. 19. Dodaj do każdej studzienki 0 l substratu TMB Sigma. Inkubuj w temperaturze pokojowej i sprawdzaj co 2- min.. Dodaj 0 l 1 N HCl w celu zatrzymania reakcji. 21. Odczytuj przy długości fali nm. 2 Ilościowy test ELISA [0341] 3 1. Powlecz 96-studzienkowe płytki Nunc-Immuno Maxisorp 0 l g/ml oczyszczonego poprzez powinowactwo swoistego wobec Fc koziego anty-ludzka IgG w roztworze do powlekania. 2. Zakryj płytki uszczelniaczami i inkubuj przez noc w 4 C. 3. Zdekantuj płytki i wystukaj pozostałości płynu. 4. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej.. Zdekantuj płytki i wystukaj pozostałości płynu. 6. Dodaj 0 l roztworu blokującego. Wytrząsaj przy 0 rpm przez 1 godzinę w temperaturze pokojowej. 7. Zdekantuj płytki i wystukaj pozostałości płynu. 8. Dodaj jako duplikat 0 l/studzienkę wystandaryzowanego stężenia oczyszczonej ludzkiej surowiczej IgG w roztworze do blokowania płytek. 80
82 1 9. Dodaj do płytek jako duplikat 0 l nadsączu z transfekcji (SOP A).. Wytrząsaj przy 0 rpm przez jedną godzinę w temperaturze pokojowej. 11. Zdekantuj płytki i wystukaj pozostałości płynu. 12. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej. 13. Powtórz etapy razy. 14. Dodaj do każdej studzienki 0 l rozcieńczenia 1:000 oczyszczonego poprzez powinowactwo koniugatu koziego anty-ludzkiego przeciwciała z HRP w roztworze blokującym. 1. Wytrząsaj przy 0 rpm przez jedną godzinę w temperaturze pokojowej. 16. Zdekantuj płytki i wystukaj pozostałości płynu. 17. Dodaj 0 l roztworu do płukania. Wytrząsaj przy 0 rpm przez min w temperaturze pokojowej. 18. Powtórz etapy razy. 19. Dodaj do każdej studzienki 0 l substratu TMB Sigma. Inkubuj w temperaturze pokojowej i sprawdzaj co 2- min.. Dodaj 0 l 1N HCl w celu zatrzymania reakcji. 21. Odczytuj przy długości fali nm. Przykład 4. Wytwarzanie i przeszukiwanie biblioteki dla Fc z wariantami kodonów pod kątem optymalnego wytwarzania przeciwciała 2 [0342] Niniejszy przykład wykonania dostarcza sposobów wytwarzania bibliotek dla Fc z wariantami kodonów oraz sposobów przeszukiwania w celu uzyskania wariantów Fc zoptymalizowanych pod względem zwiększenia wyrażania w komórkach gospodarza przy wytwarzaniu w porównaniu z postacią rodzicielską polipeptydu Fc. A. Projektowanie i konstrukcja biblioteki dla Fc z wariantami kodonów 3 [0343] Dla każdego kodonu w docelowym obszarze (w tym przypadku części Fc cząsteczki ludzkiej IgG1) zaprojektowano parę zdegenerowanych starterów (lewy i prawy), które zawierają kodon docelowy i zasad po obu stronach. Pozycja 3. docelowego kodonu (pozycja tolerancji, ang. wobble) zawiera mieszaninę zasad (Tabela 3), co umożliwia wytworzenie wszystkich cichych mutacji w pozycji docelowej przy zastosowaniu tego samego kodonu (Przykład A). Drugi zestaw dla zdegenerowanego startera jest przeznaczony dla tej samej pozycji kodonu, jeżeli odpowiedni aminokwas może być kodowany przez inny kodon (Przykład B). Odpowiednie zdegenerowane startery lewe i prawe zostały zmieszane w stosunku 1:1, przyłączone do matrycy i wydłużone do produktów pełnej długości poprzez wymianę nici z użyciem termostabilnej polimerazy DNA. Matrycę trawi się DpnI i produkty wydłużania pełnej długości transformuje do E. coli. Sekwencjonuje się do 12 kolonii na reakcję mutagenezy. Mutanty o potwierdzonej sekwencji rozmieszcza się na 96-studzienkowych 81
83 płytkach i przechowuje w glicerolu. Zapasy glicerolowe są wykorzystywane do minipreparatów plazmidowego DNA do transfekcji do komórek ssaczych i przeszukiwania. Tabela 3: Oznaczenia dla zdegenerowanych zasad w syntetycznych oligo Symbol R Y M K S W H B V D N Mieszanina zasad A, G C, T A, C G, T C, G A, T A, C, T C, G, T A, C, G A, G, T A, C, G, T Przykład A: kodon docelowy = CCC (prolina) [0344] starter lewy: CCD, starter prawy: HGG Przykład B: kodon docelowy = TCG (seryna) 1 [034] starter lewy 1: TCH, starter prawy 1: DGA starter lewy 2: AGY, starter prawy 2: RCT [0346] zasad otaczających kodon docelowy nie jest pokazanych. Całkowita długość startera: 43 zasad. B. Przeszukiwanie oparte na wyrażaniu i teście ELISA biblioteki dla Fc z wariantami kodonu 82
