Przewidywanie struktury białek: od modelowania opartego o szablony. do rekombinacji fragmentów metodą dr Frankensteina
|
|
|
- Dorota Kaźmierczak
- 8 lat temu
- Przeglądów:
Transkrypt
1 Przewidywanie struktury białek: od modelowania opartego o szablony do rekombinacji fragmentów metodą dr Frankensteina Iwona A. Cymerman, Joanna M. Sasin, Janusz M. Bujnicki Pracownia Bioinformatyki i Inżynierii Białka Międzynarodowy Instytut Biologii Molekularnej i Komórkowej ul. Ks. Trojdena 4, Warszawa, iamb@genesilico.pl Hasła do zapamiętania: bioinformatyka, struktura białka, przyrównanie sekwencji, modelowanie homologiczne, meta-serwer. Wykaz skrótów: MSA (multiple sequence alignment), PSSM (position-specific score matrix), HMM (Hidden Markov Model), ISS (Intermediate Sequence Search), FR (Fold Recognition), MR (Molecular Replacement)

2 1) Wstęp Niniejszy rozdział zawiera zwięzłą charakterystykę metody teoretycznego przewidywania trzeciorzędowej struktury białek, nazywanej modelowaniem homologicznym. Niezwykle dynamiczny rozwój informatyki jak i coraz większe możliwości obliczeniowe umożliwiły powstanie szeregu algorytmów służących analizie białek. Ze względu na ograniczoną objętość rozdziału, jak również mnogość dostępnych obecnie baz danych i serwisów, nie są one szczegółowo omawiane w tekście a jedynie zebrane w tabele zawierające odnośniki do ich lokalizacji w Internecie. Zaproponowano również tłumaczenia nowych pojęć związanych z analizą sekwencji, w nawiasach umieszczając oryginalne określenia. Zamysłem autorów było przedstawienie czytelnikowi alternatywnych programów do przewidywania struktury, jak również zwrócenie uwagi na ich ograniczenia, które należy uwzględnić podczas interpretacji wygenerowanego modelu struktury białka. Dodatkowo pragniemy zaznaczyć, że nie istnieje jedna, optymalna dla wszystkich białek metoda przewidywania ich struktury. Dlatego też autorzy mają nadzieję, że poniższy rozdział stanie się niejako drogowskazem i zastymuluje wszystkich, w których kręgu zainteresowań leży badanie białek, do sięgnięcia po niezwykle skuteczne i efektywne narzędzia, jakich dostarcza dziś bioinformatyka. 2) Struktura białka 2.1) Znajomość struktury białka Białka pełnią kluczową rolę praktycznie we wszystkich procesach zachodzących w komórce. Pełne zrozumienie mechanizmu działania badanego białka możliwe jest zazwyczaj dopiero w kontekście jego struktury trzeciorzędowej. Znajomość ta ułatwia planowanie eksperymentów, jak również interpretację otrzymanych wyników. Za przykład niech posłuży tu badanie funkcji enzymu małego białka o długości 100 aminokwasów. Aby zidentyfikować komplet aminokwasów tworzących centrum katalityczne, ale bez znajomości aminokwasów konserwowanych