Na czym skończyliśmy BLACK BOX. Sekwencjonowanie polega na odczytaniu sekwencji liter DNA/RNA badanego fragmentu genomu
|
|
|
- Karolina Kaczor
- 7 lat temu
- Przeglądów:
Transkrypt
1 ALEKSANDRA ŚWIERCZ

2 Na czym skończyliśmy BLACK BOX AAATGCCTGCCCTGAAGGCCTGCGTA GTTTTGGGAGAAGACCCACGGATA AAGGTGTAGCCCCGTAGC GGGGGGTATTATTTATTTTATACCCAC.. ACAGGAUCGUUGGAUGGTGGGA. Sekwencjonowanie polega na odczytaniu sekwencji liter DNA/RNA badanego fragmentu genomu 2

3 Czym jest mapowanie? Niedopasowania SNP Screen z mapowania IGV 3
4 4
5 5
6 Puzzle klocków Mapowanie miliony sekwencji 6
7 Co to jest genom referencyjny? zsekwencjonowany genom przedstawiciela gatunku znana jest sekwencja, ale mogą się tam znajdować dziury czyli miejsca wypełnione N -kami oznaczono [prawdopodobne] geny oznaczono miejsca, w których występują SNP, CNV, INDEL 7
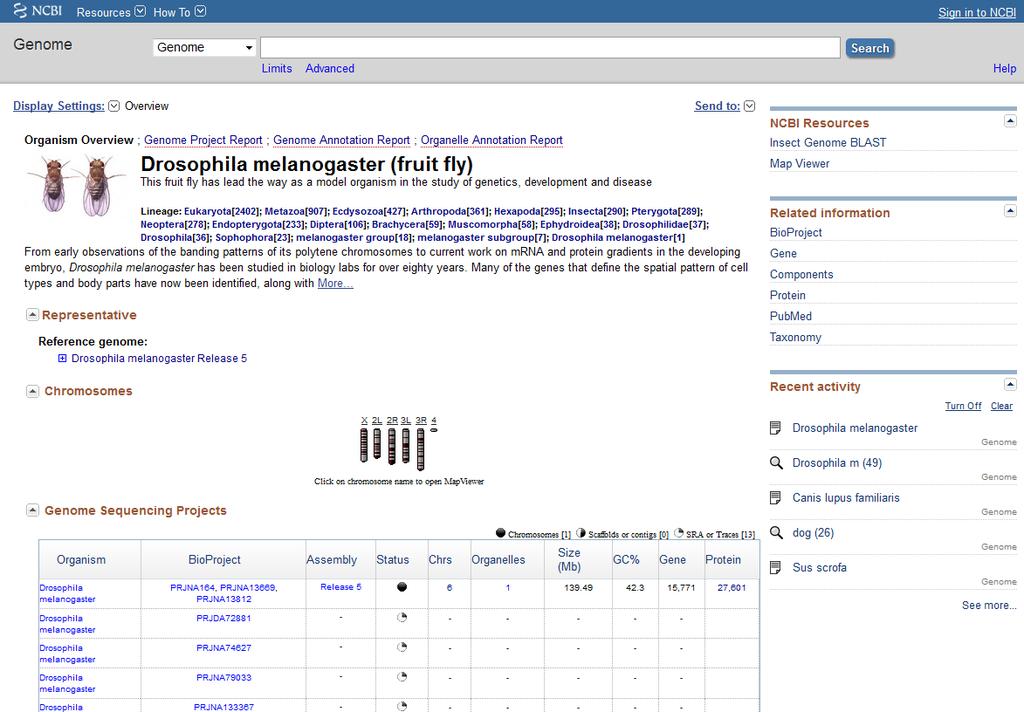
8 8

9 9
10 10
11 Po co mapować sekwencje do genomu referencyjnego? 11
12 Dopasowanie dwóch sekwencji Dopasowanie dwóch sekwencji można wyznaczyć za pomocą dokładnego algorytmu opartego na programowaniu dynamicznym. Punktowane jest każda operacja: za zgodność pary znaków punkty dodatnie, natomiast za niezgodność i każdą spację punkty ujemne. Dopasowanie globalne dopasowanie wzdłuż całej sekwencji Dopasowanie lokalne dopasowanie fragmentów sekwencji 12
13 Schemat punktacji Schemat 1: Dopasowanie (match): +1 Niedopasowanie (mismatch): -1 Przerwa (gap): -1 Schemat 2 (affine model): Dopasowanie (match): +1 Niedopasowanie (mismatch): -1 Otwarcie przerwy (gap open): G Przedłużenie przerwy (affine gap): L 13
14 Dopasowanie dwóch sekwencji Schemat 1: (match +1, mismatch -1, gap -1) ACCTCAGGTTA----CCTGAC-TATTGGACA ACCT----TTAAACACCTTACATATTCCACA score: 7 Schemat 2: (match +3, mismatch -2, gap open -7, gap continue -3) ACCTCAGGTTA----CCTGAC-TATTGGACA ACCT----TTAAACACCTTACATATTCCACA score: 12 ACCTCAGGTTACCTGAC-TATTGGACA ACCTTTAAACACCTTACATATTCCACA score: 26 14
15 Dopasowanie globalne Metoda programowania dynamicznego opiera się na strategii najlepszej ścieżki. Wynik jest w dolnym prawym narożniku tabeli. Algorytm Needelmana-Wunscha dopasowanie: +1 lub niedopasowanie: -1 Sekwencja S 2 Sekwencja S 1 M i, j = max M 0,0 = 0 i = 0.. n, M i 1, j 1 + s(i, j) M i 1, j + g M i, j 1 + g j = 0.. m spacja w jednej z sekwencji -1 SB Needelman, CD Wunsch (1970) J Mol Biol 48 15
16 Dopasowanie lokalne Wyszukiwanie wspólnych fragmentów sekwencji nie ma punktów ujemnych w tabeli. Najlepszy wynik może być w dowolnym miejscu tabeli Algorytm Smitha Watermanna Sekwencja S 2 Sekwencja S 1 M i, j = max M 0,0 = 0 i = 0.. n, M i 1, j 1 + s(i, j) M i 1, j + g M i, j 1 + g 0 j = 0.. m nie ma punktów ujemnych TF Smith, MS Waterman (1981) J Mol Biol
17 Dopasowanie semiglobalne W dopasowaniu semiglobalnym dopuszcza się przesunięcia względem siebie sekwencji. Najlepszy wyniki znajduje się w prawej kolumnie lub w ostatnim wierszu Sekwencja S 1 M i, j = max M i 1, j 1 + s(i, j) M i 1, j + g M i, j 1 + g Sekwencja S 2 M 0,0 = M 0, j = M i, 0 = 0 i = 1.. n, j = 1.. m Początkowe przesunięcie w sekwencji nie jest karane 17
18 Dopasowanie globalne -przykład -1-1 A T T G T C A ± ±1 T ±1-1 T ±1-1 ±1-1 ±1-1 G C C A
19 Dopasowanie globalne -przykład A T T G T C A T T G C C A ATTGTCA -TTGCCA score: 3 19
20 Który algorytm dopasowania można zastosować w przypadku sprawdzenia dopasowania krótkich odczytów do długiej sekwencji genomu? genom referencyjny 20
21 Zadanie match: +1 mismatch: -1 gap: -1 T T A T T A C C T G G A A A A A A A C C T G G 21
22 Teoria a praktyka Algorytmy wyznaczenia dokładnego dopasowania przy mapowaniu odczytów do genomu referencyjnego są w praktyce nierealne, gdyż * genom referencyjny jest długi * odczytów jest za dużo Jeśli długość genomu to n, długość odczytów to k, a liczba odczytów M, to wyznaczenie dopasowania wszystkich odczytów do genomu referencyjnego ma złożoność obliczeniową O(M*n*k) M kilka miliardów odczytów n dla genomu ludzkiego to 3*10 9 k długość ok. 100pz Dlatego algorytmy mapowania wstępnie przetwarzają genom referencyjny, aby w efektywny sposób wyszukiwać miejsca mapowań odczytów. 22
23 BLAST, FASTA Programy służą do porównania pewnej sekwencji lub zbioru sekwencji ze wszystkimi sekwencjami w bazie danych Można je również wykorzystać do mapowania odczytów, ale są lepsze programy, specjalizowane do tego celu (bowtie, bwa, soap, ) Jak działają BLAST, FASTA: szybkie przeglądanie bazy danych wyeliminowanie tych sekwencji które są niepodobne zestawienie najlepszych dopasowań Wykrywają lokalne dopasowania 23
24 Macierze kropkowe dot matrix A T T G T C A T * * * * T * * * G * C * C * A * * Miejsca zgodności w tabeli zaznaczone są kropkami Dopasowanie jest tam gdzie kropki tworzą linię ciągłą na przekątnej 24

25 25
