Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
|
|
|
- Bogna Skiba
- 4 lat temu
- Przeglądów:
Transkrypt

1 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno
2 Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów, jakość par zasad w odczytach Mapowanie do genomu referencyjnego: Indeksowanie genomu referencyjnego Mapowanie format fastq > SAM Zmiana formatu SAM na BAM Obróbka pliku BAM: sortowanie, indeksowanie, formatowanie Wykrywanie wariantów (generujemy plik VCF): SNP polimorfizm pojedynczego nukleotydu INDEL krótkie delecje i insercje Warianty strukturalne (np. CNV) Dalsze kroki zależnie od celu analizy
3 Najpopularniejsze (darmowe) programy do analizy danych NGS FASTQC kontrola jakości dla plików w formacie fastq Bwa mapowanie do genomu referencyjnego Samtools manipulowanie plikami w formacie SAM i BAM Samtools mpileup wykrywanie wariantów Bcftools SNP and INDEL calling Vcftools manipulowanie plikami w formacie VCF Picard manipulowanie plikami w formacie SAM i BAM GATK SNP and INDEL calling Przeglądarki genomowe: GenomeBrowse Golden Helix IGV (Integrated Genomic Viewer)

4 Przykładowy pipeline dla analizy danych NGS Pirooznia M. et al Validation and assessment of variant calling pipelines for next-generation sequenci. Hum Genomics. 2014; 8(1): 14 ng.


5 Czym jest mapowanie? Sekwencjonowanie polega na odczytaniu sekwencji DNA/RNA badanego fragmentu. Przy technologii NGS otrzymujemy miliony krótkich odczytów. Co dalej? Mapowanie do genomu referencyjnego Asemblacja de-novo

6 Genom referencyjny Zsekwencjonowany genom przedstawiciela lub kilku przedstawicieli gatunku. Genom ludzki to mieszanina 5 osób, wybranych z pośród 30 ochotników, tak aby zapewnić różnorodność etniczną. W zależności od stadium zaawansowania projektu i regionu może zawierać błędy. Dobrze jeżeli posiada informację o: lokalizacji genów oraz sekwencji regulatorowych wcześniej zidentyfikowanych polimorfizmach
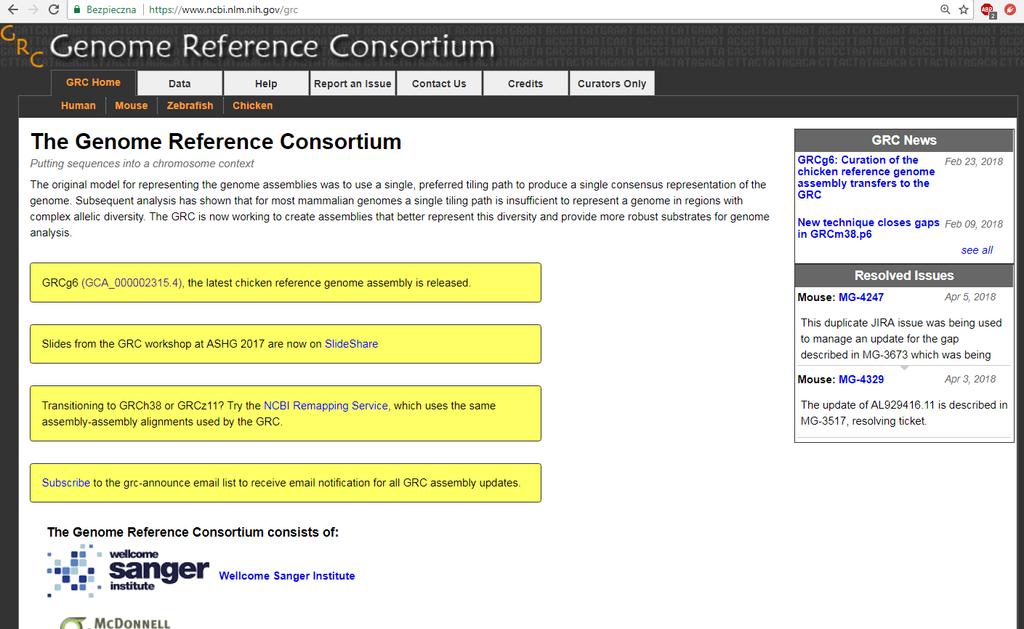
7 Genom referencyjny
8 Genom referencyjny
9 Mapowanie do genomu referencyjnego Problem: Ogromny genom referencyjny i miliony relatywnie krótkich odczytów Jak zmierzyć jakość dopasowania? Co z jakością odczytów i sekwencji referencyjnej? Kiedy uznajemy że dopasowanie jest prawidłowe? Co jeżeli odczyt pasuje w więcej niż jednym locus?

