Sekwencjonowanie DNA
|
|
|
- Feliks Barański
- 6 lat temu
- Przeglądów:
Transkrypt
1 BIOINFORMATYKA edycja 2016 / 2017 wykład 7 Sekwencjonowanie DNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl
2 Plan wykładu 1. Techniki sekwencjonowania 2. Problemy bioinformatyczne 3. Szkic algorytmów składania sekwencji 4. Przechowywanie danych slajd 2
3 Techniki sekwencjonowania slajd 3

4 Materiał genetyczny Źródło: slajd 4
5 Rozwój metod sekwencjonowania 1977 Sanger, metoda syntezy i DD-terminacji DNA; 1977 Maxam i Gilbert, metoda chemicznej degradacji; 1981 Sanger, Shotgun (hierarchiczny); 1987 automatyzacja procesu; 1995 Venter, Shotgun całego genomu; 2005 sekwencjonowanie nowej generacji; 2014 trzecia generacja slajd 5
 badanej próbki 2.")

6 Proces sekwencjonowania 1. Zwielokrotnienie (amplifikacja) badanej próbki 2. Reakcje chemiczne pozwalające na wyznakowanie poszczególnych nukleotydów 3. Rozdział na żelu (elektroforeza) lub odczyt czytnikiem fluorescencji slajd 6

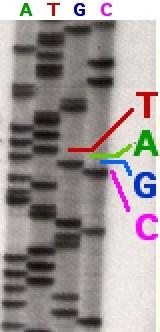
7 Odczytywanie sekwencji elektroforeza slajd 7
 capillary electrophoresis computer screen Genomes, 2nd Edition slajd 8")
8 Odczytywanie sekwencji znakowanie fluorescencyjne Fluorescent dyes ( ) capillary electrophoresis computer screen Genomes, 2nd Edition slajd 8

9 Odczytywanie sekwencji detekcja sygnału slajd 9
10 Shotgun namnożone fragmenty DNA są losowo dzielone na mniejsze odcinki, każdy z nich sekwencjonowany jest oddzielnie slajd 10
11 Shotgun poszczególne fragmenty częściowo zachodzą na siebie, co umożliwia odtworzenie pełnej sekwencji slajd 11
12 Sekwencjonowanie całego genomu czy to jest możliwe? rok liczba zmapowanych genów przewidywany czas potrzebny do zsekwencjonowania całego genomu niemożliwe ~4 mln lat ~1000 lat 2000 ~25000 wersja robocza 2005 ~30000 nowa wersja robocza ,784 / 30,384 wyzwanie $1000 genome Źródło: slajd 12
13 Sekwencjonowanie całego genomu podejścia Human Genome Project Genome race slajd 13 Celera Genomics (C.Venter)


14 Nowe techniki sekwencjonowania (next generation sequencing) 1. Technika 454 pirosekwencjonowanie (na matrycy badanej nici syntetyzowana jest druga nić, co powoduje emisję kwantów światła) 2. ION Torrent mierzona jest zmiana ph wywołana wbudowaniem nukleotydu 3. Illumina (Solexa) sekwencjonowanie w oparciu o znakowane nukleotydy slajd 14

15 Wyzwanie $1000 Projekt Sekwencjonowania Genomu Ludzkiego (HGP) kosztował 3 mld $ Czy można go zmniejszyć do 1 tys $? Wyzwanie zostało zrealizowane w 2014 roku Illumina HiSeq X Ten System (koszt odczynników: $797, amortyzacja: $137, przygotowanie próbki: $55 $65) slajd 15

16 Najnowsze techniki Sekwencjonowanie pojedynczych cząsteczek DNA slajd 16
17 Techniki sekwencjonowania porównanie Wybrane charakterystyki metod sekwencjonowania genomów slajd 17
 slajd")
18 Porównanie (2) slajd 18
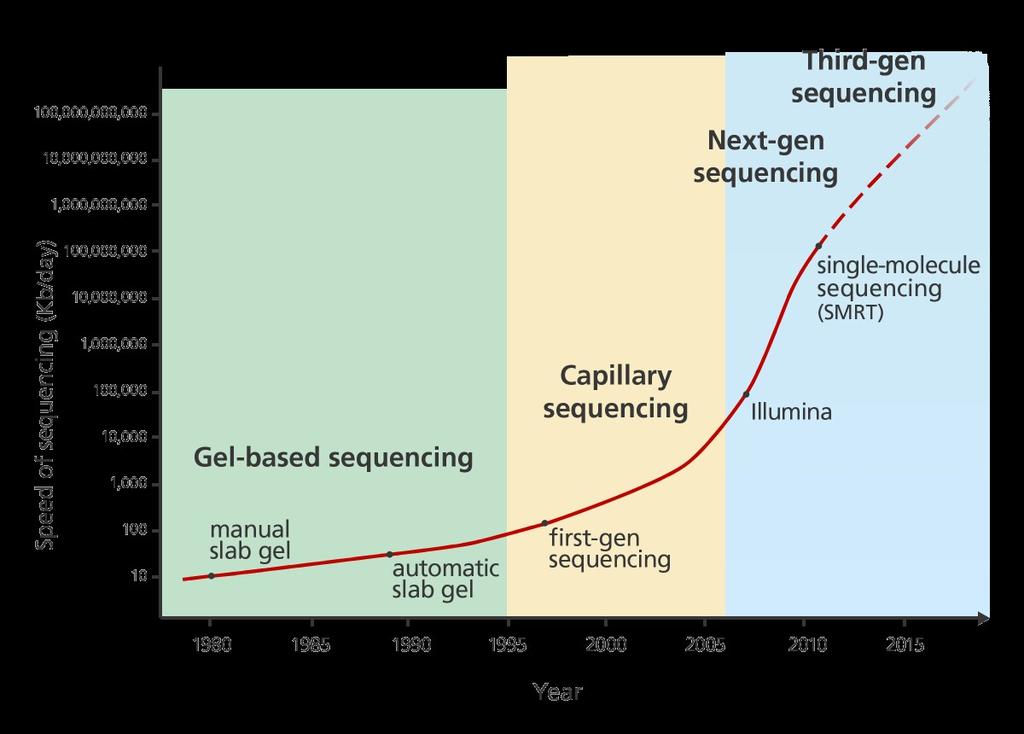

19 Prędkość sekwencjonowania slajd 19

20 Możliwości badanie zmienności genetycznej Projekt HapMap slajd 20

21 Projekt 100 genomów slajd 21

22 Możliwości medycyna personalizowana slajd 22
23 Możliwości medycyna personalizowana Określenie ryzyka zachorowania Określenie stopnia odporności Indywidualna strategia leczenia (oporność / wrażliwość na niektóre leki) HGMD Human Gene Mutation Database slajd 23
24 Problemy bioinformatyczne slajd 24
25 Etapy poznawania sekwencji Zbieranie odczytów Identyfikacja kontigów Łączenie kontigów (scaffold) Tworzenie konsensusu Kompletny chromosom Kompletny genom Etapy posekwencyjne - lokalizacja genów - przewidywanie funkcji - przechowywanie danych slajd 25
26 Składanie sekwencji idea slajd 26

27 Dlaczego to jest trudne? powtórzenia genom ludzki zawiera mnóstwo powtarzających się sekwencji, niektóre pojawiają się w genomie więcej niż razy. Przykładowo powtórzenie Alu ma długość 300 nukleotydów, pojawia się razy w ludzkim genomie. slajd 27
28 Dlaczego to jest trudne? przerwy niektórych fragmentów DNA nie da się zsekwencjonować. błędy w sekwencjonowaniu związane zarówno z ograniczeniami technologicznymi jak i z ludzkimi pomyłkami nieznana orientacja sekwencjonujemy DNA dwuniciowe; nie wiadomo, z której nici pochodzi dany odczyt slajd 28

29 Orientacja Fragment może pochodzić z dowolnego łańcucha CACGT ACGT ACTACG GTACT ACTGA CTGA CACGT -ACGT --CGTAGT -----AGTAC ACTGA CTGA jeśli odczyt pochodzi z drugiej nici, musimy składać sekwencję komplementarną czytaną od końca; ale tego, z której nici jest dany fragment, nie wiemy slajd 29
 zgubienie powtórzenia Źródło: Pop, M.; Salzberg, S.L.; Shumway, M.")