26 Metoda FASTA/FASTX 1. Znalezienie najlepiej dopasowanych regionów na przekątnej (dot matrix). Regiony to słowa o pewnej długości (4-6) 2. Sprawdzenie najlepszych regionów za pomocą macierzy substytucji (dla porównań sekwencji białkowych) 3. Połączenie wybranych regionów (próba połączenia z dopuszczeniem przerw) 4. Obliczenie optymalnego dopasowania za pomocą programowania dynamicznego dla wybranych regionów (Smith-Waterman, Needelman-Wunsch) 5. Obliczenie istotności punktacji dopasowania 26
27 Metoda BLAST Działa podobnie do metody FASTA wstępnie odfiltrowuje sekwencje niepodobne Metoda oparta jest na dokładnym dopasowaniu mniejszych fragmentów, tzw. seeds. Dopiero po ich znalezieniu próbuje w tych miejscach dopasować całą sekwencję, najpierw bezbłędnie, a jeśli nie ma takiej możliwości to z dopuszczeniem błędów. Jeśli seed jest za mały -> to będzie za dużo trafień w sekwencje Jeśli seed jest za duży -> to metoda nie będzie w stanie znaleźć dopasowań z uwzględnieniem błędów (gap, mismatch) 27
28 Haszowanie Metoda do szybkiego wyszukiwania danych w tablicach. Dane są zakodowane poprzez funkcję haszującą, która pozwala na dostęp do danych w tablicy w czasie porównywalnym z czasem stałym O(1) Podstawową wadą tego podejścia jest kolizyjność funkcja haszująca tworzy ten sam hasz dla wielu różnych danych Dlaczego? wynika to z rozmiaru dostępnej pamięci 29
29 Haszowanie cd. Załóżmy że chcemy przechowywać 5 znakowe łańcuchy. Jeśli znaki będą w kodzie ASCII, to każdy z nich będzie składał się z 8 bitów. Nasza funkcja hs() będzie łączyła bity w ciąg 40 bitowy, który określałby jednoznacznie indeks w tabeli. Nasza liczba miałaby wówczas zakres <0;2 40-1> Należy ograniczyć funkcję hs() np. poprzez sumę kodów ASCII w łańcuchu, a wynik wziąć modulo n. Np. dla n=10: s 1 = ALA hs(s 1 ) = ( ) mod 10 = 6 s 2 = MA hs(s 2 ) = (77+65) mod 10 = 2 Ale dla funkcji hs() te same hasze otrzymamy dla łańcuchów KOTA, KATO, AKOT, OKAT, RAB, ARB 30

30 Haszowanie Porcjowanie funkcja haszująca tworzy te same wartości dla określonych grup danych, zwanych porcjami. Wówczas do jednej komórki tablicy trafia nie jeden element, lecz kilka, które następnie organizowane są w postaci listy jednokierunkowej Przy odpowiednim dobraniu funkcji haszującej, rozmiar jednej porcji (czyli długość listy) nie powinien być zbyt duży 31

31 Algorytmy mapowania oparte na haszowaniu Z genomu referencyjnego tworzona jest tabela wartości haszujących: hs(k-mery) -> k-mery, pozycja w genomie k-mery to ciągi znaków o długości k ciąg znaków trzeba powtórzyć ze względu na kolizje Znalezienie mapowania odczytu: podział odczytu na k-mery wyszukanie miejsca mapowania w genomie k-merów wybranie pozycji wspólnej dla wszystkich k-merów (?) sprawdzenie dopasowania całego odczytu Budowanie struktury do szybkiego przeszukiwania genomu Przetwarzanie dla każdego odczytu 32
32 Haszowanie przykład Odczyt: ACGTTCCAATTTGCGCTGT ACGTTCCAATTTG CGTTCCAATTTGC GTTCCAATTTGCG TTCCAATTTGCGC TCCAATTTGCGCT CCAATTTGCGCTG CAATTTGCGCTGT pozycje w tablicy haszującej: chr1:2300 chr1:2301, chr5:800, chr6:568 chr1:2302 chr1:2303, chr2:9000, chr1:2700 chr1:2304 chr1:2305, chrx:444 chr1:2306, chr11:987 Dopasowanie na pozycji: chr1:
33 Wybór k-merów Jeśli sprawdzane k-mery nie nakładają się na siebie, to przyspieszamy czas obliczeń ale możemy nie znaleźć dopasowania, nawet pomimo małej liczby błędów odczyt idealny: odczyt z 2 błędami ACGTTCCAATTTGCGCTG ACGTTCCAA TTTGCGCTG ACATTCCAATTTACGCTG ACATTCCAA TTTACGCTG ACGTTCCAATTTGCGCTG ACATTCCAATTTACGCTG ACGTTCCAA ACATTCCAA CGTTCCAAT CATTCCAAT GTTCCAATT ATTCCAATT TTCCAATTT TTCCAATTT TCCAATTTG TCCAATTTA TTTGCGCTG TTTACGCTG 34
34 Rozmiar k-merów (SOAP) s seed size/długość k-meru L read length/długość odczytu s*2+3 <= min(l) Dla większych k-merów algorytm będzie działał szybciej jest mniejsza liczba trafień w genomie s=1 miliony pozycji w genomie s=8 setki s=15 kilka 35
35 Wady i zalety + łatwe w implementacji + można w łatwy sposób wyszukiwać z dopuszczeniem niedopasowań nie zwiększa to znacząco czasu wyszukiwania + łatwe generowanie indexu - zajmują dużo miejsca, w szczególności jeśli dopuszczane są niedopasowania co zwiększa czas przeszukiwania w porównaniu do innych metod - przy nawet drobnej zmianie parametrów przeszukiwania, trzeba na nowo przeliczać całą tablicę 36
36 BWT, BOWTIE Metody te opierają się na transformacji Burrowsa-Wheelera Transformacja BW służy do takiej zmiany tekstu poprzez jego posortowanie, aby łatwiej można było go skompresować Tekst po transformacji można dokładnie odtworzyć Łatwiej jest znaleźć podciągi (krótkie odczyty, lub k-mery) 38
37 Transformacja Burrowsa-Wheelera MISSISSIPI RESEKWENCJONOWANIE MISSISSIPPI$ ISSISSIPPI$M SSISSIPPI$MI SISSIPPI$MIS ISSIPPI$MISS SSIPPI$MISSI SIPPI$MISSIS IPPI$MISSISS PPI$MISSISSI PI$MISSISSIP I$MISSISSIPP $MISSISSIPPI $MISSISSIPPI I$MISSISSIPP IPPI$MISSISS ISSIPPI$MISS ISSISSIPPI$M MISSISSIPPI$ PI$MISSISSIP PPI$MISSISSI SIPPI$MISSIS SISSIPPI$MIS SSIPPI$MISSI SSISSIPPI$MI sortowanie $MISSISSIPPI I$MISSISSIPP IPPI$MISSISS ISSIPPI$MISS ISSISSIPPI$M MISSISSIPPI$ PI$MISSISSIP PPI$MISSISSI SIPPI$MISSIS SISSIPPI$MIS SSIPPI$MISSI SSISSIPPI$MI F L A. Świercz 39