10 Mapowanie do genomu referencyjnego Źródło:
11 Mapowanie do genomu referencyjnego Mapowanie - kroki: Indeksowanie genomu referencyjnego Indeksowanie odczytów Mapowanie format fastq > SAM Najpopularniejsze programy: BWA Bowtie SOAP Inne:
12 Mapowanie do genomu referencyjnego Potrzebne są algorytmy: wyszukiwania oraz kompresji Algorytm seria instrukcji, która umożliwia rozwiązanie określonego problemu Często niezbedne są algorytmy heurystyczne tzn nie gwarantujące najlepszego rozwiązania Przykład: problem komiwojażera Rozwiązanie heurystyczne: podróżujemy zawsze do najbliższego miasta. Czy zawsze pokonamy najkrótszą drogę?
13 Mapowanie do genomu referencyjnego Algorytmy dopasowania sekwencji: Macierze punktowe Programowanie dynamiczne Metody heurystyczne (BLAST, FASTA) Metody statystyczne (modele Markova, statystyka Bayesa) Obecne większość algorytmów opiera się na dwóch etapach: wyszukanie tzw ziarna - dokładnie dopasowanego fragmentu odczytu programowanie dynamiczne w celu wydłużenia dopasowania ziarno dopasowanie? dopasowanie?
 Programowanie dynamiczne polega na wyborze najlepszej ścieżki, która obrazuje najlepsze dopasowanie. Punktujemy: dopasowanie, jego brak, przerwy.")
14 Dopasowanie pary sekwencji Algorytmy oparte na programowaniu dynamicznym umożliwiające znalezienie optymalnego dopasowania sekwencji: Smith-Waterman dopasowanie lokalne Needleman-Wunsch dopasowanie globalne (wzdłuż całej sekwencji) Programowanie dynamiczne polega na wyborze najlepszej ścieżki, która obrazuje najlepsze dopasowanie. Punktujemy: dopasowanie, jego brak, przerwy. Potrzeba jest macierz substytucji oraz ustalenie wysokości kary za przerwy
15 Struktury danych przydatne przy mapowaniu Genom referencyjny jest bardzo długi, a odczytów jest bardzo dużo dlatego zastosowanie bezpośrednie algorytmów wyznaczania dokładnego dopasowania jest nierealne. Genom referencyjny musi zostać poddany wstępnej obróbce Algorytmy korzystają z metod pozwalających na szybkie wyszukiwanie informacji w tablicach, np.: Drzewa suffiksowe Tablica z haszowaniem


16 Wykrywanie wariantów Warianty: Krótkie warianty: SNP polimorfizm pojedynczego nukleotydu INDEL krótkie insercje i delecje SV Warianty strukturalne. Np. CNV warianty liczby kopii
17 Metody wykrywania krótkich wariantów Metody heurystyczne gdy dokładny algorytm jest zbyt kosztowny (obliczeniowo) lub nie jest znany Ile razy pojawia się dany wariant? Jaka jest jakość odczytów oraz zasad, które wspierają wariant? Metody probabilistyczne Wyliczamy prawdopodobieństwo pojawienia się danego wariantu Zalety: Lepsza dokładność Otrzymujemy statystyczne potwierdzenie wyniku Uwzględnia wiele źródeł informacji jednocześnie
 : prawdopodobieństwo, że w danym miejscu rzeczywiście znajduje się dany genotyp biorąc pod uwagę posiadane dane Wiarygodność (ang.")
18 Samtools oraz GATK Unified Genotyper Opiera się na modelu statystyki Bayesowskiej gdzie wyznaczamy wiarygodność (ang. likelihood) : prawdopodobieństwo, że w danym miejscu rzeczywiście znajduje się dany genotyp biorąc pod uwagę posiadane dane Wiarygodność (ang. likelihood) jest liczona korzystając z formatu pileup oraz jakości dla poszczególnych zasad (uwzględniane są jedynie odczyty i zasady dobrej jakości) Bardzo podobny algorytm wykorzystywany również przez Samtools + bcftools
19 GATK Unified Genotyper

20 HaplotypeCaller ETAPY: 1. Identyfikacja aktywnego regionu na podstawie istnienia wariantów 2. Asemblacja de novo dla aktywnego regionu (graf k-merowy) oraz identyfikacja haplotypów. Każdy haplotyp jest przyrównywany do haplotypu referencyjnego z użyciem algorytmu Smitha- Watermana. 3. Określenie wiarygodności dla każdego haplotypu 4. Określenie prawdopodobieństwa dla danego genotypu (Bayes)
21 Metody wykrywania wariantów strukturalnych Kategorie metod: Parowane odczyty (read pair) wykorzystują informację o orientacyjnym dystansie oraz orientacji względem siebie odczytów sparowanych możliwe jest wykrywanie wielu rodzajów wariantów musi być znany dokładny rozkład indeli Pokrycie (read depth) Możliwe jest wykrycie dużych insercji i delecji Wrażliwe na zmiany pokrycia powiązane z jakością sekwencji referencyjnej oraz sekwencjonowania Odczyty dzielone (split read) Strategie łączone

22 Metody wykrywania wariantów strukturalnych
23 Asemblacja de-novo Odtworzenie sekwencji poprzez dopasowywanie nakładających się nas siebie odczytów Problemy: Nierównomierne rozłożenie odczytów Odczyty pochodzą z obu nici (sensownej i antysensownej) Błędy w odczytach Powtórzenia w badanej sekwencji Odczyty są dużo krótsze niż sekwencja genomu