30 Powtórzenia Krótkie powtórzenia nie stanowią dużego problemu jeśli powtarzająca się sekwencja jest krótsza od długości analizowanego klonu, zapewne uda się właściwie dopasować końce sekwencji. Jednak powtórzenie dłuższe od długości odczytu może być błędnie złożone. a) zgubienie powtórzenia Źródło: Pop, M.; Salzberg, S.L.; Shumway, M.; Genome Sequence Assembly:Algorithms and Issues, 2002, IEEE Computer 35(7):47-54 slajd 30
31 Powtórzenia (2) b) zmiana kolejności A X A X B C X X slajd 31 C B X D X D
32 Powtórzenia (3) b) zmiana kolejności A X A X B D Y Y C C slajd 32 X X D B Y E Y E
33 Powtórzenia (4) c) odwrócone powtórzenia X X X X slajd 33
34 Błędy odczytu --ACCGT-----CGTGC TTAC-----TGCCGT TTACCGTGC --ACC-GT-----CAGTGC TTAC------TACC-GT TTACC-GTGC --ACCGT-----CGTGC TTAC-----TAC-GT TTACCGTGC błędna insercja błędna delecja błędny odczyt nukleotydu slajd 34
35 Ilustracja Problemy: - nieznana orientacja - błędy sekwencjonowania - niepełne pokrycie - powtórzenia slajd 35
36 Szkic algorytmów składania sekwencji slajd 36
37 Najkrótsze wspólne nadsłowo (SCS shortest common superstring) Definicja: Dany jest zbiór słów P = {s1, s2,..., sn} Najkrótszym wspólnym nadsłowem zbioru P nazywany słowo S, najkrótsze spośród spełniających warunek, że każde słowo si P jest podsłowem S Załóżmy dodatkowo, bez straty ogólności, że słowa z P, nie zawierają się w sobie. Przykład: P = {fabcc, efab, bccla}. Słowa bcclabccefabcc i efabccla są nadsłowami P. efabccla jest najkrótszym nadsłowem P. slajd 37
38 Złożoność problemu Problem jest NP-zupełny. Klasyczny algorytm dokładny: O(2k) czas, O(2k) pamięć (Held,Karp,1962 TSP). Zachłanny algorytm aproksymacyjny: 4-aproksymacja (Blumetal.,1991) 3,5-aproksymacja (Kaplan,Shafrir,2005) 2-aproksymacja? (hipoteza,blumetal.). Algorytm 2,5-aproksymacyjny (Breslaueretal.,1997+Kaplanetal.,2005). slajd 38
39 Jakość aproksymacji ρ-aproksymacja Algorytm A nazywamy ρ-aproksymacyjnym, jeśli dla dowolnych poprawnych danych wejściowych x, oraz Wartość ρ określa ile razy otrzymane rozwiązanie jest gorsze od optimum. W przypadku gdy algorytm zwraca rozwiązanie optymalne, ρ = 1. Jeżeli rozwiązanie może być dowolnie odległe od optimum, to wartość ρ jest nieskończonością. slajd 39
40 Algorytm zachłanny S {s1, s2,..., sk} while S > 1 do (si, sj ) para si, sj S maks. ov(si, sj ) u sklej (si, sj) S S \{si, sj} {u} Da się go zaimplementować w czasie O(n log Σ) (Tarhio, Ukkonen,1986), a nawet O(n + suf ), gdzie suf to czas konstrukcji tablicy sufiksowej (Kociumaka, 2010). slajd 40
41 Wady prostego podejścia bazującego na SCS - orientacja odczytów musi być znana - nie uwzględnia pokrycia - nie uwzględnia błędnych odczytów - zakłada kompletność sekwencji x x x x slajd 41
42 Rekonstrukcja Uwzględnia błędy odczytu i nieznaną orientację. Definicja: f jest przybliżonym podciągiem S na poziomie błędu ε, gdy ds(f, S) ε f gdzie: ds odległość mierzona jako stopień niedopasowania (np. dopasowanie: 0, niedopasowanie: 1, przerwa: 1) Zadanie: Znaleźć najkrótsze możliwe słowo S, takie że dla każdego f P: ( ( )) min d s ( f, S ), d s f, S ε f slajd 42
43 Przykład Wejście: P = {ATCAT, GTCG, CGAG, TACCA} ε = 0.25 Wyjście: ATCAT ATGAT CGAC -CGAG ----TACCA ACGATACGAC GTCG ds(cgag, ACGATACGAC) = 1 = Odległość akceptowalna dla ε = 0.25 slajd 43
44 Przykład (2) Uwzględniane są również przerwy w odczytach. AT-GA----ATCGATAGAC ds = 1 slajd 44
45 Konkluzja Ten model jednak nadal ma wady: uwzględnia błędy odczytu i nieznaną orientację, ale: - nie uwzględnia powtórzeń - nie modeluje pokrycia - zawsze generuje pojedynczy kontig (nie uwzględnia luk w sekwencji) slajd 45
46 Problem SCS dla sekwencjonowania Wejście: Zbiór słów nad alfabetem {A,C,G,T} Wyjście: Najkrótsze wspólne nadsłowo (poszukiwana sekwencja?) Przykład: {ACGTAC, CATAC, TACAT} -> TACATACGTAC Problem najkrótszego wspólnego nadsłowa (SCS) można sprowadzić do problemu komiwojażera (TSP). slajd 46
47 Konstrukcja grafu dla TSP (overlap-layout-consensus) Przykład: {ACGTAC, CATAC, TACAT} - wierzchołki grafu reprezentują odczyty (reads) -krawędzie grafu opisują nałożenia (overlaps) -wagi to długości nakładających się prefiksów/sufiksów ACGTAC CATAC 3 3 Szukamy ścieżki Hamiltona o największym koszcie. TACAT Rozwiązaniem TSP jest SCS. slajd 47
48 Podejścia bazujące na teorii grafów 1. Overlap-layout-consensus J.D. Kececioglu and E.W. Myers, Combinatorial Algorithms for DNA Sequence Assembly, Algorithmica, vol. 13, 1995, pp wierzchołki reprezentują odczyty krawędzie reprezentują nałożenia (overlaps) - Szukamy takiej ścieżki, na której każdy wierzchołek występuje przynajmniej raz 2. Eulerian path P.A. Pevzner, H. Tang, and M.S. Waterman, An Eulerian Path Approach to DNA Fragment Assembly, Proc. Nat l Academy of Science USA, vol. 98, no. 17, 2001, pp Krawędzie reprezentują odczyty. Szukamy ścieżki Eulera (ścieżki przechodzącej przez wszystkie krawędzie grafu). slajd 48
49 Grafy de Brujina k ustalony parametr Wierzchołki to (k-1)-krotki. Krawędzie to k-krotki. Zbiór k-krotek nazywamy k-spektrum. Znalezienie najkrótszego słowa dla danego k-spektrum jest równoważne rozwiązaniu problemu chińskiego listonosza (Chinesse Postman Problem) { AGC,ATC,ATT,CAG, CAT,GCA,TCA,TTC} TT TC CA GC slajd 49 AT AG
50 Problem chińskiego listonosza Dana jest sieć ulic oraz poczta. Aby listonosz dostarczył korespondencję musi przejść wzdłuż każdej ulicy co najmniej raz i powrócić do punktu wyjścia. Rozwiązaniem problemu jest cykl Eulera. Jeśli graf nie jest Eulerowski, odpowiednio go przekształcamy: { AGC,ATC,ATT,CAG,CAT, GCA,TCA,TTC} TT TC AT CA GC slajd 50 AG

51 Graf de Brujina ze spacerami Problem: znaleźć ścieżkę zawierającą wszystkie szlaki odczytów. slajd 51
52 Niedoskonałości algorytmu Wady podejścia opartego na grafach De Bruijna: - arbitralny podział na k-krotki; - algorytm wrażliwy na błędy w sekwencjonowaniu - nieefektywny pamięciowo (jeden wierzchołek na każdą k-krotkę) slajd 52
53 Kontigi Czasem nie jesteśmy w stanie połączyć wszystkich fragmentów w jedną ciągłą sekwencję.? Nie wiemy jak długi jest brakujący fragment sekwencji. slajd 53 Nie wiemy jak uporządkować te kontigi.
