MultiSETTER: web server for multiple RNA structure comparison. Sandra Sobierajska Uniwersytet Jagielloński
|
|
|
- Kamila Rudnicka
- 8 lat temu
- Przeglądów:
Transkrypt
1 MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński
2 Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G, C - struktura drugorzędowa przestrzenne ukształtowanie cząsteczki - elementy tej struktury mogą być przewidziane za pomocą obliczeń
3 - struktura trzeciorzędowa Struktury 3D RNA są znane jako struktury trzeciorzędowe i formowane z długozasięgowych interakcji pomiędzy odległymi pętlami stabilizowanymi przez kanoniczne lub niekanoniczne par zasad, kationy bądź słabe oddziaływania
4 Cel Duży wpływ struktury trzeciorzędowej i porównanie struktur 3D RNA -> skuteczne narzędzie do badania funkcji RNA i relacji ewolucyjnych Kilka metod ARTS, DIAL, iparts, SARA, SARSA, Rclick, R3Dalign, RASS, FRASS, SETTER, czy R3D-BLAST
5 Ale Żadna z tych metod nie potrafi tego, czego my potrzebujemy, czyli bezpośredniego wielokrotnego dopasowania struktur RNA Tak więc opracowano MultiSETTER!
6 Zalety Można wykryć motywy niekonserwatywne na poziomie sekwencji Może być łatwo sprawdzane wizualnie w celu wykrycia wspólnych motywów -> nie wymagają tak wielu cząsteczek jak MSA Dostępność dla szerokiej publiczności biologów i bioinformatyków Bezpłatny i otwarty dla wszystkich Brak wymagań logowania
7 Zalety cd. Umożliwia wykonanie dopasowania zarówno dla dwóch, jak i więcej struktur oraz porównania między strukturami uzyskanymi z bazy danych PDB lub dostarczonego przez użytkownika Grafika 3D jak i zbiorcze dane statystyczne -> interpretacja
8 Algorytm SETTER SEcondary structure-based TERtiary superposition Podział na fragmenty drugorzędowe struktury GSSU (generalized secondary structure units)
9 Budowa GSSU
10 Algorytm SETTER cd. Nakładanie dwóch struktur Algorytm minimalizacji RMSD w celu uzyskania superpozycji strukturalnej 3 pary punktów: - Dwie poprzez dopasowanie dwóch odpowiednich reszt szyi - Trzecia identyfikowana poprzez dopasowanie każdej możliwej pary nukleotydów pętlowych (wybór najmniejsza liczba punktów)
11 Algorytm SETTER cd. Kolejne porównywanie dwóch struktur poprzez dodatkowe RMSD superpozycji wzajemnych najbliższych sąsiadujących reszt Punktacja : S-distance
12 Algorytm MultiSETTER Heurystyczne podejście dopasowanie progresywne (jak Clustal) Każda para struktur RNA jest dopasowywana przez SETTER, wyniki S-distance powodują powstanie macierzy odległości Drzewo prowadzące jest obliczane przy pomocy metody łączenia sąsiadów (neighbor-joining)
13 Algorytm MultiSETTER Dwie najbliższe sobie struktury -> average structure -> średnia pozycja indywidualnych atomów Dopóki nie dojdziemy do korzenia
14 Paralelizacja 1) Superpozycja struktury par RNA Przetwarzanie wielokrotnych par GSSU (TYLKO ONE!!), niezależnie i każdy z nich przypisany do danej jednostki obliczeniowej 2) Macierz odległości Dla n struktur: n*(n-1)/2 niezależnie
15 Paralelizacja 3) Łączenie RNA Powstawanie przeciętnej struktury może być rozłożone na scalone dopasowanie GSSUs. Takie łączenie może zachodzić niezależnie, czyli równolegle
16 Porównanie dopasowania dwóch struktur do wielokrotnego 1NKW i 1S72 (23 S) 1NKW, 1S72, 2AWB, 2Y11 (23 S) ~3000 reszt każdy 4 rdzeniowy z Hyper-V Intel(R) Core i CP GHz, 16GB RAM, Windows 8.1 Ograniczenie liczby nici od 1-4
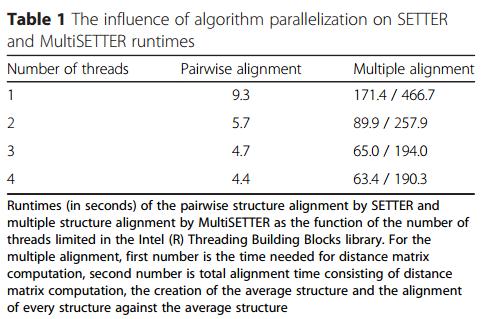
17 Wyniki
18 Języki programowania 1) Rdzeń C++ 2) Aplikacja web Python, Model-Template-View (MTV), Django version 1.4 Ze strony serweru: Python Dla klienta: jquery JavaScript version oraz JavaScript molecular viewer JSmol
19 Połączenia Dane o strukturze RNA: baza danych SETTER Relacyjna baza danych MySQL synchronizowana z PDB w każdą środę SETTER = PDB ID, ID łańcuchów, wzory wiązań wodorowych
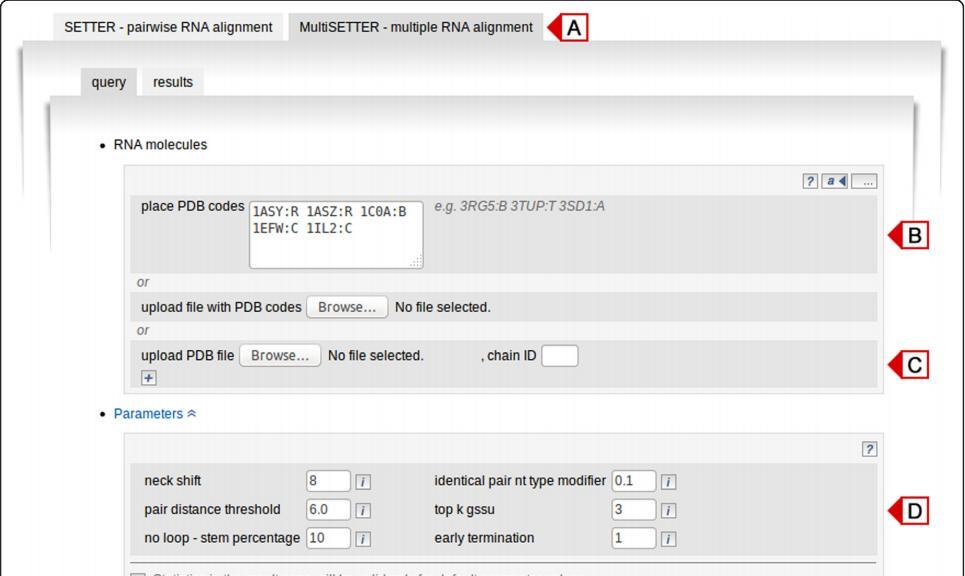
20 Input
21 Input
22 Output
23
24
25
26 Reasumując Pierwsze dostępne publicznie narzędzie do wielokrotnego dopasowania struktury RNA Wiele dokładnych dopasowań w rozsądnym czasie Wizualizacja w 3D, która może ujawnić relacje strukturalne i funkcjonalne W przyszłości łączyć dopasowania sekwencji i struktur z dużą dokładnością