84 [0347] Klony z biblioteki dla Fc z wariantami kodonu transfekowano do ssaczej linii komórkowej. IgG pełnej długości były wytwarzane i wydzielane do pożywki. Nadsącza z wyrażonymi Fc z wariantami kodonów przeszukiwano pod kątem ekspresji IgG wyższej niż klonów rodzicielskich, z zastosowaniem testu ELISA. Dane ELISA znormalizowano przy zastosowaniu testu dla beta-galaktozydazy mierzącej wydajność transfekcji. Najlepsze trafienia zidentyfikowane przy pierwszym przeszukiwaniu ponownie transfekowano i przeszukiwano ponownie trzy razy, aby potwierdzić zwiększony poziom ekspresji Zastrzeżenia patentowe 1. Sposób przeprowadzania ewolucji i wyrażania przeciwciała w gospodarzu do wytwarzaniakomórce eukariotycznej; obejmujący: 1 2 a. wyselekcjonowanie przeciwciała matrycowego; b. poddanie przeciwciała matrycowego ewolucji, aby umożliwić wytworzenie zbioru zmutowanych przeciwciał w gospodarzu do wytwarzania-komórce eukariotycznej; c. przeszukiwanie zmutowanych przeciwciał pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; d. wyselekcjonowanie ulepszonego zmutowanego przeciwciała ze zbioru zmutowanych przeciwciał w oparciu o optymalizację co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności w porównaniu do tej samej właściwości, cechy lub aktywności przeciwciała matrycowego; i e. wyrażanie ulepszonego zmutowanego przeciwciała w tym samym w gospodarzu do wytwarzania-komórce eukariotycznej co w etapie (b) dla dowolnej skali komercyjnej. 2. Sposób według zastrz. 1, w którym etap selekcjonowania obejmuje: a1. wytwarzanie biblioteki dla przeciwciała anty-antygen w gospodarzu do wytwarzaniakomórce eukariotycznej przy zastosowaniu prezentacji przeciwciała na powierzchni komórek; a2. przeszukiwanie biblioteki pod kątem co najmniej jednej określonej właściwości, cechy lub aktywności; i a3. wyselekcjonowanie przeciwciała matrycowego z biblioteki Sposób według zastrz. 1, w którym gospodarz do wytwarzania-komórka eukariotyczna jest wybierany spośród mysich komórek fibroblastów 3T3; komórek fibroblastów chomika syryjskiego BHK21; psich komórek nabłonkowych MDCK; ludzkich komórek nabłonkowych Hela; komórek nabłonkowych kanguroszczura PtK1; mysich komórek plazmatycznych SP2/0; i mysich komórek plazmatycznych NS0; ludzkich komórek embrionalnych nerki HEK 293; komórek nerkowych małpy COS; komórek jajnika chomika chińskiego CHO, CHO-S; mysich komórek embrionalnych 83
85 R1; mysich komórek embrionalnych E14.1; ludzkich komórek embrionalnych H1; ludzkich komórek embrionalnych H9; ludzkich komórek embrionalnych PER C.6, komórek drożdży S. cerevisiae; i komórek drożdży Pichia. 4. Sposób według zastrz. 3, w którym gospodarzem do wytwarzania-komórką eukariotyczną jest CHO lub HEK Sposób według zastrz. 1, w którym etapy przeszukiwania obejmują sortowanie komórek aktywowane fluorescencją (FACS). 6. Sposób według zastrz. 1, w którym etap przeprowadzania ewolucji obejmuje wytworzenie zbioru zmutowanych przeciwciał wytworzonych z przeciwciała matrycowego, mających m regionów determinujących dopasowanie (CDR), przy czym m jest liczbą całkowitą od 1 do 6, który to sposób obejmuje: a. wytworzenie m x n oddzielnych zbiorów przeciwciał, przy czym każdy zbiór obejmuje elementy-przeciwciała zawierające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji CDR; i przy czym każdy zbiór przeciwciał różni się w pojedynczej określonej uprzednio pozycji; a liczba różnych wytwarzanych elementów-przeciwciał wynosi m x n x X. 7. Sposób według zastrz. 6, w którym m wynosi Sposób według zastrz. 1, w którym przeciwciało matrycowe stanowi fragment przeciwciało wybrany spośród łańcucha ciężkiego, łańcucha lekkiego, domeny zmiennej, domeny stałej, regionu hiperzmiennego, regionu determinującego dopasowanie 1 (CDR1), regionu determinującego dopasowanie 2 (CDR2) i regionu determinującego dopasowanie 3 (CDR3) Sposób według zastrz. 1, w którym etap przeprowadzania ewolucji obejmuje h. wytworzenie n-1 odrębnych zbiorów zmutowanych polipeptydów z przeciwciała matrycowego, przy czym każdy zbiór obejmuje elementy-polipeptydy zawierające liczbę X różnych określonych uprzednio reszt aminokwasowych w jednej określonej uprzednio pozycji polipeptydu; i przy czym każdy zbiór polipeptydów różni się w pojedynczej określonej uprzednio pozycji; a liczba różnych wytwarzanych elementów-polipeptydów wynosi [n-1] x X; i w którym etap przeszukiwania (c) obejmuje: 84
86 i. testowanie każdego elementu-polipeptydu pod kątem co najmniej jednej określonej uprzednio właściwości, cechy lub aktywności; j. identyfikowanie jakiejkolwiek zmiany w tej właściwości, cesze lub aktywności elementupolipeptydu w stosunku do polipeptydu matrycowego; k. wytworzenie mapy funkcyjnej, która to mapa funkcyjna jest wykorzystywana do identyfikacji pozycji i mutacji w zmutowanym polipeptydzie, które dają w rezultacie ulepszonego mutanta i/lub mutację cichą w porównaniu do polipeptydu matrycowego.. Sposób według zastrz. 9, w którym X to Sposób według zastrz., w którym ten etap wytwarzania obejmuje: 1 i. poddanie polinukleotydu zawierającego kodony kodujące taki polipeptyd matrycowy amplifikacji opartej na polimerazie przy użyciu 64-krotnie zdegenerowanego oligonukleotydu dla każdego kodonu, który ma być mutagenizowany, przy czym każdy z tych 64-krotnie zdegenerowanych oligonukleotydów składa z pierwszej sekwencji homologicznej i zdegenerowanej sekwencji tripletu N, N, N, tak, aby wytworzyć zbiór polinukleotydów potomnych; i ii. poddanie tego zbioru polinukleotydów potomnych amplifikacji klonalnej tak, że polipeptydy mutanta kodowane przez polinukleotydy potomne są wyrażane Sposób według zastrz. 