54 Łączenie kontigów slajd 54
55 Łączenie kontigów (2) Znajdujemy linki między unikalnymi kontigami. Łączymy je ze sobą. slajd 55
56 Łączenie kontigów (3) Pozostałe luki wypełniamy powtarzającymi się sekwencjami. slajd 56
57 Etapy składania genomów Some Terminology 1. read Find overlapping reads a long word that comes out of sequencer mate pair a pair of reads from two ends 2. Merge pairs of reads of thesome same good insert fragment into longer contigs contig a contiguous sequence formed by several overlapping reads with no gaps 3. Link contigs to form supercontigs supercontig an ordered and oriented set (scaffold) of contigs, usually by mate pairs 4. consensus Derive consensus sequence sequence derived from the sequene multiple alignment of reads in a contig slajd 57..ACGATTACAATAGGTT..
58 Pokrycie Średnia liczba odczytów zawierających dany fragment DNA (oczywiście, dla poszczególnych odcinków może być ona mniejsza lub większa). inaczej łączny rozmiar sekwencji poddanych analizie w porównaniu do długości całego genomu. Lander & Waterman (1988) dla idealnego projektu (bez trudnych obszarów) pokrycie 8x-10x jest wystarczające do skompletowania genomu. // sekwencjonowanie shotgun W metodach nowej generacji, gdzie odczyty są znacznie krótsze, stosuje się większe pokrycie (np. 50x-100x) slajd 58
59 Dokańczanie - zamknięcie przerw pomiędzy kontigami; - korekcja niepoprawnych dopasowań; - powtórne sekwencjonowanie obszarów o niskim pokryciu lub niskiej jakości Zwykle jest to najbardziej czasochłonny etap prac. slajd 59
60 Metoda bazująca na grafach De Brujina podsumowanie slajd 60
61 Sekwencjonowanie podsumowanie Wejście: Duża liczba (miliony) krótkich (po kilkadziesiąt znaków) odczytów. Problemy: - nieznana orientacja (bezpośredni odczyt lub komplementarna odwrócona sekwencja) - błędy w danych wejściowych (nieprawidłowa, brak lub nadmiarowa litera) - powtórzenia (pokrywające się odczyty pochodzące z różnych miejsc genomu) - luki (nie wszystkie fragmenty są sekwencjonowane) slajd 61
62 Sekwencjonowanie podsumowanie Metody: - algorytmy słowne - podejścia grafowe: - overlap-layout-consensus (szukamy ścieżki Hamiltona) - grafy de Brujina (szukamy ścieżki Eulera) Etapy: - identyfikacja kontigów - łączenie kontigów - tworzenie konsensusu slajd 62
63 Narzędzia TIGR Assembler, G.G. Sutton et al., TIGR Assembler: A New Tool for Assembling Large Shotgun Sequencing Projects, Genome Science and Technology, 1995, vol. 1, pp Phrap, P. Green, Phrap Documentation: Algorithms, Phred/Phrap/Consed System Home Page; (current June 2002). CAP3 Euler Velvet slajd 63
64 Przewidywanie genów slajd 64
65 Przechowywanie danych slajd 65
66 Zagadnienia - Przechowywanie danych - Transmisja danych - Kompresja danych slajd 66
67 Częsta praktyka Laboratoria nie przechowują danych z sekwencjonowania, ale zamrożone próbki genu. Bardziej opłaca się ponownie zsekwencjonować daną próbkę, niż przechowywać surowe dane. slajd 67
68 Przesyłanie danych przykład z życia Laboratorium poprosiło pracowników z działu IT o zestawienie połączenia sftp, po których chcieli przesłać dane sekwencyjne do współpracującego laboratorium. IT zamiast FTP wystawił aplikację webową zdolną przesyłać maksymalnie 1 plik jednocześnie (pojedynczy eksperyment zawierał ponad plików). Ostatecznie udało się uzyskać sftp. Łącze szybko się zapchało, ponieważ allokowano tylko 120 GB transferu (roczne potrzeby: 1-2 TB). slajd 68
69 Źródło:
Sekwencjonowanie, przewidywanie genów
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Sekwencjonowanie, przewidywanie genów 1. Technologie sekwencjonowania Genomem nazywamy sekwencję
Reswkwencjonowanie vs asemblacja de novo
 ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
ALEKSANDRA ŚWIERCZ Reswkwencjonowanie vs asemblacja de novo Resekwencjonowanie to odtworzenie badanej sekwencji poprzez mapowanie odczytów do genomu/transkryptomu referencyjnego (tego samego gatunku lub
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej
 Bioinformatyka wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji genomów na trzech poziomach
Bioinformatyka wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji genomów na trzech poziomach
Algorytmy kombinatoryczne w bioinformatyce
 Algorytmy kombinatoryczne w bioinformatyce wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji
Algorytmy kombinatoryczne w bioinformatyce wykład 2: sekwencjonowanie cz. 1 prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Poznawanie sekwencji genomowej Poznawanie sekwencji
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA
 Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Metody odczytu kolejności nukleotydów - sekwencjonowania DNA 1. Metoda chemicznej degradacji DNA (metoda Maxama i Gilberta 1977) 2. Metoda terminacji syntezy łańcucha DNA - klasyczna metoda Sangera (Sanger
Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
Różnorodność osobników gatunku
 ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
ALEKSANDRA ŚWIERCZ Różnorodność osobników gatunku Single Nucleotide Polymorphism (SNP) Różnica na jednej pozycji, małe delecje, insercje (INDELs) SNP pojawia się ~1/1000 pozycji Można je znaleźć porównując
Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji
 Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu. Modele grafowe problemu sekwencjonowania
Grafy i sieci wybrane zagadnienia wykład 2: modele służące rekonstrukcji sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Plan wykładu. Modele grafowe problemu sekwencjonowania
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE
 PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
PODSTAWY BIOINFORMATYKI 12 MIKROMACIERZE WSTĘP 1. Mikromacierze ekspresyjne tworzenie macierzy przykłady zastosowań 2. Mikromacierze SNP tworzenie macierzy przykłady zastosowań MIKROMACIERZE EKSPRESYJNE
Suma dwóch grafów. Zespolenie dwóch grafów
 Suma dwóch grafów G 1 = ((G 1 ), E(G 1 )) G 2 = ((G 2 ), E(G 2 )) (G 1 ) i (G 2 ) rozłączne Suma G 1 G 2 graf ze zbiorem wierzchołków (G 1 ) (G 2 ) i rodziną krawędzi E(G 1 ) E(G 2 ) G 1 G 2 G 1 G 2 Zespolenie
Suma dwóch grafów G 1 = ((G 1 ), E(G 1 )) G 2 = ((G 2 ), E(G 2 )) (G 1 ) i (G 2 ) rozłączne Suma G 1 G 2 graf ze zbiorem wierzchołków (G 1 ) (G 2 ) i rodziną krawędzi E(G 1 ) E(G 2 ) G 1 G 2 G 1 G 2 Zespolenie
Matematyczne Podstawy Informatyki
 Matematyczne Podstawy Informatyki dr inż. Andrzej Grosser Instytut Informatyki Teoretycznej i Stosowanej Politechnika Częstochowska Rok akademicki 03/0 Przeszukiwanie w głąb i wszerz I Przeszukiwanie metodą
Matematyczne Podstawy Informatyki dr inż. Andrzej Grosser Instytut Informatyki Teoretycznej i Stosowanej Politechnika Częstochowska Rok akademicki 03/0 Przeszukiwanie w głąb i wszerz I Przeszukiwanie metodą
Przetarg nieograniczony na zakup specjalistycznej aparatury laboratoryjnej Znak sprawy: DZ-2501/6/17
 Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej
 Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej rozdzielczości jest sekwencja nukleotydowa -mapowanie fizyczne genomu
Mapowanie fizyczne genomów -konstrukcja map wyskalowanych w jednostkach fizycznych -najdokładniejszą mapą fizyczną genomu, o największej rozdzielczości jest sekwencja nukleotydowa -mapowanie fizyczne genomu
Algorytmika Problemów Trudnych
 Algorytmika Problemów Trudnych Wykład 9 Tomasz Krawczyk krawczyk@tcs.uj.edu.pl Kraków, semestr letni 2016/17 plan wykładu Algorytmy aproksymacyjne: Pojęcie algorytmu aproksymacyjnego i współczynnika aproksymowalności.