27 Dziękuję za uwagę.
Bioinformatyka wykład 8, 27.XI.2012
 Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Bioinformatyka wykład 10
 Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych
 RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych Joanna Wiśniewska Promotor: dr inż. P. Łukasiak Spis treści 1. Zakres pracy magisterskiej 2. Struktura białka 3. Struktura kwasów nukleionowych
RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych Joanna Wiśniewska Promotor: dr inż. P. Łukasiak Spis treści 1. Zakres pracy magisterskiej 2. Struktura białka 3. Struktura kwasów nukleionowych
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Genomika Porównawcza. Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski
 Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci
 Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu 1. Sieci jako modele interakcji
Grafy i sieci wybrane zagadnienia wykład 3: modele służące porównywaniu sieci prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu 1. Sieci jako modele interakcji
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das
 na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das wykonała: Marta Szynczewska bioinformatyka Uniwersytet Jagielloński Struktura I-rzędowa
na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das wykonała: Marta Szynczewska bioinformatyka Uniwersytet Jagielloński Struktura I-rzędowa
Bioinformatyka wykład 3.I.2008
 Bioinformatyka wykład 3.I.2008 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2008-01-03 1 Plan wykładu analiza i porównywanie struktur białek. doświadczalne metody badania struktur
Bioinformatyka wykład 3.I.2008 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2008-01-03 1 Plan wykładu analiza i porównywanie struktur białek. doświadczalne metody badania struktur
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
 ń ż Ą Ł ż ć ż ć ż ć Ś Ż ć ć ż ć ż ż ż Ą ż ż Ź ń Ą ź ń ź ń Ą ż Ń ż ń Ą ń ż ń Ź ć ń ż Ń Ą ż ż ż ć ń ń Ł ż ż ż ń Ź ź Ą ż Ł ż ż ć ń Ś ć Ó ż ć Ś ż ż Ą ń ż ń Ł ż Ż ń Ą Ł ć ż ń ż ń Ż ń ń Ą ż ż Ł ż ż ż ż ć ż Ń
ń ż Ą Ł ż ć ż ć ż ć Ś Ż ć ć ż ć ż ż ż Ą ż ż Ź ń Ą ź ń ź ń Ą ż Ń ż ń Ą ń ż ń Ź ć ń ż Ń Ą ż ż ż ć ń ń Ł ż ż ż ń Ź ź Ą ż Ł ż ż ć ń Ś ć Ó ż ć Ś ż ż Ą ń ż ń Ł ż Ż ń Ą Ł ć ż ń ż ń Ż ń ń Ą ż ż Ł ż ż ż ż ć ż Ń
Tematy prac dyplomowych inżynierskich
 inżynierskich Oferujemy możliwość realizowania poniższych tematów w ramach projektu realizowanego ze środków Narodowego Centrum Badań i Rozwoju. Najlepszym umożliwimy realizację pracy dyplomowej w połączeniu
inżynierskich Oferujemy możliwość realizowania poniższych tematów w ramach projektu realizowanego ze środków Narodowego Centrum Badań i Rozwoju. Najlepszym umożliwimy realizację pracy dyplomowej w połączeniu
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA. Marta Szachniuk
 Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO
 Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO Bogumil Konopka 1, Jean-Christophe Nebel 2, Malgorzata Kotulska 1 * 1 Politechnika
Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO Bogumil Konopka 1, Jean-Christophe Nebel 2, Malgorzata Kotulska 1 * 1 Politechnika
Wykład I. Wprowadzenie do baz danych
 Wykład I Wprowadzenie do baz danych Trochę historii Pierwsze znane użycie terminu baza danych miało miejsce w listopadzie w 1963 roku. W latach sześcdziesątych XX wieku został opracowany przez Charles
Wykład I Wprowadzenie do baz danych Trochę historii Pierwsze znane użycie terminu baza danych miało miejsce w listopadzie w 1963 roku. W latach sześcdziesątych XX wieku został opracowany przez Charles
WorkingDoc CostControl: Precyzyjna kontrola kosztów wydruku na urządzeniach Grupy Ricoh
 WorkingDoc CostControl WorkingDoc CostControl: Precyzyjna kontrola kosztów wydruku na urządzeniach Grupy Ricoh Agenda Omówienie rozwiązania Cechy i zalety WDCC vs. WDCC Lite Rozwiązanie techniczne Administracja/Raporty
WorkingDoc CostControl WorkingDoc CostControl: Precyzyjna kontrola kosztów wydruku na urządzeniach Grupy Ricoh Agenda Omówienie rozwiązania Cechy i zalety WDCC vs. WDCC Lite Rozwiązanie techniczne Administracja/Raporty
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d.
 Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 11.01.11 1 Dopasowanie strukturalne (alignment) odległość: d ij = (x i -x J ) 2 + (y i -y J ) 2
Bioinformatyka wykład 11, 11.I.2011 Białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 11.01.11 1 Dopasowanie strukturalne (alignment) odległość: d ij = (x i -x J ) 2 + (y i -y J ) 2
W kierunku równoległej implementacji pakietu T-Coffee
 W kierunku równoległej implementacji pakietu T-Coffee Adrian Rospondek 1 1 Wydział Inżynierii Mechanicznej i Informatyki Kierunek Informatyka, Rok V a.rospondek@poczta.fm Streszczenie Artykuł ten prezentuje
W kierunku równoległej implementacji pakietu T-Coffee Adrian Rospondek 1 1 Wydział Inżynierii Mechanicznej i Informatyki Kierunek Informatyka, Rok V a.rospondek@poczta.fm Streszczenie Artykuł ten prezentuje
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Programowanie. programowania. Klasa 3 Lekcja 9 PASCAL & C++
 Programowanie Wstęp p do programowania Klasa 3 Lekcja 9 PASCAL & C++ Język programowania Do przedstawiania algorytmów w postaci programów służą języki programowania. Tylko algorytm zapisany w postaci programu
Programowanie Wstęp p do programowania Klasa 3 Lekcja 9 PASCAL & C++ Język programowania Do przedstawiania algorytmów w postaci programów służą języki programowania. Tylko algorytm zapisany w postaci programu
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW
 PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
OMNITRACKER Wersja testowa. Szybki przewodnik instalacji
 OMNITRACKER Wersja testowa Szybki przewodnik instalacji 1 Krok 1:Rejestracja pobrania (jeżeli nie wykonana dotychczas) Proszę dokonać rejestracji na stronieomninet (www.omnitracker.com) pod Contact. Po
OMNITRACKER Wersja testowa Szybki przewodnik instalacji 1 Krok 1:Rejestracja pobrania (jeżeli nie wykonana dotychczas) Proszę dokonać rejestracji na stronieomninet (www.omnitracker.com) pod Contact. Po
Filogeneza: problem konstrukcji grafu (drzewa) zależności pomiędzy gatunkami.
 zależności pomiędzy gatunkami.") 181 Filogeneza: problem konstrukcji grafu (drzewa) zależności pomiędzy gatunkami. 3. D T(D) poprzez algorytm łączenia sąsiadów 182 D D* : macierz łącząca sąsiadów n Niech TotDist i = k=1 D i,k Definiujemy
181 Filogeneza: problem konstrukcji grafu (drzewa) zależności pomiędzy gatunkami. 3. D T(D) poprzez algorytm łączenia sąsiadów 182 D D* : macierz łącząca sąsiadów n Niech TotDist i = k=1 D i,k Definiujemy
OMNITRACKER Wersja testowa. Szybki przewodnik instalacji
 OMNITRACKER Wersja testowa Szybki przewodnik instalacji 1 Krok 1:Rejestracja pobrania (jeżeli nie wykonana dotychczas) Proszę dokonać rejestracji na stronieomninet (www.omnitracker.com) pod Contact. Po
OMNITRACKER Wersja testowa Szybki przewodnik instalacji 1 Krok 1:Rejestracja pobrania (jeżeli nie wykonana dotychczas) Proszę dokonać rejestracji na stronieomninet (www.omnitracker.com) pod Contact. Po
Generator testów 1.3.1 Bioinformatyka_zdalne wer. 1.0.13 / 0 Strona: 1
 Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Wymagania techniczne. Sage Symfonia 2.0 i Sage Symfonia Start 2.0 wersje 2019
 Wymagania techniczne Sage Symfonia 2.0 i Sage Symfonia Start 2.0 wersje 2019 Wersja krótka Wymagania techniczne Windows Windows 7/8/8.1/10 (x86/x64) Procesor 1GHz kompatybilny z x86 lub x64 (2 rdzenie)
Wymagania techniczne Sage Symfonia 2.0 i Sage Symfonia Start 2.0 wersje 2019 Wersja krótka Wymagania techniczne Windows Windows 7/8/8.1/10 (x86/x64) Procesor 1GHz kompatybilny z x86 lub x64 (2 rdzenie)
Algorytmy równoległe: ocena efektywności prostych algorytmów dla systemów wielokomputerowych
 Algorytmy równoległe: ocena efektywności prostych algorytmów dla systemów wielokomputerowych Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka 2014/15 Znajdowanie maksimum w zbiorze
Algorytmy równoległe: ocena efektywności prostych algorytmów dla systemów wielokomputerowych Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka 2014/15 Znajdowanie maksimum w zbiorze
Klaster obliczeniowy
 Warsztaty promocyjne Usług kampusowych PLATON U3 Klaster obliczeniowy czerwiec 2012 Przemysław Trzeciak Centrum Komputerowe Politechniki Łódzkiej Agenda (czas: 20min) 1) Infrastruktura sprzętowa wykorzystana
Warsztaty promocyjne Usług kampusowych PLATON U3 Klaster obliczeniowy czerwiec 2012 Przemysław Trzeciak Centrum Komputerowe Politechniki Łódzkiej Agenda (czas: 20min) 1) Infrastruktura sprzętowa wykorzystana
Analiza porównawcza wybranych własności systemów zarządzania bazami danych
 Akademia Górniczo Hutnicza im. Stanisława Staszica w Krakowie Analiza porównawcza wybranych własności systemów zarządzania bazami danych Mirosław Lach Promotor: Prof. dr hab. inŝ. Antoni Ligęza Kraków
Akademia Górniczo Hutnicza im. Stanisława Staszica w Krakowie Analiza porównawcza wybranych własności systemów zarządzania bazami danych Mirosław Lach Promotor: Prof. dr hab. inŝ. Antoni Ligęza Kraków
Porównanie szeregów czasowych z wykorzystaniem algorytmu DTW
 Zlot użytkowników R Porównanie szeregów czasowych z wykorzystaniem algorytmu DTW Paweł Teisseyre Instytut Podstaw Informatyki, Polska Akademia Nauk 21 września 2010 Miary podobieństwa między szeregami
Zlot użytkowników R Porównanie szeregów czasowych z wykorzystaniem algorytmu DTW Paweł Teisseyre Instytut Podstaw Informatyki, Polska Akademia Nauk 21 września 2010 Miary podobieństwa między szeregami
Języki skryptowe. zasady zaliczania literatura wprowadzenie
 zasady zaliczania literatura wprowadzenie Cel przedmiotu: zapoznanie z możliwościami wykorzystania języków skryptowych do obsługi danych zapoznanie z możliwościami wykorzystania języków skryptowych do
zasady zaliczania literatura wprowadzenie Cel przedmiotu: zapoznanie z możliwościami wykorzystania języków skryptowych do obsługi danych zapoznanie z możliwościami wykorzystania języków skryptowych do
Customer Attribution Models. czyli o wykorzystaniu machine learning w domu mediowym.