1, w którym określona uprzednio właściwość, cecha lub aktywność jest wybrana spośród zmniejszenia agregacji przeciwciało-białko, wzmocnienia stabilności przeciwciała, zwiększonej rozpuszczalności przeciwciała, wprowadzenia miejsc glikozylacji, wprowadzenia miejsc sprzęgania, zmniejszenia immunogenności, wzmocnienia wyrażania przeciwciała, wzrostu powinowactwa wobec antygenu, zmniejszenia powinowactwa wobec antygenu, zmiany w powinowactwie wiązania, zmiany w immunogenności lub wzmocnienia swoistości. 13. Sposób według zastrz. 2, w którym biblioteką dla przeciwciała anty-antygen jest biblioteka dla humanizowanego przeciwciała anty-antygen. 14. Sposób według zastrz. 1, w którym etap selekcjonowania (d) obejmuje: 3 wyselekcjonowanie ulepszonego zmutowanego przeciwciała w oparciu o zwiększone wyrażanie w porównaniu z przeciwciałem matrycowym; i w którym etap wyrażania (e) obejmuje wytwarzanie ulepszonego zmutowanego przeciwciała w tym samym gospodarzu do wytwarzania-komórce eukariotycznej, co w etapie przeprowadzania ewolucji (b). 8
87 1. Sposób według zastrz. 1, w którym etap przeprowadzania ewolucji (b) obejmuje jedną spośród: kompleksowej ewolucji pozycyjnej (CPE); kompleksowej insercyjnej ewolucji pozycyjnej (CPI); kompleksowej delecyjnej ewolucji pozycyjnej (CPD); kompleksowej ewolucji pozycyjnej (CPE), po której następuje kombinatoryczna synteza białka (CPS); kompleksowej delecyjnej ewolucji pozycyjnej (CPD), po której następuje kombinatoryczna synteza białka (CPS); lub kompleksowej delecyjnej ewolucji pozycyjnej (CPD), po której następuje kombinatoryczna synteza białka (CPS)
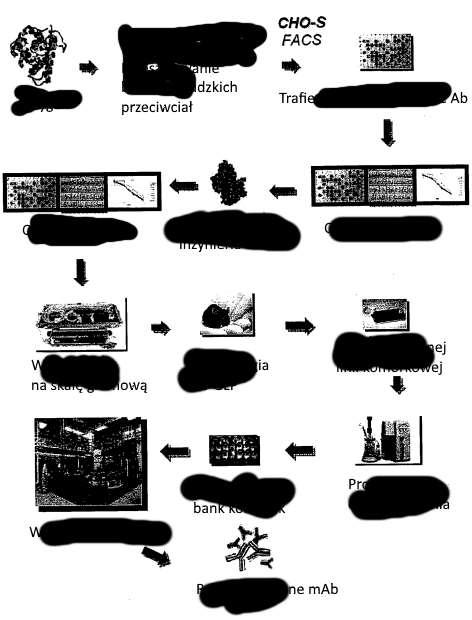
88 FIG. 1 87

89 FIG


90 FIG


91 FIG

92 FIG. 1 91


93 FIG


94 FIG. 7 1 ODNOŚNIKI CYTOWANE W OPISIE 93


95 Niniejsza lista odnośników cytowanych przez zgłaszającego podana jest tylko dla wygody czytelnika. Nie stanowi ona części europejskiego dokumentu patentowego. Nawet mimo dużej staranności przy zestawianiu odnośników nie można wykluczyć błędów lub przeoczeń, i Europejski Urząd Patentowy zrzeka się wszelkiej odpowiedzialności w tym zakresie. Literatura patentowa cytowana w opisie Literatura niepatentowa cytowana w opisie 94

96 9
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE. Ewa Waszkowska ekspert UPRP
 WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
Przegląd budowy i funkcji białek
 Przegląd budowy i funkcji białek Co piszą o białkach? Wyraz wprowadzony przez Jönsa J. Berzeliusa w 1883 r. w celu podkreślenia znaczenia tej grupy związków. Termin pochodzi od greckiego słowa proteios,
Przegląd budowy i funkcji białek Co piszą o białkach? Wyraz wprowadzony przez Jönsa J. Berzeliusa w 1883 r. w celu podkreślenia znaczenia tej grupy związków. Termin pochodzi od greckiego słowa proteios,
ROZPORZĄDZENIE KOMISJI (UE) / z dnia r.
 / z dnia r.") KOMISJA EUROPEJSKA Bruksela, dnia 29.5.2018 C(2018) 3193 final ROZPORZĄDZENIE KOMISJI (UE) / z dnia 29.5.2018 r. zmieniające rozporządzenie (WE) nr 847/2000 w odniesieniu do definicji pojęcia podobnego
KOMISJA EUROPEJSKA Bruksela, dnia 29.5.2018 C(2018) 3193 final ROZPORZĄDZENIE KOMISJI (UE) / z dnia 29.5.2018 r. zmieniające rozporządzenie (WE) nr 847/2000 w odniesieniu do definicji pojęcia podobnego
Informacje. W sprawach organizacyjnych Slajdy z wykładów
 Biochemia Informacje W sprawach organizacyjnych malgorzata.dutkiewicz@wum.edu.pl Slajdy z wykładów www.takao.pl W sprawach merytorycznych Takao Ishikawa (takao@biol.uw.edu.pl) Kiedy? Co? Kto? 24 lutego
Biochemia Informacje W sprawach organizacyjnych malgorzata.dutkiewicz@wum.edu.pl Slajdy z wykładów www.takao.pl W sprawach merytorycznych Takao Ishikawa (takao@biol.uw.edu.pl) Kiedy? Co? Kto? 24 lutego
21. Wstęp do chemii a-aminokwasów
 21. Wstęp do chemii a-aminokwasów Chemia rganiczna, dr hab. inż. Mariola Koszytkowska-Stawińska, WChem PW; 2016/2017 1 21.1. Budowa ogólna a-aminokwasów i klasyfikacja peptydów H 2 N H kwas 2-aminooctowy
21. Wstęp do chemii a-aminokwasów Chemia rganiczna, dr hab. inż. Mariola Koszytkowska-Stawińska, WChem PW; 2016/2017 1 21.1. Budowa ogólna a-aminokwasów i klasyfikacja peptydów H 2 N H kwas 2-aminooctowy
46 i 47. Wstęp do chemii -aminokwasów
 46 i 47. Wstęp do chemii -aminokwasów Chemia rganiczna, dr hab. inż. Mariola Koszytkowska-Stawińska, WChem PW; 2017/2018 1 21.1. Budowa ogólna -aminokwasów i klasyfikacja peptydów H 2 H H 2 R H R R 1 H