Algorytmika Problemów Trudnych Wykład 9 Tomasz Krawczyk krawczyk@tcs.uj.edu.pl Kraków, semestr letni 2016/17 plan wykładu Algorytmy aproksymacyjne: Pojęcie algorytmu aproksymacyjnego i współczynnika aproksymowalności.
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Ekologia molekularna. wykład 11
 Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
Ekologia molekularna wykład 11 Sekwencjonowanie nowej generacji NGS = next generation sequencing = high throughput sequencing = massive pararell sequencing =... Różne techniki i platformy Illumina (MiSeq,
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 ANALIZA DANYCH NGS SEKWENCJONOWANIE GENOMÓW NEXT GENERATION METODA NOWEJ GENERACJI Sekwencjonowanie bardzo krótkich fragmentów 50-700 bp DNA unieruchomione na płytce Szybkie
Teoria grafów - Teoria rewersali - Teoria śladów
 17 maja 2012 1 Planarność Wzór Eulera Kryterium Kuratowskiego Algorytmy testujące planarność 2 Genom i jego przekształcenia Grafy złamań Sortowanie przez odwrócenia Inne rodzaje sortowania Algorytmy sortujące
17 maja 2012 1 Planarność Wzór Eulera Kryterium Kuratowskiego Algorytmy testujące planarność 2 Genom i jego przekształcenia Grafy złamań Sortowanie przez odwrócenia Inne rodzaje sortowania Algorytmy sortujące
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Wykład 4. Droga i cykl Eulera i Hamiltona
 Wykład 4. i Hamiltona Wykład 4. i Hamiltona 1 / 35 Grafy Eulera Niech G będzie grafem spójnym. Definicja Jeżeli w grafie G istnieje zamknięta droga prosta zawierająca wszystkie krawędzie grafu, to taką
Wykład 4. i Hamiltona Wykład 4. i Hamiltona 1 / 35 Grafy Eulera Niech G będzie grafem spójnym. Definicja Jeżeli w grafie G istnieje zamknięta droga prosta zawierająca wszystkie krawędzie grafu, to taką
Wstęp do programowania
 Wstęp do programowania Algorytmy zachłanne, programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2014 P. Daniluk(Wydział Fizyki) WP w. IX Jesień 2014 1 / 26 Algorytmy zachłanne Strategia polegająca
Wstęp do programowania Algorytmy zachłanne, programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2014 P. Daniluk(Wydział Fizyki) WP w. IX Jesień 2014 1 / 26 Algorytmy zachłanne Strategia polegająca
Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
 Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Droga i cykl Eulera Przykłady zastosowania drogi i cyku Eulera Droga i cykl Hamiltona. Wykład 4. Droga i cykl Eulera i Hamiltona
 Wykład 4. Droga i cykl Eulera i Hamiltona 1 / 92 Grafy Eulera Droga i cykl Eulera Niech G będzie grafem spójnym. Definicja Jeżeli w grafie G istnieje zamknięta droga prosta zawierająca wszystkie krawędzie
Wykład 4. Droga i cykl Eulera i Hamiltona 1 / 92 Grafy Eulera Droga i cykl Eulera Niech G będzie grafem spójnym. Definicja Jeżeli w grafie G istnieje zamknięta droga prosta zawierająca wszystkie krawędzie
Wstęp. Jak programować w DNA? Idea oraz przykład. Problem FSAT charakterystyka i rozwiązanie za pomocą DNA. Jak w ogólności rozwiązywać problemy
 Ariel Zakrzewski Wstęp. Jak programować w DNA? Idea oraz przykład. Problem FSAT charakterystyka i rozwiązanie za pomocą DNA. Jak w ogólności rozwiązywać problemy matematyczne z użyciem DNA? Gdzie są problemy?
Ariel Zakrzewski Wstęp. Jak programować w DNA? Idea oraz przykład. Problem FSAT charakterystyka i rozwiązanie za pomocą DNA. Jak w ogólności rozwiązywać problemy matematyczne z użyciem DNA? Gdzie są problemy?
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:
 w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu:") Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
Przydatność technologii Sekwencjonowania Nowej Generacji (NGS) w kolekcjach Banków Genów Joanna Noceń Kinga Smolińska Marta Puchta Kierownik tematu: prof. dr hab. Jerzy H. Czembor SEKWENCJONOWANIE I generacji
PODSTAWY BIOINFORMATYKI 3 SEKWENCJONOWANIE GENOMÓW I
 PODSTAWY BIOINFORMATYKI 3 SEKWENCJONOWANIE GENOMÓW I PROJEKTY POZNANIA INNYCH GENOMÓW 1 400 2009 2010 1 200 2011 2012 1 000 800 600 400 200 986 1153 1285 914 954 717 759 757 251 254 889 0 23 ukończone
PODSTAWY BIOINFORMATYKI 3 SEKWENCJONOWANIE GENOMÓW I PROJEKTY POZNANIA INNYCH GENOMÓW 1 400 2009 2010 1 200 2011 2012 1 000 800 600 400 200 986 1153 1285 914 954 717 759 757 251 254 889 0 23 ukończone
PODSTAWY BIOINFORMATYKI 2 SEKWENCJONOWANIE GENOMÓW
 PODSTAWY BIOINFORMATYKI 2 SEKWENCJONOWANIE GENOMÓW PROJEKTY POZNANIA INNYCH GENOMÓW 300 250 Seria 1 251 254 200 150 100 50 0 23 ukończone w trakcie scalania w trakcie realizacji SEKWENCJONOWANIE GENOMÓW
PODSTAWY BIOINFORMATYKI 2 SEKWENCJONOWANIE GENOMÓW PROJEKTY POZNANIA INNYCH GENOMÓW 300 250 Seria 1 251 254 200 150 100 50 0 23 ukończone w trakcie scalania w trakcie realizacji SEKWENCJONOWANIE GENOMÓW
Metody analizy genomu
 Metody analizy genomu 1. Mapowanie restrykcyjne. 2. Sondy do rozpoznawania DNA 3. FISH 4. Odczytanie sekwencji DNA 5. Interpretacja sekwencji DNA genomu 6. Transkryptom 7. Proteom 1. Mapy restrykcyjne
Metody analizy genomu 1. Mapowanie restrykcyjne. 2. Sondy do rozpoznawania DNA 3. FISH 4. Odczytanie sekwencji DNA 5. Interpretacja sekwencji DNA genomu 6. Transkryptom 7. Proteom 1. Mapy restrykcyjne
Data Mining Wykład 9. Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster. Plan wykładu. Sformułowanie problemu
 Grupowanie hierarchiczne O-Cluster. Plan wykładu. Sformułowanie problemu") Data Mining Wykład 9 Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster Plan wykładu Wprowadzanie Definicja problemu Klasyfikacja metod grupowania Grupowanie hierarchiczne Sformułowanie problemu
Data Mining Wykład 9 Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster Plan wykładu Wprowadzanie Definicja problemu Klasyfikacja metod grupowania Grupowanie hierarchiczne Sformułowanie problemu
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Analiza stanów gry na potrzeby UCT w DVRP
 Analiza stanów gry na potrzeby UCT w DVRP Seminarium IO na MiNI 04.11.2014 Michał Okulewicz based on the decision DEC-2012/07/B/ST6/01527 Plan prezentacji Definicja problemu DVRP DVRP na potrzeby UCB Analiza
Analiza stanów gry na potrzeby UCT w DVRP Seminarium IO na MiNI 04.11.2014 Michał Okulewicz based on the decision DEC-2012/07/B/ST6/01527 Plan prezentacji Definicja problemu DVRP DVRP na potrzeby UCB Analiza
Matematyka dyskretna
 Matematyka dyskretna Wykład 13: Teoria Grafów Gniewomir Sarbicki Literatura R.J. Wilson Wprowadzenie do teorii grafów Definicja: Grafem (skończonym, nieskierowanym) G nazywamy parę zbiorów (V (G), E(G)),
Matematyka dyskretna Wykład 13: Teoria Grafów Gniewomir Sarbicki Literatura R.J. Wilson Wprowadzenie do teorii grafów Definicja: Grafem (skończonym, nieskierowanym) G nazywamy parę zbiorów (V (G), E(G)),
PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE
 D: PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE I. Strategia zachłanna II. Problem przetasowań w genomie III. Sortowanie przez odwrócenia IV. Algorytmy przybliżone V. Algorytm zachłanny
D: PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE I. Strategia zachłanna II. Problem przetasowań w genomie III. Sortowanie przez odwrócenia IV. Algorytmy przybliżone V. Algorytm zachłanny
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing. Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 6 Część 1 NGS - wstęp Dr Wioleta Drobik-Czwarno Macierze tkankowe TMA ang. Tissue microarray Technika opisana w 1987 roku (Wan i
Struktury danych i złożoność obliczeniowa Wykład 7. Prof. dr hab. inż. Jan Magott
 Struktury danych i złożoność obliczeniowa Wykład 7 Prof. dr hab. inż. Jan Magott Problemy NP-zupełne Transformacją wielomianową problemu π 2 do problemu π 1 (π 2 π 1 ) jest funkcja f: D π2 D π1 spełniająca
Struktury danych i złożoność obliczeniowa Wykład 7 Prof. dr hab. inż. Jan Magott Problemy NP-zupełne Transformacją wielomianową problemu π 2 do problemu π 1 (π 2 π 1 ) jest funkcja f: D π2 D π1 spełniająca
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
"Zapisane w genach, czyli Python a tajemnice naszego genomu."
 "Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
"Zapisane w genach, czyli Python a tajemnice naszego genomu." Dr Kaja Milanowska Instytut Biologii Molekularnej i Biotechnologii UAM VitaInSilica sp. z o.o. Warszawa, 9 lutego 2015 Dane biomedyczne 1)
Opracowanie prof. J. Domsta 1
 Opracowanie prof. J. Domsta 1 Algorytm FLEURY'ego: Twierdzenie 6.5 G-graf eulerowski. Wtedy cykl Eulera otrzymujemy nastepująco: a) Start w dowolnym wierzchołku b) Krawędzie w dowolnej kolejności po przebyciu
Opracowanie prof. J. Domsta 1 Algorytm FLEURY'ego: Twierdzenie 6.5 G-graf eulerowski. Wtedy cykl Eulera otrzymujemy nastepująco: a) Start w dowolnym wierzchołku b) Krawędzie w dowolnej kolejności po przebyciu
PODSTAWY BIOINFORMATYKI WYKŁAD 2 SEKWENCJONOWANIE GENOMÓW
 PODSTAWY BIOINFORMATYKI WYKŁAD 2 SEKWENCJONOWANIE GENOMÓW SEKWENCJONOWANIE GENOMÓW 1. Sekwencjonowanie genomów 2. Automatyzacja sekwencjonowania 3. 1 000 (human) Genomes project 4. 1 000 Bull Genomes project
PODSTAWY BIOINFORMATYKI WYKŁAD 2 SEKWENCJONOWANIE GENOMÓW SEKWENCJONOWANIE GENOMÓW 1. Sekwencjonowanie genomów 2. Automatyzacja sekwencjonowania 3. 1 000 (human) Genomes project 4. 1 000 Bull Genomes project
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA. Marta Szachniuk
 Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Substancje stosowane do osadzania enzymu na stałym podłożu Biotyna (witamina H, witamina B 7 ) Tworzenie aktywnej powierzchni biosensorów
 Tworzenie aktywnej powierzchni biosensorów") SEKWECJWAIE ASTĘPEJ GEERACJI- GS Losowa fragmentacja nici DA Dołączanie odpowiednich linkerów (konstrukcja biblioteki) Amplifikacja biblioteki na podłożu szklanym lub plastikowym biosensory Wielokrotnie
SEKWECJWAIE ASTĘPEJ GEERACJI- GS Losowa fragmentacja nici DA Dołączanie odpowiednich linkerów (konstrukcja biblioteki) Amplifikacja biblioteki na podłożu szklanym lub plastikowym biosensory Wielokrotnie
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Przybliżone algorytmy analizy ekspresji genów.
 Przybliżone algorytmy analizy ekspresji genów. Opracowanie i implementacja algorytmu analizy danych uzyskanych z eksperymentu biologicznego. 20.06.04 Seminarium - SKISR 1 Wstęp. Dane wejściowe dla programu
Przybliżone algorytmy analizy ekspresji genów. Opracowanie i implementacja algorytmu analizy danych uzyskanych z eksperymentu biologicznego. 20.06.04 Seminarium - SKISR 1 Wstęp. Dane wejściowe dla programu
Programowanie dynamiczne cz. 2
 Programowanie dynamiczne cz. 2 Wykład 7 16 kwietnia 2019 (Wykład 7) Programowanie dynamiczne cz. 2 16 kwietnia 2019 1 / 19 Outline 1 Mnożenie ciągu macierzy Konstruowanie optymalnego rozwiązania 2 Podstawy
Programowanie dynamiczne cz. 2 Wykład 7 16 kwietnia 2019 (Wykład 7) Programowanie dynamiczne cz. 2 16 kwietnia 2019 1 / 19 Outline 1 Mnożenie ciągu macierzy Konstruowanie optymalnego rozwiązania 2 Podstawy
PODSTAWY BIOINFORMATYKI
 PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
Wykład 5 Dopasowywanie lokalne
 Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Metody Programowania
 POLITECHNIKA KRAKOWSKA - WIEiK KATEDRA AUTOMATYKI i TECHNIK INFORMACYJNYCH Metody Programowania www.pk.edu.pl/~zk/mp_hp.html Wykładowca: dr inż. Zbigniew Kokosiński zk@pk.edu.pl Wykład 8: Wyszukiwanie
POLITECHNIKA KRAKOWSKA - WIEiK KATEDRA AUTOMATYKI i TECHNIK INFORMACYJNYCH Metody Programowania www.pk.edu.pl/~zk/mp_hp.html Wykładowca: dr inż. Zbigniew Kokosiński zk@pk.edu.pl Wykład 8: Wyszukiwanie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Algorytmy wyznaczania centralności w sieci Szymon Szylko
 Algorytmy wyznaczania centralności w sieci Szymon Szylko Zakład systemów Informacyjnych Wrocław 10.01.2008 Agenda prezentacji Cechy sieci Algorytmy grafowe Badanie centralności Algorytmy wyznaczania centralności
Algorytmy wyznaczania centralności w sieci Szymon Szylko Zakład systemów Informacyjnych Wrocław 10.01.2008 Agenda prezentacji Cechy sieci Algorytmy grafowe Badanie centralności Algorytmy wyznaczania centralności
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
xx + x = 1, to y = Jeśli x = 0, to y = 0 Przykładowy układ Funkcja przykładowego układu Metody poszukiwania testów Porównanie tabel prawdy
 Testowanie układów kombinacyjnych Przykładowy układ Wykrywanie błędów: 1. Sklejenie z 0 2. Sklejenie z 1 Testem danego uszkodzenia nazywa się takie wzbudzenie funkcji (wektor wejściowy), które daje błędną
Testowanie układów kombinacyjnych Przykładowy układ Wykrywanie błędów: 1. Sklejenie z 0 2. Sklejenie z 1 Testem danego uszkodzenia nazywa się takie wzbudzenie funkcji (wektor wejściowy), które daje błędną
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Równoległy algorytm wyznaczania bloków dla cyklicznego problemu przepływowego z przezbrojeniami
 Równoległy algorytm wyznaczania bloków dla cyklicznego problemu przepływowego z przezbrojeniami dr inż. Mariusz Uchroński Wrocławskie Centrum Sieciowo-Superkomputerowe Agenda Cykliczny problem przepływowy
Równoległy algorytm wyznaczania bloków dla cyklicznego problemu przepływowego z przezbrojeniami dr inż. Mariusz Uchroński Wrocławskie Centrum Sieciowo-Superkomputerowe Agenda Cykliczny problem przepływowy
Algorytmy Równoległe i Rozproszone Część X - Algorytmy samostabilizujące.