38 Suffix array Suffix tree $MISSISSIPPI I$MISSISSIPP IPPI$MISSISS ISSIPPI$MISS ISSISSIPPI$M MISSISSIPPI$ PI$MISSISSIP PPI$MISSISSI SIPPI$MISSIS SISSIPPI$MIS SSIPPI$MISSI SSISSIPPI$MI $ I P S I$ PI$ I SI 9 40
39 41
40 42
41 Wykorzystanie indeksu BW do mapowania Mapowanie wg. Algorytmu FM (Ferragina and Manzini, 2000) Wykorzystanie transformaty BT oraz posortowanego BT Odczyty sprawdzane są od końca Dla odczytu GAT, najpierw sprawdzane jest wystąpienie T, potem AT, a na końcu GAT 43
42 Wady i zalety Przy typowej implementacji drzew sufiksowych potrzeba ok. 15B/zasadę co dla genomu ludzkiego daje 45GB pamięci FM-index, oparty na BWT potrzebuje 0.5-2B/zasadę, co pozwala znacznie zaoszczędzić potrzebną pamięć + indeks zajmuje mało miejsca + dość łatwo można zaimplementować szukanie k-merów z niewielką liczbą niedopasowań - generowanie indeksu jest powolne ze względu na BWT - trudno zaprojektować przeszukiwanie z większą liczbą niedopasowań 44
43 Bowtie 45
44 Output SAM & BAM SAM plik tekstowy, w którym zapisane są wyniki mapowania - dla odczytów sparowanych drugi odczyt zapisany jest w następnej linii po pierwszym, zaznaczona jest względna pozycja względem pierwszego BAM binarna wersja pliku SAM 1:497:R: M17D24M M CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==- ==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 QNAME FLAG RNAME POS MAPQ CIGAR MRNM/RNEXT MPOS/PNEXT ISIZE/TLEN SEQ QUAL TAGs nazwa Flaga, czy fragment się zmapował, do której nici, info o sparowanych Reference sequence name Pozycja dopasowania z lewej liczona od 1 (SAM) lub 0 (BAM) Ciąg wskazujący na dopasowanie, np. 3M1I3M1D5M sekwencja Jakość sekwencji 46
45 HWI-ST201:289:C18RMACXX:1:1101:1461: chr M = GGTAGGTACAGAA...??ACAC@;CCCC... AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:97 YT:Z:UP HWI-ST201:289:C18RMACXX:1:1101:1461: chr * = AATATAGTTACTGC... DFHHHGHGHI... YT:Z:UP HWI-ST201:289:C18RMACXX:1:1101:1279: chr M = GATGGGAACCC... ###@C<8&@... AS:i:-4 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:89G7 YS:i:0 YT:Z:CP HWI-ST201:289:C18RMACXX:1:1101:1279: chr M = TCTCTCCTCCA... ###@C<8&@... AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:97 YS:i:-4 YT:Z:CP HWI-ST201:289:C18RMACXX:1:1101:1413: chr M = TATTCATCCACTG... CCACCC>C... AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:97 YS:i:0 YT:Z:CP HWI-ST201:289:C18RMACXX:1:1101:1413: chr M = TTATAGTTGCA... FHGGHHHIHJJJ.. AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:97 YS:i:0 YT:Z:CP 47

46 Haploid vs diploid 48

47 Mapowanie sparowanych odczytów 49
48 Obrazki, slajdy N. Rodriguez-Ezpelta, M. Hackenberg, AM Aransay (eds) Bioinformatics for high thorughput sequencing, Springer, 2012 Bioinformatyka dla Informatyków, J Śmietański, II UJ, 2013 Informatics on High Throughput Sequencing Data S.SCHBATH, V. MARTIN, M.ZYTNICKI, J.FAYOLLE, V.LOUX, J.F. GIBRAT, Mapping Reads on a Genomic Sequence: An Algorithmic Overview and a Practical Comparative Analysis, JOURNAL OF COMPUTATIONAL BIOLOGY Volume 19, Number 6,
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
 Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Wykład 5 Dopasowywanie lokalne
 Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przyrównywanie sekwencji
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Algorytmy i. Wykład 5: Drzewa. Dr inż. Paweł Kasprowski
 Algorytmy i struktury danych Wykład 5: Drzewa Dr inż. Paweł Kasprowski pawel@kasprowski.pl Drzewa Struktury przechowywania danych podobne do list ale z innymi zasadami wskazywania następników Szczególny
Algorytmy i struktury danych Wykład 5: Drzewa Dr inż. Paweł Kasprowski pawel@kasprowski.pl Drzewa Struktury przechowywania danych podobne do list ale z innymi zasadami wskazywania następników Szczególny
Różnorodność osobników gatunku
 ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Dopasowanie sekwencji
 Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing. Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Haszowanie (adresowanie rozpraszające, mieszające)
") Haszowanie (adresowanie rozpraszające, mieszające) Tadeusz Pankowski H. Garcia-Molina, J.D. Ullman, J. Widom, Implementacja systemów baz danych, WNT, Warszawa, Haszowanie W adresowaniu haszującym wyróżniamy
Haszowanie (adresowanie rozpraszające, mieszające) Tadeusz Pankowski H. Garcia-Molina, J.D. Ullman, J. Widom, Implementacja systemów baz danych, WNT, Warszawa, Haszowanie W adresowaniu haszującym wyróżniamy
Tablice z haszowaniem
 Tablice z haszowaniem - efektywna metoda reprezentacji słowników (zbiorów dynamicznych, na których zdefiniowane są operacje Insert, Search i Delete) - jest uogólnieniem zwykłej tablicy - przyspiesza operacje
Tablice z haszowaniem - efektywna metoda reprezentacji słowników (zbiorów dynamicznych, na których zdefiniowane są operacje Insert, Search i Delete) - jest uogólnieniem zwykłej tablicy - przyspiesza operacje
Tablice z haszowaniem