24 Asemblacja de-novo Wykorzystywany jest graf de Bruijna (graf k-merowy) Graf jest strukturą danych, umożliwiającą przedstawienie i badanie relacji pomiędzy obiektami. Składa się z dwóch zbiorów: Wierzchołków Krawędzi
25 Asemblacja de-novo Graf de Bruijna (graf k-merowy) Jest grafem skierowanym uporządkowana para zbiorów Wierzchołkami są sekwencje o długości k, a odczyty są wrysowywane w graf poprzez połączenie łukami kolejnych wierzchołków Sekwencja: ACTGCC Odczyty: ACTG CTGC CTGC TGCC Graf (k=3): ACT CTG TGC GCC Odczyty: ACTG CTGC CTGA TGCC Graf (k=3): ACT CTG TGC GCC TGA
26 Asemblacja de-novo Graf k-merowy (k=3) a SNP: TAGCCTGACT TAGCCTGACT TAGCATGACT TAGCATGACT TAG AGC GCC CCT CTG TGA GAC ACT GCA CAT ATG Narysuj graf k-merowy dla następujących odczytów: AATTGCG AAATGCG AATGCGA AATGCGAA TTGCGAA

27 Metody wykrywania krótkich wariantów użyty algorytm a otrzymany wynik Yu and Sun, Comparing a few SNP calling algorithms using low-coverage sequencing data. BMC Bioinformatics 14: 274.
28 Perspektywa z 1000 Genomes Project Przeanalizowano 629 próbek Wykryto 25 milionów unikalnych wariantów, z których 15 milionów miało frekwencję poniżej 2 % 7.9 mln wariantów znajdowało się w bazie dbsnp 129 Liczba wariantów dla pojedynczej próbki: 4 miliony Średnio 20 tysięcy znajdzie się w regionach kodujących (eksony) z nich tylko będzie powodowało zmianę funkcji biologicznej białka z nich będą to warianty już wcześniej powiązane z chorobami Khoury, 2010
29 Literatura Khoury M Dealing With the Evidence Dilemma in Genomics and Personalized Medicine. Clinical Pharmacology & Therapeutics, 87, , doi: /clpt clpt/journal/v87/n6/full/clpt20104a.html Rudy G A Hitchhiker s Guide to Next-Generation Sequencing. Higgs P.G., Attwood T.K Bioinformatyka i ewolucja molekularna. Wydawnictow naukowe PWN
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing. Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:
 w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:") Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
 Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Różnorodność osobników gatunku
 ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
Wykład 5 Dopasowywanie lokalne
 Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Pytania i odpowiedzi
 Pytania i odpowiedzi PCA PCA a MDS - PCA bazuje na macierzy kowariancji, MDS bazuje na macierzy dystansów genetycznych Będą identyczne jeśli kowariancja będzie równa odległości euklidesowej. W badaniach
Pytania i odpowiedzi PCA PCA a MDS - PCA bazuje na macierzy kowariancji, MDS bazuje na macierzy dystansów genetycznych Będą identyczne jeśli kowariancja będzie równa odległości euklidesowej. W badaniach
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
1. System analizy danych NGS z paneli genów
 1. System analizy danych NGS z paneli genów (programistyczny) Sekwenator to instrument odczytujący sekwencję DNA w kilku-kilkudziesieciu probkach na raz. Instrument zapisuje na dysku dane w skompresowanych
1. System analizy danych NGS z paneli genów (programistyczny) Sekwenator to instrument odczytujący sekwencję DNA w kilku-kilkudziesieciu probkach na raz. Instrument zapisuje na dysku dane w skompresowanych
Algorytmy kombinatoryczne w bioinformatyce
 Algorytmy kombinatoryczne w bioinformatyce wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji
Algorytmy kombinatoryczne w bioinformatyce wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji
Na czym skończyliśmy BLACK BOX. Sekwencjonowanie polega na odczytaniu sekwencji liter DNA/RNA badanego fragmentu genomu
 ALEKSANDRA ŚWIERCZ Na czym skończyliśmy BLACK BOX AAATGCCTGCCCTGAAGGCCTGCGTA GTTTTGGGAGAAGACCCACGGATA AAGGTGTAGCCCCGTAGC GGGGGGTATTATTTATTTTATACCCAC.. ACAGGAUCGUUGGAUGGTGGGA. Sekwencjonowanie polega na
ALEKSANDRA ŚWIERCZ Na czym skończyliśmy BLACK BOX AAATGCCTGCCCTGAAGGCCTGCGTA GTTTTGGGAGAAGACCCACGGATA AAGGTGTAGCCCCGTAGC GGGGGGTATTATTTATTTTATACCCAC.. ACAGGAUCGUUGGAUGGTGGGA. Sekwencjonowanie polega na
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Podstawy bioinformatyki sekwencjonowanie nowej generacji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Podstawy bioinformatyki sekwencjonowanie nowej generacji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Rozwój technologii i przyrost danych Wzrost olbrzymiej ilości i objętości
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Sekwencjonowanie, przewidywanie genów
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
"Zapisane w genach, czyli Python a tajemnice naszego genomu."