 Customer Attribution Models czyli o wykorzystaniu machine learning w domu mediowym. Proces decyzyjny MAILING SEO SEM DISPLAY RETARGETING PRZEGRANI??? ZWYCIĘZCA!!! Modelowanie atrybucja > Słowo klucz: wpływ
Customer Attribution Models czyli o wykorzystaniu machine learning w domu mediowym. Proces decyzyjny MAILING SEO SEM DISPLAY RETARGETING PRZEGRANI??? ZWYCIĘZCA!!! Modelowanie atrybucja > Słowo klucz: wpływ
WIZUALNA EKSPLORACJA DANYCH I RAPORTOWANIE W SAS VISUAL ANALYTICS ORAZ WSTĘP DO SAS VISUAL STATISTICS
 WIZUALNA EKSPLORACJA DANYCH I RAPORTOWANIE W SAS VISUAL ANALYTICS ORAZ WSTĘP DO SAS VISUAL STATISTICS WEBINARIUM, 2016.03.08 Dr Sławomir Strzykowski, Senior Business Solution Manager SAS VISUAL ANALYTICS
WIZUALNA EKSPLORACJA DANYCH I RAPORTOWANIE W SAS VISUAL ANALYTICS ORAZ WSTĘP DO SAS VISUAL STATISTICS WEBINARIUM, 2016.03.08 Dr Sławomir Strzykowski, Senior Business Solution Manager SAS VISUAL ANALYTICS
Wymagania techniczne
 Wymagania techniczne Sage Symfonia 2.0 Finanse i Księgowość 2017 Sage Symfonia 2.0 Handel 2017 Sage Symfonia 2.0 Kadry i Płace 2017 Sage Symfonia 2.0 Środki Trwałe 2017 Sage Symfonia Start 2.0 Mała Księgowość
Wymagania techniczne Sage Symfonia 2.0 Finanse i Księgowość 2017 Sage Symfonia 2.0 Handel 2017 Sage Symfonia 2.0 Kadry i Płace 2017 Sage Symfonia 2.0 Środki Trwałe 2017 Sage Symfonia Start 2.0 Mała Księgowość
"Zapisane w genach, czyli Python a tajemnice naszego genomu."
 "Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
"Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
Podstawy biologiczne - komórki. Podstawy biologiczne - cząsteczki. Model komórki eukariotycznej. Wprowadzenie do Informatyki Biomedycznej
 Wprowadzenie do Informatyki Biomedycznej Wykład 1: Podstawy bioinformatyki Wydział Informatyki PB Podstawy biologiczne - komórki Wszystkie organizmy zbudowane są z komórek komórka jest skomplikowanym systemem
Wprowadzenie do Informatyki Biomedycznej Wykład 1: Podstawy bioinformatyki Wydział Informatyki PB Podstawy biologiczne - komórki Wszystkie organizmy zbudowane są z komórek komórka jest skomplikowanym systemem
Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Generator testów Bioinformatyka wer / 0 Strona: 1
 Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
E: Rekonstrukcja ewolucji. Algorytmy filogenetyczne
 E: Rekonstrukcja ewolucji. Algorytmy filogenetyczne Przypominajka: 152 drzewo filogenetyczne to drzewo, którego liśćmi są istniejące gatunki, a węzły wewnętrzne mają stopień większy niż jeden i reprezentują
E: Rekonstrukcja ewolucji. Algorytmy filogenetyczne Przypominajka: 152 drzewo filogenetyczne to drzewo, którego liśćmi są istniejące gatunki, a węzły wewnętrzne mają stopień większy niż jeden i reprezentują
Kierunek:Informatyka- - inż., rok I specjalność: Grafika komputerowa i multimedia
 :Informatyka- - inż., rok I specjalność: Grafika komputerowa i multimedia Podstawy prawne. 1 15 1 Podstawy ekonomii. 1 15 15 2 Metody uczenia się i studiowania. 1 15 1 Środowisko programisty. 1 30 3 Komputerowy
:Informatyka- - inż., rok I specjalność: Grafika komputerowa i multimedia Podstawy prawne. 1 15 1 Podstawy ekonomii. 1 15 15 2 Metody uczenia się i studiowania. 1 15 1 Środowisko programisty. 1 30 3 Komputerowy
Kierunek:Informatyka- - inż., rok I specjalność: Grafika komputerowa
 :Informatyka- - inż., rok I specjalność: Grafika komputerowa Metody uczenia się i studiowania. 1 Podstawy prawne. 1 Podstawy ekonomii. 1 Matematyka dyskretna. 1 Wprowadzenie do informatyki. 1 Podstawy
:Informatyka- - inż., rok I specjalność: Grafika komputerowa Metody uczenia się i studiowania. 1 Podstawy prawne. 1 Podstawy ekonomii. 1 Matematyka dyskretna. 1 Wprowadzenie do informatyki. 1 Podstawy
Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania? Dlaczego? Struktura krystaliczną czy NMR (to samo białko, ta sama rozdzielczość)? Strukturę
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania? Dlaczego? Struktura krystaliczną czy NMR (to samo białko, ta sama rozdzielczość)? Strukturę
Założenia monitoringu innowacyjności województwa mazowieckiego