 Tablice z haszowaniem - efektywna metoda reprezentacji słowników (zbiorów dynamicznych, na których zdefiniowane są operacje Insert, Search i Delete) - jest uogólnieniem zwykłej tablicy - przyspiesza operacje
Tablice z haszowaniem - efektywna metoda reprezentacji słowników (zbiorów dynamicznych, na których zdefiniowane są operacje Insert, Search i Delete) - jest uogólnieniem zwykłej tablicy - przyspiesza operacje
Analiza algorytmów zadania podstawowe
 Analiza algorytmów zadania podstawowe Zadanie 1 Zliczanie Zliczaj(n) 1 r 0 2 for i 1 to n 1 3 do for j i + 1 to n 4 do for k 1 to j 5 do r r + 1 6 return r 0 Jaka wartość zostanie zwrócona przez powyższą
Analiza algorytmów zadania podstawowe Zadanie 1 Zliczanie Zliczaj(n) 1 r 0 2 for i 1 to n 1 3 do for j i + 1 to n 4 do for k 1 to j 5 do r r + 1 6 return r 0 Jaka wartość zostanie zwrócona przez powyższą
Searching for SNPs with cloud computing
 Ben Langmead, Michael C Schatz, Jimmy Lin, Mihai Pop and Steven L Salzberg Genome Biology November 20, 2009 April 7, 2010 Problem Cel Problem Bardzo dużo krótkich odczytów mapujemy na genom referencyjny
Ben Langmead, Michael C Schatz, Jimmy Lin, Mihai Pop and Steven L Salzberg Genome Biology November 20, 2009 April 7, 2010 Problem Cel Problem Bardzo dużo krótkich odczytów mapujemy na genom referencyjny
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Algorytmy i struktury danych
 Algorytmy i struktury danych ĆWICZENIE 2 - WYBRANE ZŁOŻONE STRUKTURY DANYCH - (12.3.212) Prowadząca: dr hab. inż. Małgorzata Sterna Informatyka i3, poniedziałek godz. 11:45 Adam Matuszewski, nr 1655 Oliver
Algorytmy i struktury danych ĆWICZENIE 2 - WYBRANE ZŁOŻONE STRUKTURY DANYCH - (12.3.212) Prowadząca: dr hab. inż. Małgorzata Sterna Informatyka i3, poniedziałek godz. 11:45 Adam Matuszewski, nr 1655 Oliver
INŻYNIERIA BEZPIECZEŃSTWA LABORATORIUM NR 2 ALGORYTM XOR ŁAMANIE ALGORYTMU XOR
 INŻYNIERIA BEZPIECZEŃSTWA LABORATORIUM NR 2 ALGORYTM XOR ŁAMANIE ALGORYTMU XOR 1. Algorytm XOR Operacja XOR to inaczej alternatywa wykluczająca, oznaczona symbolem ^ w języku C i symbolem w matematyce.
INŻYNIERIA BEZPIECZEŃSTWA LABORATORIUM NR 2 ALGORYTM XOR ŁAMANIE ALGORYTMU XOR 1. Algorytm XOR Operacja XOR to inaczej alternatywa wykluczająca, oznaczona symbolem ^ w języku C i symbolem w matematyce.
2 Kryptografia: algorytmy symetryczne
 1 Kryptografia: wstęp Wyróżniamy algorytmy: Kodowanie i kompresja Streszczenie Wieczorowe Studia Licencjackie Wykład 14, 12.06.2007 symetryczne: ten sam klucz jest stosowany do szyfrowania i deszyfrowania;
1 Kryptografia: wstęp Wyróżniamy algorytmy: Kodowanie i kompresja Streszczenie Wieczorowe Studia Licencjackie Wykład 14, 12.06.2007 symetryczne: ten sam klucz jest stosowany do szyfrowania i deszyfrowania;
Dopasowanie par sekwencji
 BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Def. Kod jednoznacznie definiowalny Def. Kod przedrostkowy Def. Kod optymalny. Przykłady kodów. Kody optymalne
 Załóżmy, że mamy źródło S, które generuje symbole ze zbioru S={x, x 2,..., x N } z prawdopodobieństwem P={p, p 2,..., p N }, symbolom tym odpowiadają kody P={c, c 2,..., c N }. fektywność danego sposobu
Załóżmy, że mamy źródło S, które generuje symbole ze zbioru S={x, x 2,..., x N } z prawdopodobieństwem P={p, p 2,..., p N }, symbolom tym odpowiadają kody P={c, c 2,..., c N }. fektywność danego sposobu
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Podstawy bioinformatyki sekwencjonowanie nowej generacji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Kompresja Kodowanie arytmetyczne. Dariusz Sobczuk
 Kompresja Kodowanie arytmetyczne Dariusz Sobczuk Kodowanie arytmetyczne (lata 1960-te) Pierwsze prace w tym kierunku sięgają początków lat 60-tych XX wieku Pierwszy algorytm Eliasa nie został opublikowany
Kompresja Kodowanie arytmetyczne Dariusz Sobczuk Kodowanie arytmetyczne (lata 1960-te) Pierwsze prace w tym kierunku sięgają początków lat 60-tych XX wieku Pierwszy algorytm Eliasa nie został opublikowany
Programowanie w języku Java
 Katedra Inżynierii Wiedzy laborki 3 Kilka przydatnych rzeczy String jako klasa; length() - długość łańcucha; char CharAt (int index) - sprawdzenie znaku na zadanym numerze; int compareto(string anotherstring)
Katedra Inżynierii Wiedzy laborki 3 Kilka przydatnych rzeczy String jako klasa; length() - długość łańcucha; char CharAt (int index) - sprawdzenie znaku na zadanym numerze; int compareto(string anotherstring)
Wykład 6. Wyszukiwanie wzorca w tekście
 Wykład 6 Wyszukiwanie wzorca w tekście 1 Wyszukiwanie wzorca (przegląd) Porównywanie łańcuchów Algorytm podstawowy siłowy (naive algorithm) Jak go zrealizować? Algorytm Rabina-Karpa Inteligentne wykorzystanie
Wykład 6 Wyszukiwanie wzorca w tekście 1 Wyszukiwanie wzorca (przegląd) Porównywanie łańcuchów Algorytm podstawowy siłowy (naive algorithm) Jak go zrealizować? Algorytm Rabina-Karpa Inteligentne wykorzystanie
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Zadanie 1 Przygotuj algorytm programu - sortowanie przez wstawianie.