 "Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
"Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
Przyrównywanie sekwencji
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Przetarg nieograniczony na zakup specjalistycznej aparatury laboratoryjnej Znak sprawy: DZ-2501/6/17
 Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Reswkwencjonowanie vs asemblacja de novo
 ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Dopasowanie sekwencji
 Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej
 Bioinformatyka wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji genomów na trzech poziomach
Bioinformatyka wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji genomów na trzech poziomach
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI
 ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI Podstawy Bioinformatyki lab 1 PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 1 BIOINFORMATYKA Dr Magda Mielczarek Katedra Genetyki, pokój nr 14 ul. Kożuchowska
ZAJĘCIA ORGANIZACYJNE WSTĘP DO BIOINFORMATYKI Podstawy Bioinformatyki lab 1 PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 1 BIOINFORMATYKA Dr Magda Mielczarek Katedra Genetyki, pokój nr 14 ul. Kożuchowska
Bioinformatyczna analiza danych. Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt
 Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Analiza danych pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego. - część I -
 pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego - część I - Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Plan wykładów --------------------------------------------------------
pochodzących z sekwencjonowania nowej generacji - przyrównanie do genomu referencyjnego - część I - Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Plan wykładów --------------------------------------------------------
PODSTAWY BIOINFORMATYKI
 PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
BUDOWA I FUNKCJA GENOMU LUDZKIEGO
 BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
1. Analiza asocjacyjna. Cechy ciągłe. Cechy binarne. Analiza sprzężeń. Runs of homozygosity. Signatures of selection
 BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
PODSTAWY BIOINFORMATYKI ORGANIZACJA ZAJĘĆ BIOINFORMATYKA PRZETWARZANIE I ANALIZA DANYCH
 PODSTAWY BIOINFORMATYKI ORGANIZACJA ZAJĘĆ BIOINFORMATYKA PRZETWARZANIE I ANALIZA DANYCH Magda Mielczarek Podstawy Bioinformatyki 1 Organizacja zajęć mgr Magda Mielczarek Katedra Genetyki, pokój nr 14 magda.mielczarek@up.wroc.pl
PODSTAWY BIOINFORMATYKI ORGANIZACJA ZAJĘĆ BIOINFORMATYKA PRZETWARZANIE I ANALIZA DANYCH Magda Mielczarek Podstawy Bioinformatyki 1 Organizacja zajęć mgr Magda Mielczarek Katedra Genetyki, pokój nr 14 magda.mielczarek@up.wroc.pl
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka
 Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka Słowo wstępne XIII Przedmowa XV 1. Bioinformatyka i Internet Andreas D. Baxevanis 1 1.1. Podstawy Internetu 2 1.2. Połączenie z Internetem
Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka Słowo wstępne XIII Przedmowa XV 1. Bioinformatyka i Internet Andreas D. Baxevanis 1 1.1. Podstawy Internetu 2 1.2. Połączenie z Internetem
CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1
 CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1 PRACOWNIA INFORMATYCZNA PROWADZĄCY: Dr Magda Mielczarek (biolog) Katedra Genetyki, pokój nr 21
CHARAKTERYSTYKA PRZEDMIOTU Pracownia Informatyczna 1 PRACOWNIA INFORMATYCZNA 2018/2019 MAGDA MIELCZAREK 1 PRACOWNIA INFORMATYCZNA PROWADZĄCY: Dr Magda Mielczarek (biolog) Katedra Genetyki, pokój nr 21