 Algorytmy Równoległe i Rozproszone Część X - Algorytmy samostabilizujące. Łukasz Kuszner pokój 209, WETI http://www.sphere.pl/ kuszner/ kuszner@sphere.pl Oficjalna strona wykładu http://www.sphere.pl/
Algorytmy Równoległe i Rozproszone Część X - Algorytmy samostabilizujące. Łukasz Kuszner pokój 209, WETI http://www.sphere.pl/ kuszner/ kuszner@sphere.pl Oficjalna strona wykładu http://www.sphere.pl/
EGZAMIN - Wersja A. ALGORYTMY I STRUKTURY DANYCH Lisek89 opracowanie kartki od Pani dr E. Koszelew
 1. ( pkt) Dany jest algorytm, który dla dowolnej liczby naturalnej n, powinien wyznaczyd sumę kolejnych liczb naturalnych mniejszych od n. Wynik algorytmu jest zapisany w zmiennej suma. Algorytm i=1; suma=0;
1. ( pkt) Dany jest algorytm, który dla dowolnej liczby naturalnej n, powinien wyznaczyd sumę kolejnych liczb naturalnych mniejszych od n. Wynik algorytmu jest zapisany w zmiennej suma. Algorytm i=1; suma=0;
Wstęp do programowania
 Wstęp do programowania Programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. X Jesień 2013 1 / 21 Dziel i zwyciężaj przypomnienie 1 Podział problemu na 2 lub
Wstęp do programowania Programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. X Jesień 2013 1 / 21 Dziel i zwyciężaj przypomnienie 1 Podział problemu na 2 lub
Drzewa spinające MST dla grafów ważonych Maksymalne drzewo spinające Drzewo Steinera. Wykład 6. Drzewa cz. II
 Wykład 6. Drzewa cz. II 1 / 65 drzewa spinające Drzewa spinające Zliczanie drzew spinających Drzewo T nazywamy drzewem rozpinającym (spinającym) (lub dendrytem) spójnego grafu G, jeżeli jest podgrafem
Wykład 6. Drzewa cz. II 1 / 65 drzewa spinające Drzewa spinające Zliczanie drzew spinających Drzewo T nazywamy drzewem rozpinającym (spinającym) (lub dendrytem) spójnego grafu G, jeżeli jest podgrafem
Elementy teorii grafów Elementy teorii grafów
 Spis tresci 1 Spis tresci 1 Często w zagadnieniach praktycznych rozważa się pewien zbiór obiektów wraz z zależnościami jakie łączą te obiekty. Dla przykładu można badać pewną grupę ludzi oraz strukturę
Spis tresci 1 Spis tresci 1 Często w zagadnieniach praktycznych rozważa się pewien zbiór obiektów wraz z zależnościami jakie łączą te obiekty. Dla przykładu można badać pewną grupę ludzi oraz strukturę
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Wstęp do programowania
 Wstęp do programowania Stosy, kolejki, drzewa Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. VII Jesień 2013 1 / 25 Listy Lista jest uporządkowanym zbiorem elementów. W Pythonie
Wstęp do programowania Stosy, kolejki, drzewa Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. VII Jesień 2013 1 / 25 Listy Lista jest uporządkowanym zbiorem elementów. W Pythonie
Teoria grafów dla małolatów. Andrzej Przemysław Urbański Instytut Informatyki Politechnika Poznańska
 Teoria grafów dla małolatów Andrzej Przemysław Urbański Instytut Informatyki Politechnika Poznańska Wstęp Matematyka to wiele różnych dyscyplin Bowiem świat jest bardzo skomplikowany wymaga rozważenia
Teoria grafów dla małolatów Andrzej Przemysław Urbański Instytut Informatyki Politechnika Poznańska Wstęp Matematyka to wiele różnych dyscyplin Bowiem świat jest bardzo skomplikowany wymaga rozważenia
GRADIENT TEMPERATUR TOUCH DOWN PCR. Standardowy PCR RAPD- PCR. RealTime- PCR. Nested- PCR. Digital- PCR.
 Standardowy PCR RAPD- PCR Nested- PCR Multipleks- PCR Allelospecyficzny PCR Touchdown- PCR RealTime- PCR Digital- PCR In situ (slide)- PCR Metylacyjno specyficzny- PCR Assembly- PCR Inverse- PCR GRADIENT
Standardowy PCR RAPD- PCR Nested- PCR Multipleks- PCR Allelospecyficzny PCR Touchdown- PCR RealTime- PCR Digital- PCR In situ (slide)- PCR Metylacyjno specyficzny- PCR Assembly- PCR Inverse- PCR GRADIENT
Algorytmy aproksymacyjne i parametryzowane
 Algorytmy aproksymacyjne i parametryzowane Marek Cygan Uniwersytet Warszawski 18 października 2012 Marek Cygan Algorytmy aproksymacyjne i parametryzowane 1/22 Wstęp W algorytmice problemy dzielimy na obliczeniowo
Algorytmy aproksymacyjne i parametryzowane Marek Cygan Uniwersytet Warszawski 18 października 2012 Marek Cygan Algorytmy aproksymacyjne i parametryzowane 1/22 Wstęp W algorytmice problemy dzielimy na obliczeniowo
Czy istnieje zamknięta droga spaceru przechodząca przez wszystkie mosty w Królewcu dokładnie jeden raz?
 DROGI i CYKLE EULERA w grafach Czy istnieje zamknięta droga spaceru przechodząca przez wszystkie mosty w Królewcu dokładnie jeden raz? Czy można narysować podaną figurę nie odrywając ołówka od papieru
DROGI i CYKLE EULERA w grafach Czy istnieje zamknięta droga spaceru przechodząca przez wszystkie mosty w Królewcu dokładnie jeden raz? Czy można narysować podaną figurę nie odrywając ołówka od papieru
Grafy dla każdego. dr Krzysztof Bryś. Wydział Matematyki i Nauk Informacyjnych Politechnika Warszawska.