w obrębie rodziny, do której przynależy białko, należałoby prawdopodobnie wykonać kilkadziesiąt substytucji, głównie aminokwasów o zjonizowanych lub polarnych grupach bocznych oraz histydyn i cystein, bo to one właśnie najczęściej tworzą miejsca aktywne w enzymach. Analiza sekwencji w rodzinie białek spokrewnionych z białkiem badanym pozwala zazwyczaj zredukować tę liczbę do konserwowanych pozycji. Jednak w wielu rodzinach białek także aminokwasy nienależące do centrum aktywnego są konserwowane, a czasami centrum katalityczne wykazuje różny skład aminokwasowy u różnych członków rodziny. Dysponując wiarygodnym modelem struktury trzeciorzędowej białka wybór celów do mutagenezy można zredukować do kilku aminokwasów np. tworzących kieszeń katalityczną i znajdujących się blisko siebie w przestrzeni. Struktura dostarcza także informacji o położeniu każdego z funkcjonalnie ważnych aminokwasów względem innych (co może sugerować sposób wiązania substratu i mechanizm reakcji) oraz względem reszty białka, skąd można wnioskować, czy znajduje się on na powierzchni, czy też jest położony głęboko w strukturze. Badanie cech białka, które uwidaczniają się jedynie w kontekście struktury trzeciorzędowej, jak np. rozkład potencjału elektrostatycznego lub obecność hydrofobowych grup bocznych na powierzchni białka, może być pomocne w identyfikacji regionów biorących udział w oddziaływaniu z innymi białkami, kwasami nukleinowymi, niebiałkowymi cząsteczkami będącymi inhibitorami, czy też aktywatorami. Jednym z praktycznych zastosowań informacji o strukturze trzeciorzędowej białka jest użycie jej do projektowania leków, które dzięki oddziaływaniom z jego powierzchnią modyfikują jego niepożądaną aktywność. Innym przykładem wykorzystania znajomości struktury może być lokalizacja przestrzenna mutacji odpowiedzialnych za choroby. Porównanie struktury białka funkcjonalnego i zmutowanego dostarcza nie tylko informacji o zmianie konformacyjnej, jaka

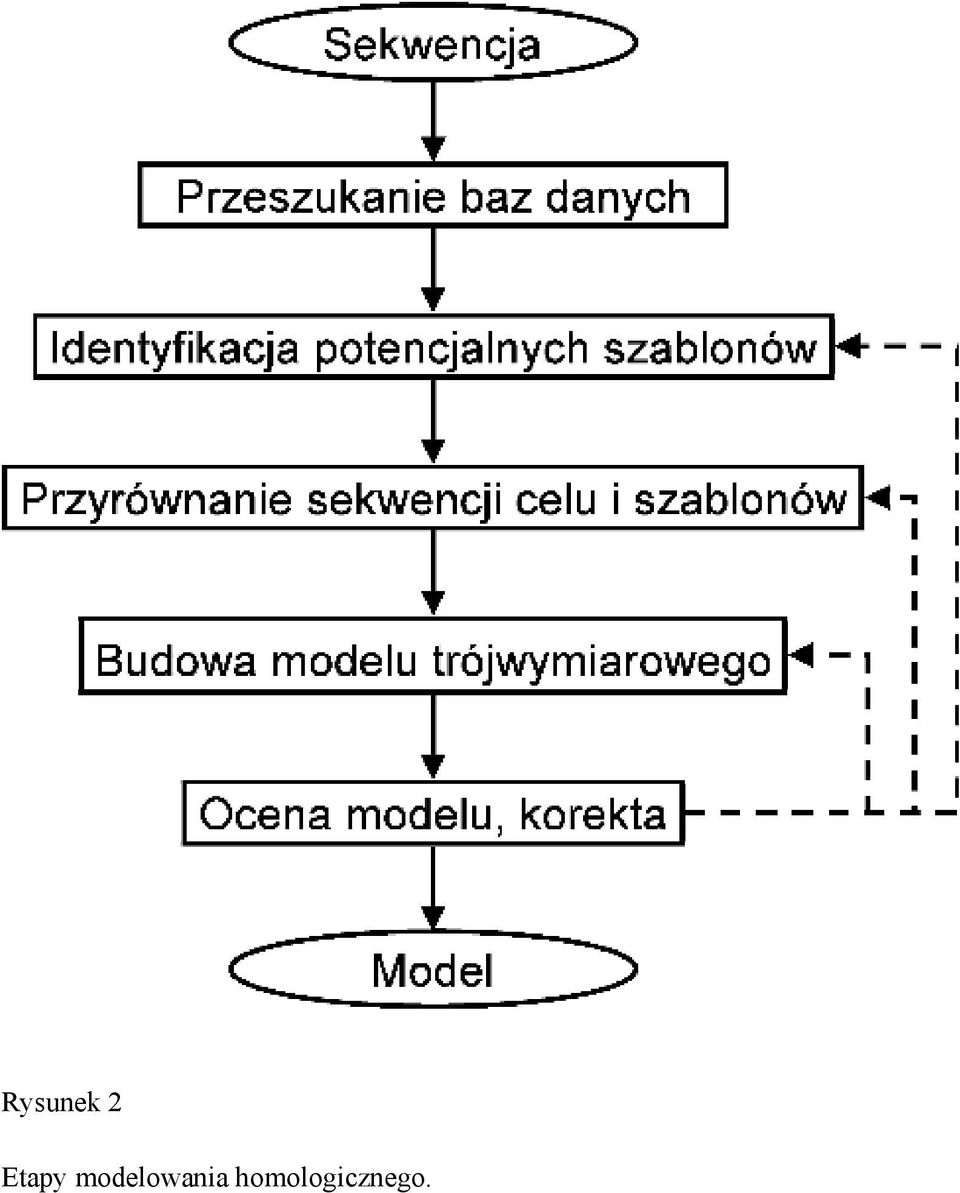
3 nastąpiła na skutek mutacji, ale również ułatwia racjonalne projektowanie potencjalnego leku, znoszącego efekt mutacji. 2.2) A jeśli struktura badanego białka nie jest znana? W chwili obecnej (lipiec 2004) w bazach danych dostępnych jest sekwencji. Większość z nich stanowią jednak hipotetyczne produkty otwartych ramek odczytu, wydedukowane w oparciu o sekwencje DNA pochodzące z projektów genomowych. Natomiast liczba struktur białek rozwiązanych doświadczalnie (przy użyciu technik krystalograficznych lub jądrowego rezonansu magnetycznego (NMR)) wynosi zaledwie Wynika to z konieczności zainwestowania dużych nakładów czasowych (otrzymanie czystego białka, hodowla kryształów, które pozwolą otrzymać