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
PODSTAWY BIOINFORMATYKI 11 BAZA DANYCH HAPMAP
 PODSTAWY BIOINFORMATYKI 11 BAZA DANYCH HAPMAP WSTĘP 1. SNP 2. haplotyp 3. równowaga sprzężeń 4. zawartość bazy HapMap 5. przykłady zastosowań Copyright 2013, Joanna Szyda HAPMAP BAZA DANYCH HAPMAP - haplotypy
PODSTAWY BIOINFORMATYKI 11 BAZA DANYCH HAPMAP WSTĘP 1. SNP 2. haplotyp 3. równowaga sprzężeń 4. zawartość bazy HapMap 5. przykłady zastosowań Copyright 2013, Joanna Szyda HAPMAP BAZA DANYCH HAPMAP - haplotypy
Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji
 Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu. Modele grafowe problemu sekwencjonowania
Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu. Modele grafowe problemu sekwencjonowania
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
Bioinformatyka. Porównywanie sekwencji
 Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Przeglądarki genomowe
 Przeglądarki genomowe Popularne typy danych w nowoczesnych naukach biologicznych obejmują: sekwencje genomu sekwencje transkryptomu sekwencje proteomu epigenom adnotacje: geny, eksony, introny, izoformy,
Przeglądarki genomowe Popularne typy danych w nowoczesnych naukach biologicznych obejmują: sekwencje genomu sekwencje transkryptomu sekwencje proteomu epigenom adnotacje: geny, eksony, introny, izoformy,
Analizy wielkoskalowe w badaniach chromatyny
 Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: dodany punkt na temat testów do sprawozdania. Biologia, bioinformatyka:
 Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,
PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (1)
") PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (1) BIOINFORMATYKA HISTORIA 1. 1982 utworzenie bazy danych GenBank (NIH) dane ogólnodostępne sekwencje nukleotydów 2. Wprowadzenie sekwencji z projektu
PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (1) BIOINFORMATYKA HISTORIA 1. 1982 utworzenie bazy danych GenBank (NIH) dane ogólnodostępne sekwencje nukleotydów 2. Wprowadzenie sekwencji z projektu
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (2)
") PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (2) BIOINFORMATYKA HISTORIA 1. 1982 utworzenie bazy danych GenBank (NIH) dane ogólnodostępne sekwencje nukleotydów 2. Wprowadzenie sekwencji z projektu
PODSTAWY BIOINFORMATYKI WYKŁAD 3 BIOLOGICZNE BAZY DANYCH (2) BIOINFORMATYKA HISTORIA 1. 1982 utworzenie bazy danych GenBank (NIH) dane ogólnodostępne sekwencje nukleotydów 2. Wprowadzenie sekwencji z projektu
MODELOWANIE STOCHASTYCZNE CZĘŚĆ II - ŁAŃCUCHY MARKOWA. Biomatematyka Dr Wioleta Drobik-Czwarno
 MODELOWANIE STOCHASTYCZNE CZĘŚĆ II - ŁAŃCUCHY MARKOWA Biomatematyka Dr Wioleta Drobik-Czwarno Polecane Łańcuchy Markowa wizualnie: http://setosa.io/ev/markov-chains/ Procesy stochastyczne Procesem stochastycznym
MODELOWANIE STOCHASTYCZNE CZĘŚĆ II - ŁAŃCUCHY MARKOWA Biomatematyka Dr Wioleta Drobik-Czwarno Polecane Łańcuchy Markowa wizualnie: http://setosa.io/ev/markov-chains/ Procesy stochastyczne Procesem stochastycznym
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
Rak tarczycy - prognostyka
 Rak tarczycy - prognostyka Wariant rs966423-tt w genie DIRC3 jest czynnikiem rokowniczo niekorzystnym, związanym ze zwiększonym ryzykiem zgonu w przebiegu raka zróżnicowanego tarczycy (Świerniak et al.
Rak tarczycy - prognostyka Wariant rs966423-tt w genie DIRC3 jest czynnikiem rokowniczo niekorzystnym, związanym ze zwiększonym ryzykiem zgonu w przebiegu raka zróżnicowanego tarczycy (Świerniak et al.