 Grafy dla każdego dr Krzysztof Bryś brys@mini.pw.edu.pl Wydział Matematyki i Nauk Informacyjnych Politechnika Warszawska www.mini.pw.edu.pl Warszawa, 28 marca 2015 Graf składa się z elementów pewnego zbioru
Grafy dla każdego dr Krzysztof Bryś brys@mini.pw.edu.pl Wydział Matematyki i Nauk Informacyjnych Politechnika Warszawska www.mini.pw.edu.pl Warszawa, 28 marca 2015 Graf składa się z elementów pewnego zbioru
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów:
 Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Powodzenie reakcji PCR wymaga właściwego doboru szeregu parametrów: dobór warunków samej reakcji PCR (temperatury, czas trwania cykli, ilości cykli itp.) dobór odpowiednich starterów do reakcji amplifikacji
Glimmer umożliwia znalezienie regionów kodujących
 Narzędzia ułatwiające identyfikację właściwych genów GLIMMER TaxPlot Narzędzia ułatwiające amplifikację tych genów techniki PCR Primer3, Primer3plus PrimerBLAST Reverse Complement Narzędzia ułatwiające
Narzędzia ułatwiające identyfikację właściwych genów GLIMMER TaxPlot Narzędzia ułatwiające amplifikację tych genów techniki PCR Primer3, Primer3plus PrimerBLAST Reverse Complement Narzędzia ułatwiające
Podstawy techniki cyfrowej. Układy asynchroniczne Opracował: R.Walkowiak Styczeń 2014
 Podstawy techniki cyfrowej Układy asynchroniczne Opracował: R.Walkowiak Styczeń 2014 Charakterystyka układów asynchronicznych Brak wejścia: zegarowego, synchronizującego. Natychmiastowa (niesynchronizowana)
Podstawy techniki cyfrowej Układy asynchroniczne Opracował: R.Walkowiak Styczeń 2014 Charakterystyka układów asynchronicznych Brak wejścia: zegarowego, synchronizującego. Natychmiastowa (niesynchronizowana)
XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery
 http://xqtav.sourceforge.net XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery dr hab. Jerzy Tyszkiewicz dr Andrzej Kierzek mgr Jacek Sroka Grzegorz Kaczor praca mgr pod
http://xqtav.sourceforge.net XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery dr hab. Jerzy Tyszkiewicz dr Andrzej Kierzek mgr Jacek Sroka Grzegorz Kaczor praca mgr pod
Algorytmy Równoległe i Rozproszone Część V - Model PRAM II
 Algorytmy Równoległe i Rozproszone Część V - Model PRAM II Łukasz Kuszner pokój 209, WETI http://www.sphere.pl/ kuszner/ kuszner@sphere.pl Oficjalna strona wykładu http://www.sphere.pl/ kuszner/arir/ 2005/06
Algorytmy Równoległe i Rozproszone Część V - Model PRAM II Łukasz Kuszner pokój 209, WETI http://www.sphere.pl/ kuszner/ kuszner@sphere.pl Oficjalna strona wykładu http://www.sphere.pl/ kuszner/arir/ 2005/06
Analizy wielkoskalowe w badaniach chromatyny
 Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Analizy wielkoskalowe w badaniach chromatyny Analizy wielkoskalowe wykorzystujące mikromacierze DNA Genotypowanie: zróżnicowane wewnątrz genów RNA Komórka eukariotyczna Ekspresja genów: Które geny? Poziom
Bioinformatyczna analiza danych. Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt
 Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
Bioinformatyczna analiza danych Wykład 1 Dr Wioleta Drobik-Czwarno Katedra Genetyki i Ogólnej Hodowli Zwierząt Sprawy organizacyjne Prowadzący przedmiot: Dr Wioleta Drobik-Czwarno koordynator przedmiotu,
UNIKANIE IMPASÓW W SYSTEMACH PROCESÓW WSPÓŁBIEŻNYCH
 UNIKANIE IMPASÓW W SYSTEMACH PROCESÓW WSPÓŁBIEŻNYCH Robert Wójcik Instytut Cybernetyki Technicznej Politechniki Wrocławskiej 1. Impasy w systemach procesów współbieżnych 2. Klasyczne algorytmy unikania
UNIKANIE IMPASÓW W SYSTEMACH PROCESÓW WSPÓŁBIEŻNYCH Robert Wójcik Instytut Cybernetyki Technicznej Politechniki Wrocławskiej 1. Impasy w systemach procesów współbieżnych 2. Klasyczne algorytmy unikania
Historia Bioinformatyki
 Historia Bioinformatyki 1859 Darwin i Wallace opublikowali O powstaniu gatunku 1865 Mendel eksperymentując z grochem, wykazuje, że cechy dziedziczą się w odrębnych jednostkach 1869 Meischer wyizolował
Historia Bioinformatyki 1859 Darwin i Wallace opublikowali O powstaniu gatunku 1865 Mendel eksperymentując z grochem, wykazuje, że cechy dziedziczą się w odrębnych jednostkach 1869 Meischer wyizolował
Algorytmy i Struktury Danych
 Algorytmy i Struktury Danych Podstawowe informacje Prowadzący: Jan Tuziemski Email: jan.tuziemski@pg.edu.pl Konsultacje: pokój 412 GB (do ustalenia 412 GB) Podstawowe informacje literatura K. Goczyła Struktury
Algorytmy i Struktury Danych Podstawowe informacje Prowadzący: Jan Tuziemski Email: jan.tuziemski@pg.edu.pl Konsultacje: pokój 412 GB (do ustalenia 412 GB) Podstawowe informacje literatura K. Goczyła Struktury
Przykłady grafów. Graf prosty, to graf bez pętli i bez krawędzi wielokrotnych.
 Grafy Graf Graf (ang. graph) to zbiór wierzchołków (ang. vertices), które mogą być połączone krawędziami (ang. edges) w taki sposób, że każda krawędź kończy się i zaczyna w którymś z wierzchołków. Graf
Grafy Graf Graf (ang. graph) to zbiór wierzchołków (ang. vertices), które mogą być połączone krawędziami (ang. edges) w taki sposób, że każda krawędź kończy się i zaczyna w którymś z wierzchołków. Graf
Algorytmy stochastyczne, wykład 08 Sieci bayesowskie
 Algorytmy stochastyczne, wykład 08 Jarosław Piersa Wydział Matematyki i Informatyki, Uniwersytet Mikołaja Kopernika 2014-04-10 Prawdopodobieństwo Prawdopodobieństwo Prawdopodobieństwo warunkowe Zmienne
Algorytmy stochastyczne, wykład 08 Jarosław Piersa Wydział Matematyki i Informatyki, Uniwersytet Mikołaja Kopernika 2014-04-10 Prawdopodobieństwo Prawdopodobieństwo Prawdopodobieństwo warunkowe Zmienne
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: dodany punkt na temat testów do sprawozdania. Biologia, bioinformatyka:
 Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,
Zofia Kruczkiewicz, Algorytmu i struktury danych, Wykład 14, 1
 Wykład Algorytmy grafowe metoda zachłanna. Właściwości algorytmu zachłannego:. W przeciwieństwie do metody programowania dynamicznego nie występuje etap dzielenia na mniejsze realizacje z wykorzystaniem
Wykład Algorytmy grafowe metoda zachłanna. Właściwości algorytmu zachłannego:. W przeciwieństwie do metody programowania dynamicznego nie występuje etap dzielenia na mniejsze realizacje z wykorzystaniem
Routing. mgr inż. Krzysztof Szałajko
 Routing mgr inż. Krzysztof Szałajko Modele odniesienia 7 Aplikacji 6 Prezentacji 5 Sesji 4 Transportowa 3 Sieciowa 2 Łącza danych 1 Fizyczna Aplikacji Transportowa Internetowa Dostępu do sieci Wersja 1.0
Routing mgr inż. Krzysztof Szałajko Modele odniesienia 7 Aplikacji 6 Prezentacji 5 Sesji 4 Transportowa 3 Sieciowa 2 Łącza danych 1 Fizyczna Aplikacji Transportowa Internetowa Dostępu do sieci Wersja 1.0