 Sortowanie Dane wejściowe: ciąg n-liczb (kluczy) (a 1, a 2, a 3,..., a n 1, a n ) Dane wyjściowe: permutacja ciągu wejściowego (a 1, a 2, a 3,..., a n 1, a n) taka, że a 1 a 2 a 3... a n 1 a n. Będziemy
Sortowanie Dane wejściowe: ciąg n-liczb (kluczy) (a 1, a 2, a 3,..., a n 1, a n ) Dane wyjściowe: permutacja ciągu wejściowego (a 1, a 2, a 3,..., a n 1, a n) taka, że a 1 a 2 a 3... a n 1 a n. Będziemy
Techniki wyszukiwania danych haszowanie
 Algorytmy i struktury danych Instytut Sterowania i Systemów Informatycznych Wydział Elektrotechniki, Informatyki i Telekomunikacji Uniwersytet Zielonogórski Techniki wyszukiwania danych haszowanie 1 Cel
Algorytmy i struktury danych Instytut Sterowania i Systemów Informatycznych Wydział Elektrotechniki, Informatyki i Telekomunikacji Uniwersytet Zielonogórski Techniki wyszukiwania danych haszowanie 1 Cel
Sortowanie. Bartman Jacek Algorytmy i struktury
 Sortowanie Bartman Jacek jbartman@univ.rzeszow.pl Algorytmy i struktury danych Sortowanie przez proste wstawianie przykład 41 56 17 39 88 24 03 72 41 56 17 39 88 24 03 72 17 41 56 39 88 24 03 72 17 39
Sortowanie Bartman Jacek jbartman@univ.rzeszow.pl Algorytmy i struktury danych Sortowanie przez proste wstawianie przykład 41 56 17 39 88 24 03 72 41 56 17 39 88 24 03 72 17 41 56 39 88 24 03 72 17 39
Algorytmy i struktury danych. Wykład 6 Tablice rozproszone cz. 2
 Algorytmy i struktury danych Wykład 6 Tablice rozproszone cz. 2 Na poprzednim wykładzie Wiele problemów wymaga dynamicznych zbiorów danych, na których można wykonywać operacje: wstawiania (Insert) szukania
Algorytmy i struktury danych Wykład 6 Tablice rozproszone cz. 2 Na poprzednim wykładzie Wiele problemów wymaga dynamicznych zbiorów danych, na których można wykonywać operacje: wstawiania (Insert) szukania
Krzysztof Leszczyński Adam Sosnowski Michał Winiarski. Projekt UCYF
 Krzysztof Leszczyński Adam Sosnowski Michał Winiarski Projekt UCYF Temat: Dekodowanie kodów 2D. 1. Opis zagadnienia Kody dwuwymiarowe nazywane często kodami 2D stanowią uporządkowany zbiór jasnych i ciemnych
Krzysztof Leszczyński Adam Sosnowski Michał Winiarski Projekt UCYF Temat: Dekodowanie kodów 2D. 1. Opis zagadnienia Kody dwuwymiarowe nazywane często kodami 2D stanowią uporządkowany zbiór jasnych i ciemnych
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
Bioinformatyka. Porównywanie sekwencji
 Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Pomorski Czarodziej 2016 Zadania. Kategoria C
 Pomorski Czarodziej 2016 Zadania. Kategoria C Poniżej znajduje się 5 zadań. Za poprawne rozwiązanie każdego z nich możesz otrzymać 10 punktów. Jeżeli otrzymasz za zadanie maksymalną liczbę punktów, możesz
Pomorski Czarodziej 2016 Zadania. Kategoria C Poniżej znajduje się 5 zadań. Za poprawne rozwiązanie każdego z nich możesz otrzymać 10 punktów. Jeżeli otrzymasz za zadanie maksymalną liczbę punktów, możesz
ang. file) Pojęcie pliku (ang( Typy plików Atrybuty pliku Fragmentacja wewnętrzna w systemie plików Struktura pliku
 Pojęcie pliku (ang( Typy plików Atrybuty pliku Fragmentacja wewnętrzna w systemie plików Struktura pliku") System plików 1. Pojęcie pliku 2. Typy i struktury plików 3. etody dostępu do plików 4. Katalogi 5. Budowa systemu plików Pojęcie pliku (ang( ang. file)! Plik jest abstrakcyjnym obrazem informacji gromadzonej
System plików 1. Pojęcie pliku 2. Typy i struktury plików 3. etody dostępu do plików 4. Katalogi 5. Budowa systemu plików Pojęcie pliku (ang( ang. file)! Plik jest abstrakcyjnym obrazem informacji gromadzonej
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
System plików warstwa fizyczna
 System plików warstwa fizyczna Dariusz Wawrzyniak Przydział miejsca na dysku Przydział ciągły (ang. contiguous allocation) cały plik zajmuje ciąg kolejnych bloków Przydział listowy (łańcuchowy, ang. linked
System plików warstwa fizyczna Dariusz Wawrzyniak Przydział miejsca na dysku Przydział ciągły (ang. contiguous allocation) cały plik zajmuje ciąg kolejnych bloków Przydział listowy (łańcuchowy, ang. linked
System plików warstwa fizyczna
 System plików warstwa fizyczna Dariusz Wawrzyniak Plan wykładu Przydział miejsca na dysku Zarządzanie wolną przestrzenią Implementacja katalogu Przechowywanie podręczne Integralność systemu plików Semantyka
System plików warstwa fizyczna Dariusz Wawrzyniak Plan wykładu Przydział miejsca na dysku Zarządzanie wolną przestrzenią Implementacja katalogu Przechowywanie podręczne Integralność systemu plików Semantyka
System plików warstwa fizyczna
 System plików warstwa fizyczna Dariusz Wawrzyniak Przydział miejsca na dysku Zarządzanie wolną przestrzenią Implementacja katalogu Przechowywanie podręczne Integralność systemu plików Semantyka spójności
System plików warstwa fizyczna Dariusz Wawrzyniak Przydział miejsca na dysku Zarządzanie wolną przestrzenią Implementacja katalogu Przechowywanie podręczne Integralność systemu plików Semantyka spójności
3. Opracować program kodowania/dekodowania pliku tekstowego. Algorytm kodowania:
 Zadania-7 1. Opracować program prowadzący spis pracowników firmy (max.. 50 pracowników). Każdy pracownik opisany jest za pomocą struktury zawierającej nazwisko i pensję. Program realizuje następujące polecenia:
Zadania-7 1. Opracować program prowadzący spis pracowników firmy (max.. 50 pracowników). Każdy pracownik opisany jest za pomocą struktury zawierającej nazwisko i pensję. Program realizuje następujące polecenia:
Algorytmy przeszukiwania wzorca
 Algorytmy i struktury danych Instytut Sterowania i Systemów Informatycznych Wydział Elektrotechniki, Informatyki i Telekomunikacji Uniwersytet Zielonogórski Algorytmy przeszukiwania wzorca 1 Wstęp Algorytmy
Algorytmy i struktury danych Instytut Sterowania i Systemów Informatycznych Wydział Elektrotechniki, Informatyki i Telekomunikacji Uniwersytet Zielonogórski Algorytmy przeszukiwania wzorca 1 Wstęp Algorytmy
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Definicja. Ciąg wejściowy: Funkcja uporządkowująca: Sortowanie polega na: a 1, a 2,, a n-1, a n. f(a 1 ) f(a 2 ) f(a n )
 f(a 2 ) f(a n )") SORTOWANIE 1 SORTOWANIE Proces ustawiania zbioru elementów w określonym porządku. Stosuje się w celu ułatwienia późniejszego wyszukiwania elementów sortowanego zbioru. 2 Definicja Ciąg wejściowy: a 1,
SORTOWANIE 1 SORTOWANIE Proces ustawiania zbioru elementów w określonym porządku. Stosuje się w celu ułatwienia późniejszego wyszukiwania elementów sortowanego zbioru. 2 Definicja Ciąg wejściowy: a 1,
Algorytmy sortujące i wyszukujące
 Algorytmy sortujące i wyszukujące Zadaniem algorytmów sortujących jest ułożenie elementów danego zbioru w ściśle określonej kolejności. Najczęściej wykorzystywany jest porządek numeryczny lub leksykograficzny.