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Ćwiczenie 12. Diagnostyka molekularna. Poszukiwanie SNPs Odczytywanie danych z sekwencjonowania. Prof. dr hab. Roman Zieliński
 Ćwiczenie 12 Diagnostyka molekularna. Poszukiwanie SNPs Odczytywanie danych z sekwencjonowania Prof. dr hab. Roman Zieliński 1. Diagnostyka molekularna 1.1. Pytania i zagadnienia 1.1.1. Jak definiujemy
Ćwiczenie 12 Diagnostyka molekularna. Poszukiwanie SNPs Odczytywanie danych z sekwencjonowania Prof. dr hab. Roman Zieliński 1. Diagnostyka molekularna 1.1. Pytania i zagadnienia 1.1.1. Jak definiujemy
Oznaczenie polimorfizmu genetycznego cytochromu CYP2D6: wykrywanie liczby kopii genu
 Ćwiczenie 4 Oznaczenie polimorfizmu genetycznego cytochromu CYP2D6: wykrywanie liczby kopii genu Wstęp CYP2D6 kodowany przez gen występujący w co najmniej w 78 allelicznych formach związanych ze zmniejszoną
Ćwiczenie 4 Oznaczenie polimorfizmu genetycznego cytochromu CYP2D6: wykrywanie liczby kopii genu Wstęp CYP2D6 kodowany przez gen występujący w co najmniej w 78 allelicznych formach związanych ze zmniejszoną
STATYSTYKA MATEMATYCZNA
 STATYSTYKA MATEMATYCZNA 1. Wykład wstępny 2. Teoria prawdopodobieństwa i elementy kombinatoryki 3. Zmienne losowe 4. Populacje i próby danych 5. Testowanie hipotez i estymacja parametrów 6. Test t 7. Test
STATYSTYKA MATEMATYCZNA 1. Wykład wstępny 2. Teoria prawdopodobieństwa i elementy kombinatoryki 3. Zmienne losowe 4. Populacje i próby danych 5. Testowanie hipotez i estymacja parametrów 6. Test t 7. Test
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA
 Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19
 WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
Podstawy bioinformatyki - biologiczne bazy danych
 Podstawy bioinformatyki - biologiczne bazy danych Czym jest bioinformatyka? Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie metod obliczeniowych do badania
Podstawy bioinformatyki - biologiczne bazy danych Czym jest bioinformatyka? Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie metod obliczeniowych do badania
Acrodermatitis enteropathica
 Acrodermatitis enteropathica Acrodermatitis enteropathica (zespół Brandta) to choroba związana z uszkodzeniem białka odpowiedzialnego za wchłanianie cynku. Objawy choroby ujawniają się w pierwszych miesiącach
Acrodermatitis enteropathica Acrodermatitis enteropathica (zespół Brandta) to choroba związana z uszkodzeniem białka odpowiedzialnego za wchłanianie cynku. Objawy choroby ujawniają się w pierwszych miesiącach
Pytania i odpowiedzi
 Pytania i odpowiedzi Czy kontrola jakości płytek w programach analizy danych jest dostosowywana do przeprowadzanego badania, czy też przyjmuje się jednakową jej wartość dla różnych analiz? We wstępnym
Pytania i odpowiedzi Czy kontrola jakości płytek w programach analizy danych jest dostosowywana do przeprowadzanego badania, czy też przyjmuje się jednakową jej wartość dla różnych analiz? We wstępnym
Choroba syropu klonowego
 Choroba syropu klonowego Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze BCKDHA Maple syrup urine disease AR 40 BCKDHB Maple syrup urine disease AR 64 DBT
Choroba syropu klonowego Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze BCKDHA Maple syrup urine disease AR 40 BCKDHB Maple syrup urine disease AR 64 DBT
PODSTAWY BIOINFORMATYKI 6 BAZA DANYCH NCBI - II
 PODSTAWY BIOINFORMATYKI 6 BAZA DANYCH NCBI - II BAZA DANYCH NCBI 1. NCBI 2. Dane gromadzone przez NCBI 3. Przegląd baz danych NCBI: Publikacje naukowe Projekty analizy genomów OMIM: fenotypy człowieka
PODSTAWY BIOINFORMATYKI 6 BAZA DANYCH NCBI - II BAZA DANYCH NCBI 1. NCBI 2. Dane gromadzone przez NCBI 3. Przegląd baz danych NCBI: Publikacje naukowe Projekty analizy genomów OMIM: fenotypy człowieka
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE
 PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
Choroba Niemanna-Picka, typ C
 Choroba Niemanna-Picka, typ C Choroba Niemanna-Picka typu C jest dziedziczną neurodegeneracyjną chorobą. Jest ona spowodowana mutacjami w genach NPC1 lub NPC2. Brak funkcjonalnego białka kodowanego przez