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
PROBLEM: SEKWENCJONOWANIE DNA METODA: ALGORYTMY GRAFOWE
 F : PROBLEM: SEKWENCJONOWANIE DNA METODA: ALGORYTMY GRAFOWE I. Grafy i genetyka II. Sekwencjonowanie DNA III. Macierze DNA IV. Sekwencjonowanie przez hybrydyzacje DNA V. Sekwencjonowanie i identyfikacja
F : PROBLEM: SEKWENCJONOWANIE DNA METODA: ALGORYTMY GRAFOWE I. Grafy i genetyka II. Sekwencjonowanie DNA III. Macierze DNA IV. Sekwencjonowanie przez hybrydyzacje DNA V. Sekwencjonowanie i identyfikacja
Kalibracja. W obu przypadkach jeśli mamy dane, to możemy znaleźć równowagę: Konwesatorium z Ekonometrii, IV rok, WNE UW 1
 Kalibracja Kalibracja - nazwa pochodzi z nauk ścisłych - kalibrowanie instrumentu oznacza wyznaczanie jego skali (np. kalibrowanie termometru polega na wyznaczeniu 0C i 100C tak by oznaczały punkt zamarzania
Kalibracja Kalibracja - nazwa pochodzi z nauk ścisłych - kalibrowanie instrumentu oznacza wyznaczanie jego skali (np. kalibrowanie termometru polega na wyznaczeniu 0C i 100C tak by oznaczały punkt zamarzania
Klonowanie molekularne Kurs doskonalący. Zakład Geriatrii i Gerontologii CMKP
 Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Klonowanie molekularne Kurs doskonalący Zakład Geriatrii i Gerontologii CMKP Etapy klonowania molekularnego 1. Wybór wektora i organizmu gospodarza Po co klonuję (do namnożenia DNA [czy ma być metylowane
Biologia medyczna, materiały dla studentów
 Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
Zasada reakcji PCR Reakcja PCR (replikacja in vitro) obejmuje denaturację DNA, przyłączanie starterów (annealing) i syntezę nowych nici DNA (elongacja). 1. Denaturacja: rozplecenie nici DNA, temp. 94 o
Programowanie dynamiczne
 Programowanie dynamiczne Patryk Żywica 5 maja 2008 1 Spis treści 1 Problem wydawania reszty 3 1.1 Sformułowanie problemu...................... 3 1.2 Algorytm.............................. 3 1.2.1 Prosty
Programowanie dynamiczne Patryk Żywica 5 maja 2008 1 Spis treści 1 Problem wydawania reszty 3 1.1 Sformułowanie problemu...................... 3 1.2 Algorytm.............................. 3 1.2.1 Prosty
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Drzewa. Jeżeli graf G jest lasem, który ma n wierzchołków i k składowych, to G ma n k krawędzi. Własności drzew
 Drzewa Las - graf, który nie zawiera cykli Drzewo - las spójny Jeżeli graf G jest lasem, który ma n wierzchołków i k składowych, to G ma n k krawędzi. Własności drzew Niech T graf o n wierzchołkach będący
Drzewa Las - graf, który nie zawiera cykli Drzewo - las spójny Jeżeli graf G jest lasem, który ma n wierzchołków i k składowych, to G ma n k krawędzi. Własności drzew Niech T graf o n wierzchołkach będący
TEORETYCZNE PODSTAWY INFORMATYKI
 1 TEORETYCZNE PODSTAWY INFORMATYKI WFAiS UJ, Informatyka Stosowana I rok studiów, I stopień Wykład 14c 2 Definicje indukcyjne Twierdzenia dowodzone przez indukcje Definicje indukcyjne Definicja drzewa
1 TEORETYCZNE PODSTAWY INFORMATYKI WFAiS UJ, Informatyka Stosowana I rok studiów, I stopień Wykład 14c 2 Definicje indukcyjne Twierdzenia dowodzone przez indukcje Definicje indukcyjne Definicja drzewa
Porównanie czasów działania algorytmów sortowania przez wstawianie i scalanie
 Więcej o sprawności algorytmów Porównanie czasów działania algorytmów sortowania przez wstawianie i scalanie Załóżmy, że możemy wykonać dane zadanie przy użyciu dwóch algorytmów: jednego o złożoności czasowej
Więcej o sprawności algorytmów Porównanie czasów działania algorytmów sortowania przez wstawianie i scalanie Załóżmy, że możemy wykonać dane zadanie przy użyciu dwóch algorytmów: jednego o złożoności czasowej
Aproksymacja funkcji a regresja symboliczna
 Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
Kodowanie i kompresja Streszczenie Studia dzienne Wykład 9,
 1 Kody Tunstalla Kodowanie i kompresja Streszczenie Studia dzienne Wykład 9, 14.04.2005 Inne podejście: słowa kodowe mają ustaloną długość, lecz mogą kodować ciągi liter z alfabetu wejściowego o różnej
1 Kody Tunstalla Kodowanie i kompresja Streszczenie Studia dzienne Wykład 9, 14.04.2005 Inne podejście: słowa kodowe mają ustaloną długość, lecz mogą kodować ciągi liter z alfabetu wejściowego o różnej
BUDOWA I FUNKCJA GENOMU LUDZKIEGO
 BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
BUDOWA I FUNKCJA GENOMU LUDZKIEGO Magdalena Mayer Katedra i Zakład Genetyki Medycznej UM w Poznaniu 1. Projekt poznania genomu człowieka: Cele programu: - skonstruowanie szczegółowych map fizycznych i
Działanie algorytmu oparte jest na minimalizacji funkcji celu jako suma funkcji kosztu ( ) oraz funkcji heurystycznej ( ).
 oraz funkcji heurystycznej ( ).") Algorytm A* Opracowanie: Joanna Raczyńska 1.Wstęp Algorytm A* jest heurystycznym algorytmem służącym do znajdowania najkrótszej ścieżki w grafie. Jest to algorytm zupełny i optymalny, co oznacza, że zawsze
Algorytm A* Opracowanie: Joanna Raczyńska 1.Wstęp Algorytm A* jest heurystycznym algorytmem służącym do znajdowania najkrótszej ścieżki w grafie. Jest to algorytm zupełny i optymalny, co oznacza, że zawsze
Wstęp do Techniki Cyfrowej... Teoria automatów
 Wstęp do Techniki Cyfrowej... Teoria automatów Alfabety i litery Układ logiczny opisywany jest przez wektory, których wartości reprezentowane są przez ciągi kombinacji zerojedynkowych. Zwiększenie stopnia
Wstęp do Techniki Cyfrowej... Teoria automatów Alfabety i litery Układ logiczny opisywany jest przez wektory, których wartości reprezentowane są przez ciągi kombinacji zerojedynkowych. Zwiększenie stopnia
WYŻSZA SZKOŁA INFORMATYKI STOSOWANEJ I ZARZĄDZANIA
 DROGI i CYKLE w grafach Dla grafu (nieskierowanego) G = ( V, E ) drogą z wierzchołka v 0 V do v t V nazywamy ciąg (naprzemienny) wierzchołków i krawędzi grafu: ( v 0, e, v, e,..., v t, e t, v t ), spełniający
DROGI i CYKLE w grafach Dla grafu (nieskierowanego) G = ( V, E ) drogą z wierzchołka v 0 V do v t V nazywamy ciąg (naprzemienny) wierzchołków i krawędzi grafu: ( v 0, e, v, e,..., v t, e t, v t ), spełniający
Badania operacyjne: Wykład Zastosowanie kolorowania grafów w planowaniu produkcji typu no-idle
 Badania operacyjne: Wykład Zastosowanie kolorowania grafów w planowaniu produkcji typu no-idle Paweł Szołtysek 12 czerwca 2008 Streszczenie Planowanie produkcji jest jednym z problemów optymalizacji dyskretnej,
Badania operacyjne: Wykład Zastosowanie kolorowania grafów w planowaniu produkcji typu no-idle Paweł Szołtysek 12 czerwca 2008 Streszczenie Planowanie produkcji jest jednym z problemów optymalizacji dyskretnej,