Algorytmy sortujące i wyszukujące Zadaniem algorytmów sortujących jest ułożenie elementów danego zbioru w ściśle określonej kolejności. Najczęściej wykorzystywany jest porządek numeryczny lub leksykograficzny.
INFORMATYKA W ZARZĄDZANIU Arkusz kalkulacyjny MS EXCEL. Ćwiczenie 5 MS EXCEL. Zmiana rodzajów odwołania podczas kolejnych naciśnięć klawisza F4
 Ćwiczenie 5 MS EXCEL 1. ODWOŁANIA WZGLĘDNE I BEZWZGLĘDNE Zmiana rodzajów odwołania podczas kolejnych naciśnięć klawisza F4 Odwołanie względne С6 Odwołanie złożone Bezwzględne odwołanie do kolumny i względne
Ćwiczenie 5 MS EXCEL 1. ODWOŁANIA WZGLĘDNE I BEZWZGLĘDNE Zmiana rodzajów odwołania podczas kolejnych naciśnięć klawisza F4 Odwołanie względne С6 Odwołanie złożone Bezwzględne odwołanie do kolumny i względne
Plan wykładów. A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Plan wykładów Wprowadzenie do różnych metod sekwencjonowania Resekwencjonowanie mapowanie do genomu referencyjnego Sekwencjonowanie de novo asemblacja Różnica w ekspresji genów, alternatywny
ALEKSANDRA ŚWIERCZ Plan wykładów Wprowadzenie do różnych metod sekwencjonowania Resekwencjonowanie mapowanie do genomu referencyjnego Sekwencjonowanie de novo asemblacja Różnica w ekspresji genów, alternatywny
Algorytmy i struktury danych. wykład 8
 Plan wykładu: Kodowanie. : wyszukiwanie wzorca w tekście, odległość edycyjna. Kodowanie Kodowanie Kodowanie jest to proces przekształcania informacji wybranego typu w informację innego typu. Kod: jest
Plan wykładu: Kodowanie. : wyszukiwanie wzorca w tekście, odległość edycyjna. Kodowanie Kodowanie Kodowanie jest to proces przekształcania informacji wybranego typu w informację innego typu. Kod: jest
wykład Organizacja plików Opracował: dr inż. Janusz DUDCZYK
 wykład Organizacja plików Opracował: dr inż. Janusz DUDCZYK 1 2 3 Pamięć zewnętrzna Pamięć zewnętrzna organizacja plikowa. Pamięć operacyjna organizacja blokowa. 4 Bufory bazy danych. STRUKTURA PROSTA
wykład Organizacja plików Opracował: dr inż. Janusz DUDCZYK 1 2 3 Pamięć zewnętrzna Pamięć zewnętrzna organizacja plikowa. Pamięć operacyjna organizacja blokowa. 4 Bufory bazy danych. STRUKTURA PROSTA
Lista, Stos, Kolejka, Tablica Asocjacyjna
 Lista, Stos, Kolejka, Tablica Asocjacyjna Listy Lista zbiór elementów tego samego typu może dynamicznie zmieniać rozmiar, pozwala na dostęp do poszczególnych elementów Typowo dwie implementacje: tablicowa,
Lista, Stos, Kolejka, Tablica Asocjacyjna Listy Lista zbiór elementów tego samego typu może dynamicznie zmieniać rozmiar, pozwala na dostęp do poszczególnych elementów Typowo dwie implementacje: tablicowa,
OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) Algorytmy i Struktury Danych PIŁA
 PAWLICKI Piotr (55567) Algorytmy i Struktury Danych PIŁA") OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) 20.11.2002 Algorytmy i Struktury Danych PIŁA ZŁOŻONE STRUKTURY DANYCH C za s tw or ze nia s tr uk tur y (m s ) TWORZENIE ZŁOŻONYCH STRUKTUR DANYCH: 00 0
OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) 20.11.2002 Algorytmy i Struktury Danych PIŁA ZŁOŻONE STRUKTURY DANYCH C za s tw or ze nia s tr uk tur y (m s ) TWORZENIE ZŁOŻONYCH STRUKTUR DANYCH: 00 0
Architektura komputerów
 Architektura komputerów Wykład 7 Jan Kazimirski 1 Pamięć podręczna 2 Pamięć komputera - charakterystyka Położenie Procesor rejestry, pamięć podręczna Pamięć wewnętrzna pamięć podręczna, główna Pamięć zewnętrzna
Architektura komputerów Wykład 7 Jan Kazimirski 1 Pamięć podręczna 2 Pamięć komputera - charakterystyka Położenie Procesor rejestry, pamięć podręczna Pamięć wewnętrzna pamięć podręczna, główna Pamięć zewnętrzna
Wielkości liczbowe. Wykład z Podstaw Informatyki dla I roku BO. Piotr Mika
 Wielkości liczbowe Wykład z Podstaw Informatyki dla I roku BO Piotr Mika Wprowadzenie, liczby naturalne Komputer to podstawowe narzędzie do wykonywania obliczeń Jeden bajt reprezentuje 0 oraz liczby naturalne
Wielkości liczbowe Wykład z Podstaw Informatyki dla I roku BO Piotr Mika Wprowadzenie, liczby naturalne Komputer to podstawowe narzędzie do wykonywania obliczeń Jeden bajt reprezentuje 0 oraz liczby naturalne
BASH - WPROWADZENIE Bioinformatyka 4
 BASH - WPROWADZENIE Bioinformatyka 4 DLACZEGO BASH? Praca na klastrach obliczeniowych Brak GUI Środowisko programistyczne Szybkie przetwarzanie danych Pisanie własnych skryptów W praktyce przetwarzanie
BASH - WPROWADZENIE Bioinformatyka 4 DLACZEGO BASH? Praca na klastrach obliczeniowych Brak GUI Środowisko programistyczne Szybkie przetwarzanie danych Pisanie własnych skryptów W praktyce przetwarzanie
Funkcje wyszukiwania i adresu PODAJ.POZYCJĘ
 Funkcje wyszukiwania i adresu PODAJ.POZYCJĘ Mariusz Jankowski autor strony internetowej poświęconej Excelowi i programowaniu w VBA; Bogdan Gilarski właściciel firmy szkoleniowej Perfect And Practical;
Funkcje wyszukiwania i adresu PODAJ.POZYCJĘ Mariusz Jankowski autor strony internetowej poświęconej Excelowi i programowaniu w VBA; Bogdan Gilarski właściciel firmy szkoleniowej Perfect And Practical;
Materiał Typy zmiennych Instrukcje warunkowe Pętle Tablice statyczne Wskaźniki Tablice dynamiczne Referencje Funkcje
 Podstawy informatyki Informatyka stosowana - studia niestacjonarne - Zajęcia nr 4 Grzegorz Smyk Wydział Inżynierii Metali i Informatyki Przemysłowej Akademia Górniczo Hutnicza im. Stanisława Staszica w
Podstawy informatyki Informatyka stosowana - studia niestacjonarne - Zajęcia nr 4 Grzegorz Smyk Wydział Inżynierii Metali i Informatyki Przemysłowej Akademia Górniczo Hutnicza im. Stanisława Staszica w
Pamięć. Jan Tuziemski Źródło części materiałów: os-book.com
 Pamięć Jan Tuziemski Źródło części materiałów: os-book.com Cele wykładu Przedstawienie sposobów organizacji pamięci komputera Przedstawienie technik zarządzania pamięcią Podstawy Przed uruchomieniem program
Pamięć Jan Tuziemski Źródło części materiałów: os-book.com Cele wykładu Przedstawienie sposobów organizacji pamięci komputera Przedstawienie technik zarządzania pamięcią Podstawy Przed uruchomieniem program
Wielkości liczbowe. Wykład z Podstaw Informatyki. Piotr Mika