 Założenia monitoringu innowacyjności województwa mazowieckiego Wojciech Dziemianowicz prezentacja składa się z materiałów przygotowanych przez firmy GEOPROFIT i ECORYS Polska sp. z o.o. na zlecenie Urzędu
Założenia monitoringu innowacyjności województwa mazowieckiego Wojciech Dziemianowicz prezentacja składa się z materiałów przygotowanych przez firmy GEOPROFIT i ECORYS Polska sp. z o.o. na zlecenie Urzędu
MongoDB. wprowadzenie. dr inż. Paweł Boiński, Politechnika Poznańska
 MongoDB wprowadzenie dr inż. Paweł Boiński, Politechnika Poznańska Plan Historia Podstawowe pojęcia: Dokument Kolekcja Generowanie identyfikatora Model danych Dokumenty zagnieżdżone Dokumenty z referencjami
MongoDB wprowadzenie dr inż. Paweł Boiński, Politechnika Poznańska Plan Historia Podstawowe pojęcia: Dokument Kolekcja Generowanie identyfikatora Model danych Dokumenty zagnieżdżone Dokumenty z referencjami
Algorytmy równoległe. Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka 2010
 Algorytmy równoległe Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka Znajdowanie maksimum w zbiorze n liczb węzły - maksimum liczb głębokość = 3 praca = 4++ = 7 (operacji) n - liczność
Algorytmy równoległe Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka Znajdowanie maksimum w zbiorze n liczb węzły - maksimum liczb głębokość = 3 praca = 4++ = 7 (operacji) n - liczność
Efekt kształcenia. Wiedza
 Efekty dla studiów drugiego stopnia profil ogólnoakademicki na kierunku Informatyka na specjalności Przetwarzanie i analiza danych, na Wydziale Matematyki i Nauk Informacyjnych, gdzie: * Odniesienie oznacza
Efekty dla studiów drugiego stopnia profil ogólnoakademicki na kierunku Informatyka na specjalności Przetwarzanie i analiza danych, na Wydziale Matematyki i Nauk Informacyjnych, gdzie: * Odniesienie oznacza
Wykład V. Rzut okiem na języki programowania. Studia Podyplomowe INFORMATYKA Podstawy Informatyki
 Studia Podyplomowe INFORMATYKA Podstawy Informatyki Wykład V Rzut okiem na języki programowania 1 Kompilacja vs. interpretacja KOMPILACJA Proces, który przetwarza program zapisany w języku programowania,
Studia Podyplomowe INFORMATYKA Podstawy Informatyki Wykład V Rzut okiem na języki programowania 1 Kompilacja vs. interpretacja KOMPILACJA Proces, który przetwarza program zapisany w języku programowania,
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Praca magisterska Jakub Reczycki. Opiekun : dr inż. Jacek Rumiński. Katedra Inżynierii Biomedycznej Wydział ETI Politechnika Gdańska
 System gromadzenia, indeksowania i opisu słownikowego norm i rekomendacji Praca magisterska Jakub Reczycki Opiekun : dr inż. Jacek Rumiński Katedra Inżynierii Biomedycznej Wydział ETI Politechnika Gdańska
System gromadzenia, indeksowania i opisu słownikowego norm i rekomendacji Praca magisterska Jakub Reczycki Opiekun : dr inż. Jacek Rumiński Katedra Inżynierii Biomedycznej Wydział ETI Politechnika Gdańska
WYMAGANIA EDUKACYJNE. Programowanie Aplikacji Internetowych klasa III
 WYMAGANIA EDUKACYJNE Programowanie Aplikacji Internetowych klasa III Dopuszczający Zna historię języka PHP Zna witryny internetowe oferujące darmowe skrypty PHP Potrafi wyświetlad dokument php na lokalnym
WYMAGANIA EDUKACYJNE Programowanie Aplikacji Internetowych klasa III Dopuszczający Zna historię języka PHP Zna witryny internetowe oferujące darmowe skrypty PHP Potrafi wyświetlad dokument php na lokalnym
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
KARTA KURSU. Języki hipertekstowe i tworzenie stron WWW. Opis kursu (cele kształcenia) Warunki wstępne. Efekty kształcenia. Nazwa
 Warunki wstępne. Efekty kształcenia. Nazwa") KARTA KURSU Nazwa Nazwa w j. ang. Języki hipertekstowe i tworzenie stron WWW Hypertext languages and web page design Kod Punktacja ECTS* 4 Koordynator dr inż. Marcin Piekarczyk Zespół dydaktyczny: dr inż.
KARTA KURSU Nazwa Nazwa w j. ang. Języki hipertekstowe i tworzenie stron WWW Hypertext languages and web page design Kod Punktacja ECTS* 4 Koordynator dr inż. Marcin Piekarczyk Zespół dydaktyczny: dr inż.