46 i 47. Wstęp do chemii -aminokwasów Chemia rganiczna, dr hab. inż. Mariola Koszytkowska-Stawińska, WChem PW; 2017/2018 1 21.1. Budowa ogólna -aminokwasów i klasyfikacja peptydów H 2 H H 2 R H R R 1 H
Nowoczesne systemy ekspresji genów
 Nowoczesne systemy ekspresji genów Ekspresja genów w organizmach żywych GEN - pojęcia podstawowe promotor sekwencja kodująca RNA terminator gen Gen - odcinek DNA zawierający zakodowaną informację wystarczającą
Nowoczesne systemy ekspresji genów Ekspresja genów w organizmach żywych GEN - pojęcia podstawowe promotor sekwencja kodująca RNA terminator gen Gen - odcinek DNA zawierający zakodowaną informację wystarczającą
Chemiczne składniki komórek
 Chemiczne składniki komórek Pierwiastki chemiczne w komórkach: - makroelementy (pierwiastki biogenne) H, O, C, N, S, P Ca, Mg, K, Na, Cl >1% suchej masy - mikroelementy Fe, Cu, Mn, Mo, B, Zn, Co, J, F
Chemiczne składniki komórek Pierwiastki chemiczne w komórkach: - makroelementy (pierwiastki biogenne) H, O, C, N, S, P Ca, Mg, K, Na, Cl >1% suchej masy - mikroelementy Fe, Cu, Mn, Mo, B, Zn, Co, J, F
etyloamina Aminy mają właściwości zasadowe i w roztworach kwaśnych tworzą jon alkinowy
 Temat: Białka Aminy Pochodne węglowodorów zawierające grupę NH 2 Wzór ogólny amin: R NH 2 Przykład: CH 3 -CH 2 -NH 2 etyloamina Aminy mają właściwości zasadowe i w roztworach kwaśnych tworzą jon alkinowy
Temat: Białka Aminy Pochodne węglowodorów zawierające grupę NH 2 Wzór ogólny amin: R NH 2 Przykład: CH 3 -CH 2 -NH 2 etyloamina Aminy mają właściwości zasadowe i w roztworach kwaśnych tworzą jon alkinowy
Klonowanie molekularne Kurs doskonalący. Zakład Geriatrii i Gerontologii CMKP
 Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii
 Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych aspekty praktyczne dr Małgorzata Kozłowska Ekspert
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych aspekty praktyczne dr Małgorzata Kozłowska Ekspert
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych
 Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2068922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2007 07868510.4
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2068922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2007 07868510.4
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
WYKŁAD 4: MOLEKULARNE MECHANIZMY BIOSYNTEZY BIAŁEK. Prof. dr hab. n. med. Małgorzata Milkiewicz Zakład Biologii Medycznej.
 Pierwsza litera Trzecia litera 2018-10-26 WYKŁAD 4: MOLEKULARNE MECHANIZMY BIOSYNTEZY BIAŁEK Prof. dr hab. n. med. Małgorzata Milkiewicz Zakład Biologii Medycznej Druga litera 1 Enancjomery para nienakładalnych
Pierwsza litera Trzecia litera 2018-10-26 WYKŁAD 4: MOLEKULARNE MECHANIZMY BIOSYNTEZY BIAŁEK Prof. dr hab. n. med. Małgorzata Milkiewicz Zakład Biologii Medycznej Druga litera 1 Enancjomery para nienakładalnych
starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg
 STRESZCZENIE Przewlekła białaczka limfocytowa (PBL) jest najczęstszą białaczką ludzi starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg kliniczny, zróżnicowane rokowanie. Etiologia
STRESZCZENIE Przewlekła białaczka limfocytowa (PBL) jest najczęstszą białaczką ludzi starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg kliniczny, zróżnicowane rokowanie. Etiologia
Materiały pochodzą z Platformy Edukacyjnej Portalu www.szkolnictwo.pl
 Materiały pochodzą z Platformy Edukacyjnej Portalu www.szkolnictwo.pl Wszelkie treści i zasoby edukacyjne publikowane na łamach Portalu www.szkolnictwo.pl mogą byd wykorzystywane przez jego Użytkowników
Materiały pochodzą z Platformy Edukacyjnej Portalu www.szkolnictwo.pl Wszelkie treści i zasoby edukacyjne publikowane na łamach Portalu www.szkolnictwo.pl mogą byd wykorzystywane przez jego Użytkowników
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
Agenda: Wynalazek jako własnośd intelektualna. Co może byd wynalazkiem. Wynalazek biotechnologiczny. Ochrona patentowa
 dr Bartosz Walter Agenda: Wynalazek jako własnośd intelektualna Co może byd wynalazkiem Wynalazek biotechnologiczny Ochrona patentowa Procedura zgłoszenia patentowego Gdzie, kiedy i jak warto patentowad
dr Bartosz Walter Agenda: Wynalazek jako własnośd intelektualna Co może byd wynalazkiem Wynalazek biotechnologiczny Ochrona patentowa Procedura zgłoszenia patentowego Gdzie, kiedy i jak warto patentowad
wykład dla studentów II roku biotechnologii Andrzej Wierzbicki
 Genetyka ogólna wykład dla studentów II roku biotechnologii Andrzej Wierzbicki Uniwersytet Warszawski Wydział Biologii andw@ibb.waw.pl http://arete.ibb.waw.pl/private/genetyka/ Wykład 4 Jak działają geny?
Genetyka ogólna wykład dla studentów II roku biotechnologii Andrzej Wierzbicki Uniwersytet Warszawski Wydział Biologii andw@ibb.waw.pl http://arete.ibb.waw.pl/private/genetyka/ Wykład 4 Jak działają geny?