 Wielkości liczbowe Wykład z Podstaw Informatyki Piotr Mika Wprowadzenie, liczby naturalne Komputer to podstawowe narzędzie do wykonywania obliczeń Jeden bajt reprezentuje oraz liczby naturalne od do 255
Wielkości liczbowe Wykład z Podstaw Informatyki Piotr Mika Wprowadzenie, liczby naturalne Komputer to podstawowe narzędzie do wykonywania obliczeń Jeden bajt reprezentuje oraz liczby naturalne od do 255
Reswkwencjonowanie vs asemblacja de novo
 ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
znalezienia elementu w zbiorze, gdy w nim jest; dołączenia nowego elementu w odpowiednie miejsce, aby zbiór pozostał nadal uporządkowany.
 Przedstawiamy algorytmy porządkowania dowolnej liczby elementów, którymi mogą być liczby, jak również elementy o bardziej złożonej postaci (takie jak słowa i daty). Porządkowanie, nazywane również często
Przedstawiamy algorytmy porządkowania dowolnej liczby elementów, którymi mogą być liczby, jak również elementy o bardziej złożonej postaci (takie jak słowa i daty). Porządkowanie, nazywane również często
Wstęp do Informatyki
 Wstęp do Informatyki Bożena Woźna-Szcześniak bwozna@gmail.com Jan Długosz University, Poland Wykład 4 Bożena Woźna-Szcześniak (AJD) Wstęp do Informatyki Wykład 4 1 / 1 DZIELENIE LICZB BINARNYCH Dzielenie
Wstęp do Informatyki Bożena Woźna-Szcześniak bwozna@gmail.com Jan Długosz University, Poland Wykład 4 Bożena Woźna-Szcześniak (AJD) Wstęp do Informatyki Wykład 4 1 / 1 DZIELENIE LICZB BINARNYCH Dzielenie
Algorytmy i struktury danych. Drzewa: BST, kopce. Letnie Warsztaty Matematyczno-Informatyczne
 Algorytmy i struktury danych Drzewa: BST, kopce Letnie Warsztaty Matematyczno-Informatyczne Drzewa: BST, kopce Definicja drzewa Drzewo (ang. tree) to nieskierowany, acykliczny, spójny graf. Drzewo może
Algorytmy i struktury danych Drzewa: BST, kopce Letnie Warsztaty Matematyczno-Informatyczne Drzewa: BST, kopce Definicja drzewa Drzewo (ang. tree) to nieskierowany, acykliczny, spójny graf. Drzewo może
Tadeusz Pankowski www.put.poznan.pl/~tadeusz.pankowski
 : idea Indeksowanie: Drzewo decyzyjne, przeszukiwania binarnego: F = {5, 7, 10, 12, 13, 15, 17, 30, 34, 35, 37, 40, 45, 50, 60} 30 12 40 7 15 35 50 Tadeusz Pankowski www.put.poznan.pl/~tadeusz.pankowski
: idea Indeksowanie: Drzewo decyzyjne, przeszukiwania binarnego: F = {5, 7, 10, 12, 13, 15, 17, 30, 34, 35, 37, 40, 45, 50, 60} 30 12 40 7 15 35 50 Tadeusz Pankowski www.put.poznan.pl/~tadeusz.pankowski
POPULARNE POLECENIA SKRYPTY. Pracownia Informatyczna 2
 SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
Technologie cyfrowe. Artur Kalinowski. Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15
 Technologie cyfrowe Artur Kalinowski Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15 Artur.Kalinowski@fuw.edu.pl Semestr letni 2014/2015 Zadanie algorytmiczne: wyszukiwanie dane wejściowe:
Technologie cyfrowe Artur Kalinowski Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15 Artur.Kalinowski@fuw.edu.pl Semestr letni 2014/2015 Zadanie algorytmiczne: wyszukiwanie dane wejściowe:
Podstawy programowania w języku C i C++
 Podstawy programowania w języku C i C++ Część czwarta Operatory i wyrażenia Autor Roman Simiński Kontakt roman.siminski@us.edu.pl www.us.edu.pl/~siminski Niniejsze opracowanie zawiera skrót treści wykładu,
Podstawy programowania w języku C i C++ Część czwarta Operatory i wyrażenia Autor Roman Simiński Kontakt roman.siminski@us.edu.pl www.us.edu.pl/~siminski Niniejsze opracowanie zawiera skrót treści wykładu,
Tabela wewnętrzna - definicja
 ABAP/4 Tabela wewnętrzna - definicja Temporalna tabela przechowywana w pamięci operacyjnej serwera aplikacji Tworzona, wypełniana i modyfikowana jest przez program podczas jego wykonywania i usuwana, gdy
ABAP/4 Tabela wewnętrzna - definicja Temporalna tabela przechowywana w pamięci operacyjnej serwera aplikacji Tworzona, wypełniana i modyfikowana jest przez program podczas jego wykonywania i usuwana, gdy
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Kodowanie i kompresja Tomasz Jurdziński Studia Wieczorowe Wykład Kody liniowe - kodowanie w oparciu o macierz parzystości
 Kodowanie i kompresja Tomasz Jurdziński Studia Wieczorowe Wykład 13 1 Kody liniowe - kodowanie w oparciu o macierz parzystości Przykład Różne macierze parzystości dla kodu powtórzeniowego. Co wiemy z algebry
Kodowanie i kompresja Tomasz Jurdziński Studia Wieczorowe Wykład 13 1 Kody liniowe - kodowanie w oparciu o macierz parzystości Przykład Różne macierze parzystości dla kodu powtórzeniowego. Co wiemy z algebry
Według raportu ISO z 1988 roku algorytm JPEG składa się z następujących kroków: 0.5, = V i, j. /Q i, j
 Kompresja transformacyjna. Opis standardu JPEG. Algorytm JPEG powstał w wyniku prac prowadzonych przez grupę ekspertów (ang. Joint Photographic Expert Group). Prace te zakończyły się w 1991 roku, kiedy
Kompresja transformacyjna. Opis standardu JPEG. Algorytm JPEG powstał w wyniku prac prowadzonych przez grupę ekspertów (ang. Joint Photographic Expert Group). Prace te zakończyły się w 1991 roku, kiedy
utworz tworzącą w pamięci dynamicznej tablicę dwuwymiarową liczb rzeczywistych, a następnie zerującą jej wszystkie elementy,
 Lista 3 Zestaw I Zadanie 1. Zaprojektować i zaimplementować funkcje: utworz tworzącą w pamięci dynamicznej tablicę dwuwymiarową liczb rzeczywistych, a następnie zerującą jej wszystkie elementy, zapisz
Lista 3 Zestaw I Zadanie 1. Zaprojektować i zaimplementować funkcje: utworz tworzącą w pamięci dynamicznej tablicę dwuwymiarową liczb rzeczywistych, a następnie zerującą jej wszystkie elementy, zapisz
Haszowanie. dr inż. Urszula Gałązka
 Haszowanie dr inż. Urszula Gałązka Problem Potrzebujemy struktury do Wstawiania usuwania wyszukiwania Liczb, napisów, rekordów w Bazach danych, sieciach komputerowych, innych Rozwiązanie Tablice z haszowaniem
Haszowanie dr inż. Urszula Gałązka Problem Potrzebujemy struktury do Wstawiania usuwania wyszukiwania Liczb, napisów, rekordów w Bazach danych, sieciach komputerowych, innych Rozwiązanie Tablice z haszowaniem
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Zadania do wykonania. Rozwiązując poniższe zadania użyj pętlę for.