Choroba Niemanna-Picka, typ C Choroba Niemanna-Picka typu C jest dziedziczną neurodegeneracyjną chorobą. Jest ona spowodowana mutacjami w genach NPC1 lub NPC2. Brak funkcjonalnego białka kodowanego przez
1.7. Eksploracja danych: pogłębianie, przeszukiwanie i wyławianie
 Wykaz tabel Wykaz rysunków Przedmowa 1. Wprowadzenie 1.1. Wprowadzenie do eksploracji danych 1.2. Natura zbiorów danych 1.3. Rodzaje struktur: modele i wzorce 1.4. Zadania eksploracji danych 1.5. Komponenty
Wykaz tabel Wykaz rysunków Przedmowa 1. Wprowadzenie 1.1. Wprowadzenie do eksploracji danych 1.2. Natura zbiorów danych 1.3. Rodzaje struktur: modele i wzorce 1.4. Zadania eksploracji danych 1.5. Komponenty
Stwardnienie guzowate
 Stwardnienie guzowate Stwardnienie guzowate jest zespołem nerwowo-skórnym, w przebiegu którego na skórze, nerkach, sercu, kościach, płucach i mózgowiu powstają hamartoma - guzy o charakterze nienowotworowym,
Stwardnienie guzowate Stwardnienie guzowate jest zespołem nerwowo-skórnym, w przebiegu którego na skórze, nerkach, sercu, kościach, płucach i mózgowiu powstają hamartoma - guzy o charakterze nienowotworowym,
BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH
 http://theta.edu.pl/ Podstawy Bioinformatyki II BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH 1 Czym jest bioinformatyka? 2 Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie
http://theta.edu.pl/ Podstawy Bioinformatyki II BIOINFORMATYKA BIOLOGICZNE BAZY DANYCH 1 Czym jest bioinformatyka? 2 Bioinformatyka Bioinformatyka jest interdyscyplinarną dziedziną nauki obejmującą wykorzystanie
Genomika Porównawcza. Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski
 Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Bioinformatyczne bazy danych - część 2. -przeszukiwanie baz danych -pobieranie danych
 Bioinformatyczne bazy danych - część 2 -przeszukiwanie baz danych -pobieranie danych Numery dostępowe baz danych (accession number) to ciąg liter i cyfr służących jako etykieta identyfikująca sekwencję
Bioinformatyczne bazy danych - część 2 -przeszukiwanie baz danych -pobieranie danych Numery dostępowe baz danych (accession number) to ciąg liter i cyfr służących jako etykieta identyfikująca sekwencję
Ekologia molekularna. wykład 11
 Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
Zespół hemolityczno-mocznicowy
 Zespół hemolityczno-mocznicowy Badanie obejmuje: analizę sekwencji genów ADAMTS13; C3; CD46; CFB; CFH; CFI; THBD; CFHR1; CFHR2; CFHR3; CFHR4; CFHR5 metodą NGS analizę poziomu przeciwciał przeciw czynnikowi
Zespół hemolityczno-mocznicowy Badanie obejmuje: analizę sekwencji genów ADAMTS13; C3; CD46; CFB; CFH; CFI; THBD; CFHR1; CFHR2; CFHR3; CFHR4; CFHR5 metodą NGS analizę poziomu przeciwciał przeciw czynnikowi
BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE. Podstawy Bioinformatyki wykład 4
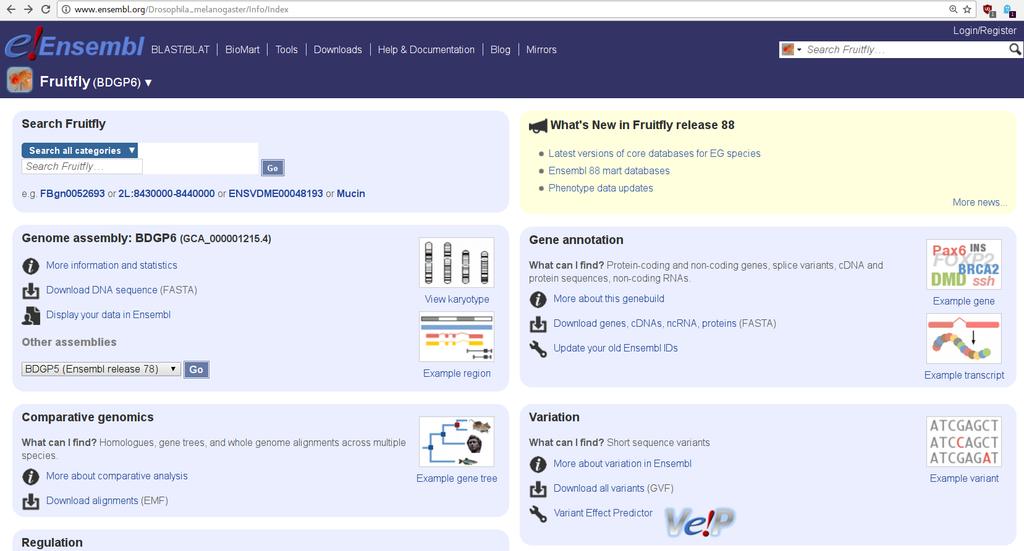
 GENOMY I ICH ADNOTACJE. Podstawy Bioinformatyki wykład 4") BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 4 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 2 GENOMY I ICH ADNOTACJE
BIOLOGICZNE BAZY DANYCH (2) GENOMY I ICH ADNOTACJE Podstawy Bioinformatyki wykład 4 GENOMY I ICH ADNOTACJE NCBI Ensembl UCSC PODSTAWY BIOINFORMATYKI 2017/2018 MAGDA MIELCZAREK 2 GENOMY I ICH ADNOTACJE
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Programowanie dynamiczne i algorytmy zachłanne
 Programowanie dynamiczne i algorytmy zachłanne Tomasz Głowacki tglowacki@cs.put.poznan.pl Zajęcia finansowane z projektu "Rozwój i doskonalenie kształcenia na Politechnice Poznańskiej w zakresie technologii
Programowanie dynamiczne i algorytmy zachłanne Tomasz Głowacki tglowacki@cs.put.poznan.pl Zajęcia finansowane z projektu "Rozwój i doskonalenie kształcenia na Politechnice Poznańskiej w zakresie technologii
Przytarczyce, zaburzenia metabolizmu wapnia
 Przytarczyce, zaburzenia metabolizmu wapnia Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze BSND Zespół Barttera, Głuchota czuciowo-nerwowa AR 10 CASR Nadczynność