Serwer SSH. Wprowadzenie do serwera SSH Instalacja i konfiguracja Zarządzanie kluczami
 Serwer SSH Serwer SSH Wprowadzenie do serwera SSH Instalacja i konfiguracja Zarządzanie kluczami Serwer SSH - Wprowadzenie do serwera SSH Praca na odległość potrzeby w zakresie bezpieczeństwa Identyfikacja
Serwer SSH Serwer SSH Wprowadzenie do serwera SSH Instalacja i konfiguracja Zarządzanie kluczami Serwer SSH - Wprowadzenie do serwera SSH Praca na odległość potrzeby w zakresie bezpieczeństwa Identyfikacja
ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI
 ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI Podstawy Bioinformatyki lab 1 PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 1 BIOINFORMATYKA Dr Magda Mielczarek Katedra Genetyki, pokój nr 14 ul. Kożuchowska
ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI Podstawy Bioinformatyki lab 1 PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 1 BIOINFORMATYKA Dr Magda Mielczarek Katedra Genetyki, pokój nr 14 ul. Kożuchowska
BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH
 http://theta.edu.pl/ Podstawy Bioinformatyki II BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH 1 Czym jest bioinformatyka? 2 Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie
http://theta.edu.pl/ Podstawy Bioinformatyki II BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH 1 Czym jest bioinformatyka? 2 Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie
Prof. dr hab. Joanna Trylska Rada Naukowa Instytutu Biochemii i Biofizyki Polskiej Akademii Nauk w Warszawie
 ul. S. Banacha 2c, 02-097 Warszawa TEL.: + 48 22 55 43 600, FAX: +48 22 55 43 606, E-MAIL: sekretariat@cent.uw.edu.pl www.cent.uw.edu.pl Warszawa, 10 października 2017 Prof. dr hab. Joanna Trylska e-mail:
ul. S. Banacha 2c, 02-097 Warszawa TEL.: + 48 22 55 43 600, FAX: +48 22 55 43 606, E-MAIL: sekretariat@cent.uw.edu.pl www.cent.uw.edu.pl Warszawa, 10 października 2017 Prof. dr hab. Joanna Trylska e-mail:
Algorytmiczne aspekty modelowania i ewaluacji biomolekuł Algorithmic aspects of modeling and evaluation of biomolecules
 Algorytmiczne aspekty modelowania i ewaluacji biomolekuł Algorithmic aspects of modeling and evaluation of biomolecules Maciej Antczak Streszczenie Rozprawy Doktorskiej Promotor: dr hab inż. Marta Kasprzak,
Algorytmiczne aspekty modelowania i ewaluacji biomolekuł Algorithmic aspects of modeling and evaluation of biomolecules Maciej Antczak Streszczenie Rozprawy Doktorskiej Promotor: dr hab inż. Marta Kasprzak,
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Dopasowanie sekwencji
 Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Translacja i proteom komórki
 Translacja i proteom komórki 1. Kod genetyczny 2. Budowa rybosomów 3. Inicjacja translacji 4. Elongacja translacji 5. Terminacja translacji 6. Potranslacyjne zmiany polipeptydów 7. Translacja a retikulum
Translacja i proteom komórki 1. Kod genetyczny 2. Budowa rybosomów 3. Inicjacja translacji 4. Elongacja translacji 5. Terminacja translacji 6. Potranslacyjne zmiany polipeptydów 7. Translacja a retikulum
Pracownia Inżynierii Procesowej
 Pracownia Inżynierii Procesowej Aktualizacja oferty styczeń 2016 WŁAŚCICIEL mgr inż. Alicja Wróbel Absolwent Politechniki Opolskiej, Wydziału Zarzadzania i Inżynierii Produkcji Rysunek techniczny 2D 3D
Pracownia Inżynierii Procesowej Aktualizacja oferty styczeń 2016 WŁAŚCICIEL mgr inż. Alicja Wróbel Absolwent Politechniki Opolskiej, Wydziału Zarzadzania i Inżynierii Produkcji Rysunek techniczny 2D 3D
UCHWAŁA NR 46/2013. Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 19 września 2013 roku
 UCHWAŁA NR 46/2013 Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 19 września 2013 roku w sprawie: korekty efektów kształcenia dla kierunku informatyka Na podstawie ustawy z dnia
UCHWAŁA NR 46/2013 Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 19 września 2013 roku w sprawie: korekty efektów kształcenia dla kierunku informatyka Na podstawie ustawy z dnia
która metoda jest najlepsza
 która metoda jest najlepsza dr inż. Marek Żabka Instytut Matematyki Wydział Matematyki Stosowanej Politechnika Śląska 20 września 2012r Nowa metoda tworzenia grafiki na stronie internetowej: element,,canvas
która metoda jest najlepsza dr inż. Marek Żabka Instytut Matematyki Wydział Matematyki Stosowanej Politechnika Śląska 20 września 2012r Nowa metoda tworzenia grafiki na stronie internetowej: element,,canvas
2011-11-04. Instalacja SQL Server Konfiguracja SQL Server Logowanie - opcje SQL Server Management Studio. Microsoft Access Oracle Sybase DB2 MySQL