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2814723 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2013 13704452.5 (13) (51) T3 Int.Cl. B63G 8/39 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2814723 (96) Data i numer zgłoszenia patentu europejskiego: 15.02.2013 13704452.5 (13) (51) T3 Int.Cl. B63G 8/39 (2006.01)
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii
 Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
Dr. habil. Anna Salek International Bio-Consulting 1 Germany
 1 2 3 Drożdże są najprostszymi Eukariontami 4 Eucaryota Procaryota 5 6 Informacja genetyczna dla każdej komórki drożdży jest identyczna A zatem każda komórka koduje w DNA wszystkie swoje substancje 7 Przy
1 2 3 Drożdże są najprostszymi Eukariontami 4 Eucaryota Procaryota 5 6 Informacja genetyczna dla każdej komórki drożdży jest identyczna A zatem każda komórka koduje w DNA wszystkie swoje substancje 7 Przy
Transformacja pośrednia składa się z trzech etapów:
 Transformacja pośrednia składa się z trzech etapów: 1. Otrzymanie pożądanego odcinka DNA z materiału genetycznego dawcy 2. Wprowadzenie obcego DNA do wektora 3. Wprowadzenie wektora, niosącego w sobie
Transformacja pośrednia składa się z trzech etapów: 1. Otrzymanie pożądanego odcinka DNA z materiału genetycznego dawcy 2. Wprowadzenie obcego DNA do wektora 3. Wprowadzenie wektora, niosącego w sobie
WPROWADZENIE DO GENETYKI MOLEKULARNEJ
 WPROWADZENIE DO GENETYKI MOLEKULARNEJ Replikacja organizacja widełek replikacyjnych Transkrypcja i biosynteza białek Operon regulacja ekspresji genów Prowadzący wykład: prof. dr hab. Jarosław Burczyk REPLIKACJA
WPROWADZENIE DO GENETYKI MOLEKULARNEJ Replikacja organizacja widełek replikacyjnych Transkrypcja i biosynteza białek Operon regulacja ekspresji genów Prowadzący wykład: prof. dr hab. Jarosław Burczyk REPLIKACJA
Inżynieria Genetyczna ćw. 3
 Materiały do ćwiczeń z przedmiotu Genetyka z inżynierią genetyczną D - blok Inżynieria Genetyczna ćw. 3 Instytut Genetyki i Biotechnologii, Wydział Biologii, Uniwersytet Warszawski, rok akad. 2018/2019
Materiały do ćwiczeń z przedmiotu Genetyka z inżynierią genetyczną D - blok Inżynieria Genetyczna ćw. 3 Instytut Genetyki i Biotechnologii, Wydział Biologii, Uniwersytet Warszawski, rok akad. 2018/2019
TATA box. Enhancery. CGCG ekson intron ekson intron ekson CZĘŚĆ KODUJĄCA GENU TERMINATOR. Elementy regulatorowe
 Promotory genu Promotor bliski leży w odległości do 40 pz od miejsca startu transkrypcji, zawiera kasetę TATA. Kaseta TATA to silnie konserwowana sekwencja TATAAAA, występująca w większości promotorów
Promotory genu Promotor bliski leży w odległości do 40 pz od miejsca startu transkrypcji, zawiera kasetę TATA. Kaseta TATA to silnie konserwowana sekwencja TATAAAA, występująca w większości promotorów
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii
 Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Analizy wielkoskalowe w badaniach chromatyny
 Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
BUDOWA I FUNKCJA GENOMU LUDZKIEGO
 BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
Ćwiczenia 1 Wirtualne Klonowanie Prowadzący: mgr inż. Joanna Tymeck-Mulik i mgr Lidia Gaffke. Część teoretyczna:
 Uniwersytet Gdański, Wydział Biologii Katedra Biologii Molekularnej Przedmiot: Biologia Molekularna z Biotechnologią Biologia II rok ===============================================================================================
Uniwersytet Gdański, Wydział Biologii Katedra Biologii Molekularnej Przedmiot: Biologia Molekularna z Biotechnologią Biologia II rok ===============================================================================================
Budowa aminokwasów i białek
 Biofizyka Ćwiczenia 1. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas Budowa aminokwasów i białek E.Banachowicz 1 Ogólna budowa aminokwasów α w neutralnym p α N 2 COO N
Biofizyka Ćwiczenia 1. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas Budowa aminokwasów i białek E.Banachowicz 1 Ogólna budowa aminokwasów α w neutralnym p α N 2 COO N
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
WPROWADZENIE DO GENETYKI MOLEKULARNEJ
 WPROWADZENIE DO GENETYKI MOLEKULARNEJ Replikacja organizacja widełek replikacyjnych Transkrypcja i biosynteza białek Operon regulacja ekspresji genów Prowadzący wykład: prof. dr hab. Jarosław Burczyk REPLIKACJA
WPROWADZENIE DO GENETYKI MOLEKULARNEJ Replikacja organizacja widełek replikacyjnych Transkrypcja i biosynteza białek Operon regulacja ekspresji genów Prowadzący wykład: prof. dr hab. Jarosław Burczyk REPLIKACJA
spektroskopia elektronowa (UV-vis)
") spektroskopia elektronowa (UV-vis) rodzaje przejść elektronowych Energia σ* π* 3 n 3 π σ σ σ* daleki nadfiolet (λ max < 200 nm) π π* bliski nadfiolet jednostki energii atomowa jednostka energii = energia
spektroskopia elektronowa (UV-vis) rodzaje przejść elektronowych Energia σ* π* 3 n 3 π σ σ σ* daleki nadfiolet (λ max < 200 nm) π π* bliski nadfiolet jednostki energii atomowa jednostka energii = energia
Biologia medyczna, materiały dla studentów
 Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
Pytania Egzamin magisterski
 Pytania Egzamin magisterski Międzyuczelniany Wydział Biotechnologii UG i GUMed 1. Krótko omów jakie informacje powinny być zawarte w typowych rozdziałach publikacji naukowej: Wstęp, Materiały i Metody,
Pytania Egzamin magisterski Międzyuczelniany Wydział Biotechnologii UG i GUMed 1. Krótko omów jakie informacje powinny być zawarte w typowych rozdziałach publikacji naukowej: Wstęp, Materiały i Metody,
protos (gr.) pierwszy protein/proteins (ang.)