 Zadania do wykonania Rozwiązując poniższe zadania użyj pętlę for. 1. apisz program, który przesuwa w prawo o dwie pozycje zawartość tablicy 10-cio elementowej liczb całkowitych tzn. element t[i] dla i=2,..,9
Zadania do wykonania Rozwiązując poniższe zadania użyj pętlę for. 1. apisz program, który przesuwa w prawo o dwie pozycje zawartość tablicy 10-cio elementowej liczb całkowitych tzn. element t[i] dla i=2,..,9
Algorytmy i złożoności Wykład 5. Haszowanie (hashowanie, mieszanie)
") Algorytmy i złożoności Wykład 5. Haszowanie (hashowanie, mieszanie) Wprowadzenie Haszowanie jest to pewna technika rozwiązywania ogólnego problemu słownika. Przez problem słownika rozumiemy tutaj takie
Algorytmy i złożoności Wykład 5. Haszowanie (hashowanie, mieszanie) Wprowadzenie Haszowanie jest to pewna technika rozwiązywania ogólnego problemu słownika. Przez problem słownika rozumiemy tutaj takie
- - Ocena wykonaniu zad3. Brak zad3
 Indeks Zad1 Zad2 Zad3 Zad4 Zad Ocena 20986 218129 ocena 4 Zadanie składa się z Cw3_2_a oraz Cw3_2_b Brak opcjonalnego wywołania operacji na tablicy. Brak pętli Ocena 2 Brak zad3 Ocena wykonaniu zad3 po
Indeks Zad1 Zad2 Zad3 Zad4 Zad Ocena 20986 218129 ocena 4 Zadanie składa się z Cw3_2_a oraz Cw3_2_b Brak opcjonalnego wywołania operacji na tablicy. Brak pętli Ocena 2 Brak zad3 Ocena wykonaniu zad3 po
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:
 w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:") Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Modelowanie motywów łańcuchami Markowa wyższego rzędu
 Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
MultiSETTER: web server for multiple RNA structure comparison. Sandra Sobierajska Uniwersytet Jagielloński
 MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
ARCHITEKRURA KOMPUTERÓW Kodowanie liczb ze znakiem 27.10.2010
 ARCHITEKRURA KOMPUTERÓW Kodowanie liczb ze znakiem 27.10.2010 Do zapisu liczby ze znakiem mamy tylko 8 bitów, pierwszy od lewej bit to bit znakowy, a pozostałem 7 to bity na liczbę. bit znakowy 1 0 1 1
ARCHITEKRURA KOMPUTERÓW Kodowanie liczb ze znakiem 27.10.2010 Do zapisu liczby ze znakiem mamy tylko 8 bitów, pierwszy od lewej bit to bit znakowy, a pozostałem 7 to bity na liczbę. bit znakowy 1 0 1 1
Złożoność obliczeniowa zadania, zestaw 2
 Złożoność obliczeniowa zadania, zestaw 2 Określanie złożoności obliczeniowej algorytmów, obliczanie pesymistycznej i oczekiwanej złożoności obliczeniowej 1. Dana jest tablica jednowymiarowa A o rozmiarze
Złożoność obliczeniowa zadania, zestaw 2 Określanie złożoności obliczeniowej algorytmów, obliczanie pesymistycznej i oczekiwanej złożoności obliczeniowej 1. Dana jest tablica jednowymiarowa A o rozmiarze
Programowanie dynamiczne
 Programowanie dynamiczne Ciąg Fibonacciego fib(0)=1 fib(1)=1 fib(n)=fib(n-1)+fib(n-2), gdzie n 2 Elementy tego ciągu stanowią liczby naturalne tworzące ciąg o takiej własności, że kolejny wyraz (z wyjątkiem
Programowanie dynamiczne Ciąg Fibonacciego fib(0)=1 fib(1)=1 fib(n)=fib(n-1)+fib(n-2), gdzie n 2 Elementy tego ciągu stanowią liczby naturalne tworzące ciąg o takiej własności, że kolejny wyraz (z wyjątkiem
Mapowanie sekwencji na genom (Ultrafast and memory-efficient alignment of short DNA sequences to the human gemone)
") Mapowanie sekwencji na genom (Ultrafast and memory-efficient alignment of short DNA sequences to the human gemone) Uniwersytet Warszawski 1 kwietnia 2010 Referowana praca Problem Problem Wstęp Referowana
Mapowanie sekwencji na genom (Ultrafast and memory-efficient alignment of short DNA sequences to the human gemone) Uniwersytet Warszawski 1 kwietnia 2010 Referowana praca Problem Problem Wstęp Referowana
Dane, informacja, programy. Kodowanie danych, kompresja stratna i bezstratna
 Dane, informacja, programy Kodowanie danych, kompresja stratna i bezstratna DANE Uporządkowane, zorganizowane fakty. Główne grupy danych: tekstowe (znaki alfanumeryczne, znaki specjalne) graficzne (ilustracje,
Dane, informacja, programy Kodowanie danych, kompresja stratna i bezstratna DANE Uporządkowane, zorganizowane fakty. Główne grupy danych: tekstowe (znaki alfanumeryczne, znaki specjalne) graficzne (ilustracje,