 Instalacja, konfiguracja Dr inŝ. Dziwiński Piotr Katedra InŜynierii Komputerowej Kontakt: piotr.dziwinski@kik.pcz.pl 2 Instalacja SQL Server Konfiguracja SQL Server Logowanie - opcje SQL Server Management
Instalacja, konfiguracja Dr inŝ. Dziwiński Piotr Katedra InŜynierii Komputerowej Kontakt: piotr.dziwinski@kik.pcz.pl 2 Instalacja SQL Server Konfiguracja SQL Server Logowanie - opcje SQL Server Management
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Akademia Górniczo-Hutnicza im. Stanisława Staszica w Krakowie. dr inż. Adam Piórkowski. Jakub Osiadacz Marcin Wróbel
 Akademia Górniczo-Hutnicza im. Stanisława Staszica w Krakowie Problem magazynowania i przetwarzania wielkoformatowych map i planów geologicznych. Promotor: dr inż. Adam Piórkowski Autorzy: Jakub Osiadacz
Akademia Górniczo-Hutnicza im. Stanisława Staszica w Krakowie Problem magazynowania i przetwarzania wielkoformatowych map i planów geologicznych. Promotor: dr inż. Adam Piórkowski Autorzy: Jakub Osiadacz
Automatyzacja Testowania w WEB 2.0
 Automatyzacja Testowania w WEB 2.0 Wojciech Pająk, Radosław Smilgin XXIV Jesienne Spotkania PTI Wisła, 20-24 października 2008 Agenda Wprowadzenie do automatyzacji testowania Technologie WEB 2.0 Narzędzia
Automatyzacja Testowania w WEB 2.0 Wojciech Pająk, Radosław Smilgin XXIV Jesienne Spotkania PTI Wisła, 20-24 października 2008 Agenda Wprowadzenie do automatyzacji testowania Technologie WEB 2.0 Narzędzia
Wymagania sprzętowe i systemowe
 System obsługi sprawozdawczości Wymagania i systemowe wersja 5.13.1 Wrocław 07.2015r. Wszelkie prawa zastrzeżone. Dokument może być reprodukowany lub przechowywany bez ograniczeń tylko w całości. Żadna
System obsługi sprawozdawczości Wymagania i systemowe wersja 5.13.1 Wrocław 07.2015r. Wszelkie prawa zastrzeżone. Dokument może być reprodukowany lub przechowywany bez ograniczeń tylko w całości. Żadna
Analiza biznesowa studium przypadku
 2012 Analiza biznesowa studium przypadku Sławomir Kurowski Nowoczesna Firma S.A. Analiza biznesowa integracji B2B Bydgoszcz, 26 września 2012 Agenda Nowoczesna Firma S.A. historia, działalność, partnerzy
2012 Analiza biznesowa studium przypadku Sławomir Kurowski Nowoczesna Firma S.A. Analiza biznesowa integracji B2B Bydgoszcz, 26 września 2012 Agenda Nowoczesna Firma S.A. historia, działalność, partnerzy
Procesy integracji modeli danych do jednolitej struktury WBD. Tadeusz Chrobak, Krystian Kozioł, Artur Krawczyk, Michał Lupa
 Procesy integracji modeli danych do jednolitej struktury WBD Tadeusz Chrobak, Krystian Kozioł, Artur Krawczyk, Michał Lupa Koncepcja Wielorozdzielczej Bazy Danych Kluczowe uwarunkowania systemu generalizacji:
Procesy integracji modeli danych do jednolitej struktury WBD Tadeusz Chrobak, Krystian Kozioł, Artur Krawczyk, Michał Lupa Koncepcja Wielorozdzielczej Bazy Danych Kluczowe uwarunkowania systemu generalizacji:
Dodatkowo planowane jest przeprowadzenie oceny algorytmów w praktycznym wykorzystaniu przez kilku niezależnych użytkowników ukończonej aplikacji.
 Spis Treści 1. Wprowadzenie... 2 1.1 Wstęp... 2 1.2 Cel pracy... 2 1.3 Zakres pracy... 2 1.4 Użyte technologie... 2 1.4.1 Unity 3D... 3 2. Sztuczna inteligencja w grach komputerowych... 4 2.1 Zadanie sztucznej
Spis Treści 1. Wprowadzenie... 2 1.1 Wstęp... 2 1.2 Cel pracy... 2 1.3 Zakres pracy... 2 1.4 Użyte technologie... 2 1.4.1 Unity 3D... 3 2. Sztuczna inteligencja w grach komputerowych... 4 2.1 Zadanie sztucznej
Technologie cyfrowe. Artur Kalinowski. Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15
 Technologie cyfrowe Artur Kalinowski Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15 Artur.Kalinowski@fuw.edu.pl Semestr letni 2014/2015 Zadanie algorytmiczne: wyszukiwanie dane wejściowe:
Technologie cyfrowe Artur Kalinowski Zakład Cząstek i Oddziaływań Fundamentalnych Pasteura 5, pokój 4.15 Artur.Kalinowski@fuw.edu.pl Semestr letni 2014/2015 Zadanie algorytmiczne: wyszukiwanie dane wejściowe:
SYSTEM HYDROGRAFICZNY RZGW W SZCZECINIE
 SYSTEM HYDROGRAFICZNY RZGW W SZCZECINIE. GROMADZENIE, PRZETWARZANIE ORAZ WIZUALIZACJA CIĄGŁEJ INFORMACJI BATYMETRYCZNEJ RZEKI ODRY KRZYSZTOF IWAN, TOMASZ ZAWADZKI REGIONALNY ZARZĄD GOSPODARKI WODNEJ W
SYSTEM HYDROGRAFICZNY RZGW W SZCZECINIE. GROMADZENIE, PRZETWARZANIE ORAZ WIZUALIZACJA CIĄGŁEJ INFORMACJI BATYMETRYCZNEJ RZEKI ODRY KRZYSZTOF IWAN, TOMASZ ZAWADZKI REGIONALNY ZARZĄD GOSPODARKI WODNEJ W
TOK STUDIÓW Kierunek: informatyka rok studiów: I studia stacjonarne pierwszego stopnia, rok akademicki 2014/2015. Forma zaliczen ia. egz. lab.