 pierwszy protein/proteins (ang.)") Białka 1 protos (gr.) pierwszy protein/proteins (ang.) cząsteczki życia materiał budulcowy materii ożywionej oraz wirusów wielkocząsteczkowe biopolimery o masie od kilku tysięcy do kilku milionów jednostek
Białka 1 protos (gr.) pierwszy protein/proteins (ang.) cząsteczki życia materiał budulcowy materii ożywionej oraz wirusów wielkocząsteczkowe biopolimery o masie od kilku tysięcy do kilku milionów jednostek
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
Przeglądanie bibliotek
 Przeglądanie bibliotek Czyli jak złapać (i sklonować) ciekawy gen? Klonowanie genów w oparciu o identyczność lub podobieństwo ich sekwencji do znanego już DNA Sonda homologiczna (komplementarna w 100%)
Przeglądanie bibliotek Czyli jak złapać (i sklonować) ciekawy gen? Klonowanie genów w oparciu o identyczność lub podobieństwo ich sekwencji do znanego już DNA Sonda homologiczna (komplementarna w 100%)
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Biologia Molekularna z Biotechnologią ===============================================================================================
 Uniwersytet Gdański, Wydział Biologii Biologia i Biologia Medyczna II rok Katedra Genetyki Molekularnej Bakterii Katedra Biologii i Genetyki Medycznej Przedmiot: Katedra Biologii Molekularnej Biologia
Uniwersytet Gdański, Wydział Biologii Biologia i Biologia Medyczna II rok Katedra Genetyki Molekularnej Bakterii Katedra Biologii i Genetyki Medycznej Przedmiot: Katedra Biologii Molekularnej Biologia
Test kwalifikacyjny Lifescience dla licealistów 2015
 Test kwalifikacyjny Lifescience dla licealistów 2015 Imię nazwisko (pseudonim): 1. Daltonizm (d) jest cechą recesywną sprzężoną z płcią. Rudy kolor włosów (r) jest cechą autosomalną i recesywną w stosunku
Test kwalifikacyjny Lifescience dla licealistów 2015 Imię nazwisko (pseudonim): 1. Daltonizm (d) jest cechą recesywną sprzężoną z płcią. Rudy kolor włosów (r) jest cechą autosomalną i recesywną w stosunku
KLONOWANIE DNA REKOMBINACJA DNA WEKTORY
 KLONOWANIE DNA Klonowanie DNA jest techniką powielania fragmentów DNA DNA można powielać w komórkach (replikacja in vivo) W probówce (PCR) Do przeniesienia fragmentu DNA do komórek gospodarza potrzebny
KLONOWANIE DNA Klonowanie DNA jest techniką powielania fragmentów DNA DNA można powielać w komórkach (replikacja in vivo) W probówce (PCR) Do przeniesienia fragmentu DNA do komórek gospodarza potrzebny
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych???
 Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Wykład 14 Biosynteza białek
 BIOCHEMIA Kierunek: Technologia Żywności i Żywienie Człowieka semestr III Wykład 14 Biosynteza białek WYDZIAŁ NAUK O ŻYWNOŚCI I RYBACTWA CENTRUM BIOIMMOBILIZACJI I INNOWACYJNYCH MATERIAŁÓW OPAKOWANIOWYCH
BIOCHEMIA Kierunek: Technologia Żywności i Żywienie Człowieka semestr III Wykład 14 Biosynteza białek WYDZIAŁ NAUK O ŻYWNOŚCI I RYBACTWA CENTRUM BIOIMMOBILIZACJI I INNOWACYJNYCH MATERIAŁÓW OPAKOWANIOWYCH
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
Inżynieria genetyczna
 Inżynieria genetyczna i technologia rekombinowanego DNA Dr n. biol. Urszula Wasik Zakład Biologii Medycznej Inżynieria genetyczna świadoma, celowa, kontrolowana ingerencja w materiał genetyczny organizmów
Inżynieria genetyczna i technologia rekombinowanego DNA Dr n. biol. Urszula Wasik Zakład Biologii Medycznej Inżynieria genetyczna świadoma, celowa, kontrolowana ingerencja w materiał genetyczny organizmów
Slajd 1. Slajd 2. Proteiny. Peptydy i białka są polimerami aminokwasów połączonych wiązaniem amidowym (peptydowym) Kwas α-aminokarboksylowy aminokwas
 Kwas α-aminokarboksylowy aminokwas") Slajd 1 Proteiny Slajd 2 Peptydy i białka są polimerami aminokwasów połączonych wiązaniem amidowym (peptydowym) wiązanie amidowe Kwas α-aminokarboksylowy aminokwas Slajd 3 Aminokwasy z alifatycznym łańcuchem
Slajd 1 Proteiny Slajd 2 Peptydy i białka są polimerami aminokwasów połączonych wiązaniem amidowym (peptydowym) wiązanie amidowe Kwas α-aminokarboksylowy aminokwas Slajd 3 Aminokwasy z alifatycznym łańcuchem
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
października 2013: Elementarz biologii molekularnej. Wykład nr 2 BIOINFORMATYKA rok II
 10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
WYKŁAD: Klasyczny przepływ informacji ( Dogmat) Klasyczny przepływ informacji. Ekspresja genów realizacja informacji zawartej w genach
 Klasyczny przepływ informacji. Ekspresja genów realizacja informacji zawartej w genach") WYKŁAD: Ekspresja genów realizacja informacji zawartej w genach Prof. hab. n. med. Małgorzata Milkiewicz Zakład Biologii Medycznej Klasyczny przepływ informacji ( Dogmat) Białka Retrowirusy Białka Klasyczny
WYKŁAD: Ekspresja genów realizacja informacji zawartej w genach Prof. hab. n. med. Małgorzata Milkiewicz Zakład Biologii Medycznej Klasyczny przepływ informacji ( Dogmat) Białka Retrowirusy Białka Klasyczny
wykład dla studentów II roku biotechnologii Andrzej Wierzbicki