Przytarczyce, zaburzenia metabolizmu wapnia Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze BSND Zespół Barttera, Głuchota czuciowo-nerwowa AR 10 CASR Nadczynność
POPULARNE POLECENIA SKRYPTY. Pracownia Informatyczna 2
 SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
Zespół Alporta. Gen Choroba/objawy Sposób dziedziczenia. COL4A3 Zespół Alporta AD/AR 100. COL4A4 Zespół Alporta AD/AR 84. COL4A5 Zespół Alporta XL 583
 Zespół Alporta Zespół Alporta jest przykładem zespołu wad wrodzonych ze współistniejącym niedosłuchem. Charakteryzuje się występowaniem nefropatii (choroby nerek), powodowanej zaburzeniami powstawania
Zespół Alporta Zespół Alporta jest przykładem zespołu wad wrodzonych ze współistniejącym niedosłuchem. Charakteryzuje się występowaniem nefropatii (choroby nerek), powodowanej zaburzeniami powstawania
Sekwencje akinezji płodu
 Sekwencje akinezji płodu Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze CHRNA1 Myasthenic syndrome, congenital AD/AR 19 CHRND Myasthenic syndrome AD/AR
Sekwencje akinezji płodu Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze CHRNA1 Myasthenic syndrome, congenital AD/AR 19 CHRND Myasthenic syndrome AD/AR
Galaktozemia. Gen Choroba/objawy Sposób dziedziczenia GALE AR 12. GALK1 Niedobór galaktokinazy AR 14. GALT Galaktozemia AR 233
 Galaktozemia Galaktozemia jest genetycznie uwarunkowanym zaburzeniem metabolizmu galaktozy cukru będącego elementem budulcowym laktozy i powszechnie występującym w jedzeniu. W postaci klasycznej pierwsze
Galaktozemia Galaktozemia jest genetycznie uwarunkowanym zaburzeniem metabolizmu galaktozy cukru będącego elementem budulcowym laktozy i powszechnie występującym w jedzeniu. W postaci klasycznej pierwsze
CZĘŚĆ III OPISPRZEDMIOTU ZAMÓWIENIA (OPZ)
") INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ im. Ludwika Hirszfelda Polska Akademia Nauk ul. Rudolfa Weigla 12, 53-114 Wrocław tel. / fax. (4871) 37-09-997, http://www.iitd.pan.wroc.pl NIP: 896-000-56-96;
INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ im. Ludwika Hirszfelda Polska Akademia Nauk ul. Rudolfa Weigla 12, 53-114 Wrocław tel. / fax. (4871) 37-09-997, http://www.iitd.pan.wroc.pl NIP: 896-000-56-96;
Choroba Leśniowskiego i Crohna
 Choroba Leśniowskiego i Crohna Choroba Leśniowskiego i Crohna należy do nieswoistych chorób zapalnych jelita, może dotyczyć każdego odcinka przewodu pokarmowego. Rokrocznie zapada na nią 1 na 10 000 osób;
Choroba Leśniowskiego i Crohna Choroba Leśniowskiego i Crohna należy do nieswoistych chorób zapalnych jelita, może dotyczyć każdego odcinka przewodu pokarmowego. Rokrocznie zapada na nią 1 na 10 000 osób;
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Bioinformatyka. Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl
 Bioinformatyka Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Bazy danych biologicznych Bazy danych sekwencji nukleotydowych Pierwotne bazy danych (ang. primary database) Wykorzystywane do zbierania
Bioinformatyka Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Bazy danych biologicznych Bazy danych sekwencji nukleotydowych Pierwotne bazy danych (ang. primary database) Wykorzystywane do zbierania
Przewlekła choroba ziarniniakowa
 Przewlekła choroba ziarniniakowa Przewlekła choroba ziarniniakowa związana jest z defektami aktywności granulocytów obojętnochłonnych. Choroba typowo objawia się nawracającymi, ciężkimi infekcjami bakteryjnymi
Przewlekła choroba ziarniniakowa Przewlekła choroba ziarniniakowa związana jest z defektami aktywności granulocytów obojętnochłonnych. Choroba typowo objawia się nawracającymi, ciężkimi infekcjami bakteryjnymi
Wrodzony przerost nadnerczy
 Wrodzony przerost nadnerczy Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze CYP11B1 Wrodzony przerost nadnerczy AD/AR 22 CYP17A1 Wrodzony przerost nadnerczy
Wrodzony przerost nadnerczy Geny i zespoły genetyczne Gen Choroba/objawy Sposób dziedziczenia Znane warianty chorobotwórcze CYP11B1 Wrodzony przerost nadnerczy AD/AR 22 CYP17A1 Wrodzony przerost nadnerczy