 Lp TOK TUDIÓW rok studiów: I studia stacjonarne pierwszego stopnia, rok akademicki 2014/2015 w ć w ko n lab EC T 1 Podstawy prawno-etyczne 15 1 x 2 Podstawy ekonomii 15 1 x 3 Repetytorium z matematyki
Lp TOK TUDIÓW rok studiów: I studia stacjonarne pierwszego stopnia, rok akademicki 2014/2015 w ć w ko n lab EC T 1 Podstawy prawno-etyczne 15 1 x 2 Podstawy ekonomii 15 1 x 3 Repetytorium z matematyki
Przewidywanie struktur białek
 Łukasz Ołdziejewski Wydział Chemii UW Przewidywanie struktur białek czyli droga do projektowania indywidualnych leków Sprawozdanie studenckie 2007/2008 1 Indywidualność jednostki KaŜdy człowiek jest indywidualnym
Łukasz Ołdziejewski Wydział Chemii UW Przewidywanie struktur białek czyli droga do projektowania indywidualnych leków Sprawozdanie studenckie 2007/2008 1 Indywidualność jednostki KaŜdy człowiek jest indywidualnym
UCHWAŁA NR 60/2013 Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 21 listopada 2013 roku
 UCHWAŁA NR 60/2013 Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 21 listopada 2013 roku w sprawie: korekty efektów kształcenia dla kierunku informatyka Na podstawie ustawy z dnia
UCHWAŁA NR 60/2013 Senatu Akademii Marynarki Wojennej im. Bohaterów Westerplatte z dnia 21 listopada 2013 roku w sprawie: korekty efektów kształcenia dla kierunku informatyka Na podstawie ustawy z dnia
Przewidywanie struktury kanału białkowego z wykorzystaniem probabilistycznych gramatyk formalnych oraz modelu ciągłego przepływu jonów
 Przewidywanie struktury kanału białkowego z wykorzystaniem probabilistycznych gramatyk formalnych oraz modelu ciągłego przepływu jonów Witold Dyrka Instytut Inżynierii Biomedycznej i Pomiarowej, Politechnika
Przewidywanie struktury kanału białkowego z wykorzystaniem probabilistycznych gramatyk formalnych oraz modelu ciągłego przepływu jonów Witold Dyrka Instytut Inżynierii Biomedycznej i Pomiarowej, Politechnika
Ćwiczenie 5/6. Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST.
 Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Modelowanie motywów łańcuchami Markowa wyższego rzędu
 Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
Podstawy bioinformatyki - biologiczne bazy danych
 Podstawy bioinformatyki - biologiczne bazy danych Czym jest bioinformatyka? Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie metod obliczeniowych do badania
Podstawy bioinformatyki - biologiczne bazy danych Czym jest bioinformatyka? Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie metod obliczeniowych do badania
Algorytmy równoległe: prezentacja i ocena efektywności prostych algorytmów dla systemów równoległych
 Algorytmy równoległe: prezentacja i ocena efektywności prostych algorytmów dla systemów równoległych Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka 2018/19 Problem: znajdowanie
Algorytmy równoległe: prezentacja i ocena efektywności prostych algorytmów dla systemów równoległych Rafał Walkowiak Politechnika Poznańska Studia inżynierskie Informatyka 2018/19 Problem: znajdowanie
Program MC. Obliczyć radialną funkcję korelacji. Zrobić jej wykres. Odczytać z wykresu wartość radialnej funkcji korelacji w punkcie r=
 Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
BIM jako techniczna platforma Zintegrowanej Realizacji Przedsięwzięcia (IPD - Integrated Project Delivery)
") BIM jako techniczna platforma Zintegrowanej Realizacji Przedsięwzięcia (IPD - Integrated Project Delivery) Dr inż. Michał Juszczyk Politechnika Krakowska Wydział Inżynierii Lądowej Zakład Technologii i
BIM jako techniczna platforma Zintegrowanej Realizacji Przedsięwzięcia (IPD - Integrated Project Delivery) Dr inż. Michał Juszczyk Politechnika Krakowska Wydział Inżynierii Lądowej Zakład Technologii i
Stochastyczna dynamika z opóźnieniem czasowym w grach ewolucyjnych oraz modelach ekspresji i regulacji genów
 Stochastyczna dynamika z opóźnieniem czasowym w grach ewolucyjnych oraz modelach ekspresji i regulacji genów Jacek Miękisz Instytut Matematyki Stosowanej i Mechaniki Uniwersytet Warszawski Warszawa 14
Stochastyczna dynamika z opóźnieniem czasowym w grach ewolucyjnych oraz modelach ekspresji i regulacji genów Jacek Miękisz Instytut Matematyki Stosowanej i Mechaniki Uniwersytet Warszawski Warszawa 14
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
Aplikacje webowe z wykorzystaniem Node.js oraz Express
 Aplikacje webowe z wykorzystaniem Node.js oraz Express Adresaci szkolenia: Kurs przeznaczony jest dla programistów pragnących tworzyć skalowalne aplikacje z wykorzystaniem Node.js. Parametry szkolenia:
Aplikacje webowe z wykorzystaniem Node.js oraz Express Adresaci szkolenia: Kurs przeznaczony jest dla programistów pragnących tworzyć skalowalne aplikacje z wykorzystaniem Node.js. Parametry szkolenia:
KONCEPCJA WYKORZYSTANIA TECHNOLOGII APPLET- JAVA W TWORZENIU
 KONCEPCJA WYKORZYSTANIA TECHNOLOGII APPLET- JAVA W TWORZENIU TORINGU PRZEMIESZCZA I ICH WIZUALIZACJI NA MAPIE CYFROWEJ 05-130 Zegrze, ul. Warszawska 22A Appletu przy projektowaniu i tworzeniu systemu Applet-
KONCEPCJA WYKORZYSTANIA TECHNOLOGII APPLET- JAVA W TWORZENIU TORINGU PRZEMIESZCZA I ICH WIZUALIZACJI NA MAPIE CYFROWEJ 05-130 Zegrze, ul. Warszawska 22A Appletu przy projektowaniu i tworzeniu systemu Applet-