 Genetyka ogólna wykład dla studentów II roku biotechnologii Andrzej Wierzbicki Uniwersytet Warszawski Wydział Biologii andw@ibb.waw.pl http://arete.ibb.waw.pl/private/genetyka/ Ekspresja genów jest regulowana
Genetyka ogólna wykład dla studentów II roku biotechnologii Andrzej Wierzbicki Uniwersytet Warszawski Wydział Biologii andw@ibb.waw.pl http://arete.ibb.waw.pl/private/genetyka/ Ekspresja genów jest regulowana
Aminokwasy, peptydy i białka. Związki wielofunkcyjne
 Aminokwasy, peptydy i białka Związki wielofunkcyjne Aminokwasy, peptydy i białka Aminokwasy, peptydy i białka: - wiadomości ogólne Aminokwasy: - ogólna charakterystyka - budowa i nazewnictwo - właściwości
Aminokwasy, peptydy i białka Związki wielofunkcyjne Aminokwasy, peptydy i białka Aminokwasy, peptydy i białka: - wiadomości ogólne Aminokwasy: - ogólna charakterystyka - budowa i nazewnictwo - właściwości
Geny i działania na nich
 Metody bioinformatyki Geny i działania na nich prof. dr hab. Jan Mulawka Trzy królestwa w biologii Prokaryota organizmy, których komórki nie zawierają jądra, np. bakterie Eukaryota - organizmy, których
Metody bioinformatyki Geny i działania na nich prof. dr hab. Jan Mulawka Trzy królestwa w biologii Prokaryota organizmy, których komórki nie zawierają jądra, np. bakterie Eukaryota - organizmy, których
TRANSKRYPCJA - I etap ekspresji genów
 Eksparesja genów TRANSKRYPCJA - I etap ekspresji genów Przepisywanie informacji genetycznej z makrocząsteczki DNA na mniejsze i bardziej funkcjonalne cząsteczki pre-mrna Polimeraza RNA ETAP I Inicjacja
Eksparesja genów TRANSKRYPCJA - I etap ekspresji genów Przepisywanie informacji genetycznej z makrocząsteczki DNA na mniejsze i bardziej funkcjonalne cząsteczki pre-mrna Polimeraza RNA ETAP I Inicjacja
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
SYLABUS. Wydział Biologiczno-Rolniczy. Katedra Biochemii i Biologii Komórki
 SYLABUS 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Biologia molekularna Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej kierunek) Nazwa jednostki realizującej
SYLABUS 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Biologia molekularna Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej kierunek) Nazwa jednostki realizującej
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1613347 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 12.03.04 0473.4 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1613347 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 12.03.04 0473.4 (97)
Hybrydyzacja kwasów nukleinowych
 Hybrydyzacja kwasów nukleinowych Jaka jest lokalizacja genu na chromosomie? Jakie jest jego sąsiedztwo? Hybrydyzacja - powstawanie stabilnych struktur dwuniciowych z cząsteczek jednoniciowych o komplementarnych
Hybrydyzacja kwasów nukleinowych Jaka jest lokalizacja genu na chromosomie? Jakie jest jego sąsiedztwo? Hybrydyzacja - powstawanie stabilnych struktur dwuniciowych z cząsteczek jednoniciowych o komplementarnych
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1924600. (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2006 06791878.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1924600. (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2006 06791878.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1924600 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 06.09.06 06791878.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1924600 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 06.09.06 06791878.9
Patentowanie wynalazków z dziedziny biotechnologii
 chroń swoje pomysły 3 Kongres Świata Przemysłu Farmaceutycznego Poznań, 16-17 czerwca 2011 Patentowanie wynalazków z dziedziny biotechnologii dr Izabela Milczarek Czym jest biotechnologia? Biotechnologia,
chroń swoje pomysły 3 Kongres Świata Przemysłu Farmaceutycznego Poznań, 16-17 czerwca 2011 Patentowanie wynalazków z dziedziny biotechnologii dr Izabela Milczarek Czym jest biotechnologia? Biotechnologia,
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
(96) Data i numer zgłoszenia patentu europejskiego:
 Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 222280 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2008 08864822.
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 222280 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2008 08864822.
Zawartość. Wstęp 1. Historia wirusologii. 2. Klasyfikacja wirusów
 Zawartość 139585 Wstęp 1. Historia wirusologii 2. Klasyfikacja wirusów 3. Struktura cząstek wirusowych 3.1. Metody określania struktury cząstek wirusowych 3.2. Budowa cząstek wirusowych o strukturze helikalnej
Zawartość 139585 Wstęp 1. Historia wirusologii 2. Klasyfikacja wirusów 3. Struktura cząstek wirusowych 3.1. Metody określania struktury cząstek wirusowych 3.2. Budowa cząstek wirusowych o strukturze helikalnej
Informacje dotyczące pracy kontrolnej
 Informacje dotyczące pracy kontrolnej Słuchacze, którzy z przyczyn usprawiedliwionych nie przystąpili do pracy kontrolnej lub otrzymali z niej ocenę negatywną zobowiązani są do dnia 06 grudnia 2015 r.
Informacje dotyczące pracy kontrolnej Słuchacze, którzy z przyczyn usprawiedliwionych nie przystąpili do pracy kontrolnej lub otrzymali z niej ocenę negatywną zobowiązani są do dnia 06 grudnia 2015 r.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152748 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.06.2008 08768087.2
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152748 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.06.2008 08768087.2
Biotechnologia jest dyscypliną nauk technicznych, która wykorzystuje procesy biologiczne na skalę przemysłową. Inaczej są to wszelkie działania na
 Biotechnologia jest dyscypliną nauk technicznych, która wykorzystuje procesy biologiczne na skalę przemysłową. Inaczej są to wszelkie działania na żywych organizmach prowadzące do uzyskania konkretnych
Biotechnologia jest dyscypliną nauk technicznych, która wykorzystuje procesy biologiczne na skalę przemysłową. Inaczej są to wszelkie działania na żywych organizmach prowadzące do uzyskania konkretnych
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152872. (96) Data i numer zgłoszenia patentu europejskiego: 21.05.2008 08748815.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2152872. (96) Data i numer zgłoszenia patentu europejskiego: 21.05.2008 08748815.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 212872 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 21.0.08 0874881.1 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 212872 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 21.0.08 0874881.1 (97)
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów:
 Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2764103 (96) Data i numer zgłoszenia patentu europejskiego: 12.12.2013 13824232.6 (13) (51) T3 Int.Cl. C12N 15/63 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2764103 (96) Data i numer zgłoszenia patentu europejskiego: 12.12.2013 13824232.6 (13) (51) T3 Int.Cl. C12N 15/63 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2207808. (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2008 08843849.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2207808. (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2008 08843849.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 27808 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27..08 08843849.4 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 27808 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27..08 08843849.4 (97)
S YL AB US MODUŁ U ( PRZEDMIOTU) I nforma c j e ogólne
 I nforma c j e ogólne") Załącznik Nr 3 do Uchwały Senatu PUM 14/2012 S YL AB US MODUŁ U ( PRZEDMIOTU) I nforma c j e ogólne Kod modułu Rodzaj modułu Wydział PUM Kierunek studiów Specjalność Poziom studiów Nazwa modułu INŻYNIERIA
Załącznik Nr 3 do Uchwały Senatu PUM 14/2012 S YL AB US MODUŁ U ( PRZEDMIOTU) I nforma c j e ogólne Kod modułu Rodzaj modułu Wydział PUM Kierunek studiów Specjalność Poziom studiów Nazwa modułu INŻYNIERIA
(12) OPIS PATENTOWY (19)μl
 OPIS PATENTOWY (19)μl") RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19)μl (21) Numer zgłoszenia: 313491 ( 2) Data zgłoszenia: 07.09.1994 (86) Data i numer zgłoszenia międzynarodowego:
RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19)μl (21) Numer zgłoszenia: 313491 ( 2) Data zgłoszenia: 07.09.1994 (86) Data i numer zgłoszenia międzynarodowego:
Bioinformatyka Laboratorium, 30h. Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały Bioinformatyka i ewolucja
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej)
: wprowadzenie (komórki, receptory, rozwój odporności nabytej)") PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej) Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt do wykładu
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej) Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt do wykładu
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA
 Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Bioinformatyka. z sylabusu... (wykład monograficzny) wykład 1. E. Banachowicz. Wykład monograficzny Bioinformatyka.
 wykład 1. E. Banachowicz. Wykład monograficzny Bioinformatyka.") Bioinformatyka (wykład monograficzny) wykład 1. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas z sylabusu... Wykład 1, 2006 1 Co to jest Bioinformatyka? Zastosowanie technologii
Bioinformatyka (wykład monograficzny) wykład 1. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas z sylabusu... Wykład 1, 2006 1 Co to jest Bioinformatyka? Zastosowanie technologii
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
Lek od pomysłu do wdrożenia
 Lek od pomysłu do wdrożenia Lek od pomysłu do wdrożenia KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU
Lek od pomysłu do wdrożenia Lek od pomysłu do wdrożenia KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU KRÓTKA HISTORIA LEKU
SYLABUS. Wydział Biologiczno-Rolniczy. Katedra Biochemii i Biologii Komórki
 SYLABUS 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Biologia molekularna z elementami inżynierii genetycznej Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej kierunek)
SYLABUS 1.1. PODSTAWOWE INFORMACJE O PRZEDMIOCIE/MODULE Nazwa przedmiotu/ modułu Biologia molekularna z elementami inżynierii genetycznej Kod przedmiotu/ modułu* Wydział (nazwa jednostki prowadzącej kierunek)
Model : - SCITEC 100% Whey Protein Professional 920g
 Białka > Model : - Producent : Scitec 100% Whey Protein Professional - jest najwyższej jakości, wolnym od laktozy, czystym koncentratem i izolat białek serwatkowych (WPC + WPI) o bardzo dobrej rozpuszczalności
Białka > Model : - Producent : Scitec 100% Whey Protein Professional - jest najwyższej jakości, wolnym od laktozy, czystym koncentratem i izolat białek serwatkowych (WPC + WPI) o bardzo dobrej rozpuszczalności
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii
 Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Transgeneza - genetycznie zmodyfikowane oraganizmy 2. Medycyna i ochrona zdrowia 3. Genomika poznawanie genomów Przełom XX i
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Transgeneza - genetycznie zmodyfikowane oraganizmy 2. Medycyna i ochrona zdrowia 3. Genomika poznawanie genomów Przełom XX i
TECHNIKI ANALIZY RNA TECHNIKI ANALIZY RNA TECHNIKI ANALIZY RNA
 DNA 28SRNA 18/16S RNA 5SRNA mrna Ilościowa analiza mrna aktywność genów w zależności od wybranych czynników: o rodzaju tkanki o rodzaju czynnika zewnętrznego o rodzaju upośledzenia szlaku metabolicznego
DNA 28SRNA 18/16S RNA 5SRNA mrna Ilościowa analiza mrna aktywność genów w zależności od wybranych czynników: o rodzaju tkanki o rodzaju czynnika zewnętrznego o rodzaju upośledzenia szlaku metabolicznego
Tematyka zajęć z biologii
 Tematyka zajęć z biologii klasy: I Lp. Temat zajęć Zakres treści 1 Zapoznanie z przedmiotowym systemem oceniania, wymaganiami edukacyjnymi i podstawą programową Podstawowe zagadnienia materiału nauczania
Tematyka zajęć z biologii klasy: I Lp. Temat zajęć Zakres treści 1 Zapoznanie z przedmiotowym systemem oceniania, wymaganiami edukacyjnymi i podstawą programową Podstawowe zagadnienia materiału nauczania
Wprowadzenie. DNA i białka. W uproszczeniu: program działania żywego organizmu zapisany jest w nici DNA i wykonuje się na maszynie białkowej.
 Wprowadzenie DNA i białka W uproszczeniu: program działania żywego organizmu zapisany jest w nici DNA i wykonuje się na maszynie białkowej. Białka: łańcuchy złożone z aminokwasów (kilkadziesiąt kilkadziesiąt
Wprowadzenie DNA i białka W uproszczeniu: program działania żywego organizmu zapisany jest w nici DNA i wykonuje się na maszynie białkowej. Białka: łańcuchy złożone z aminokwasów (kilkadziesiąt kilkadziesiąt
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)