wzór dyfrakcyjny o odpowiedniej rozdzielczości) i finansowych (koszt sprzętu, w przypadku krystalografii - dyfraktometr, cyklotron, w przypadku NMR spektroskop, drogie znakowanie białka) a i to nie zawsze gwarantuje rozwiązanie struktury. Naprzeciw ograniczeniom metod doświadczalnych wychodzą metody teoretyczne, pozwalające na zbudowanie modelu białka in silico. Zgodnie z hipotezą Anfinsena [1] sekwencja aminokwasowa białka ściśle determinuje jego strukturę przestrzenną, która w danych warunkach fizjologicznych odpowiada globalnemu minimum energii swobodnej. Można sobie zatem wyobrazić, że idealna metoda przewidywania struktury białka polegałaby na symulacji komputerowej procesu zwijania się łańcucha polipeptydowego. Jednak obecny stan wiedzy o fizyko-chemicznych procesach kierujących zwijaniem się białek nie pozwala na stworzenie wiarygodnej funkcji umożliwiającej wystarczająco dokładne obliczenie energii swobodnej dowolnej konformacji białka. Nawet gdyby taka funkcja została stworzona, współcześnie istniejące komputery są o wiele za wolne do przeanalizowania wystarczającej liczby konformacji, aby z dużym prawdopodobieństwem trafić na konformację na tyle bliską strukturze natywnej, by możliwe bylo odróżnienie jej od struktur nienatywnych, przy zastosowaniu kryteriów energetycznych. Dlatego też poszukuje się innych metod, które umożliwiłyby zaproponowanie hipotetycznej trójwymiarowej struktury białka w oparciu o inne zasady, niż prawa fizyki. Porównania struktur rozwiązanych doświadczalnie wykazały, że białka homologiczne (tj. pochodzące od wspólnego przodka), zazwyczaj zachowują podobną strukturę pomimo nagromadzenia substytucji aminokwasowych (Rys. 1). Także insercje i delecje najczęściej zachodzą w pętlach na powierzchni białka, pozostawiając niemal niezmienionym rdzeń strukturalny składający się zazwyczaj z α-helis i/lub β-wstęg bogatych w aminokwasy hydrofobowe. Jeżeli sekwencję badanego białka o nieznanej strukturze ( celu ang. target) przyrówna się (ang. align) do sekwencji innego, spokrewnionego białka o znanej strukturze ( szablonu ang. template), zamiast symulacji fizycznego procesu zwijania się łańcucha polipeptydowego można zasymulować proces ewolucji białka-celu z homologicznego białkaszablonu. Poprzez wprowadzenie substytucji, insercji i delecji do sekwencji szablonu można otrzymać sekwencję celu, której konformacja będzie bardzo przypominać konformację wyjściową. Metoda symulacji procesu ewolucji sekwencji i struktur białek homologicznych została nazwana modelowaniem homologicznym (ang. homology modeling) i jest obecnie najbardziej skuteczną strategią przewidywania struktury białka. 3) Budowa modelu w praktyce (krok po kroku) Modelowanie homologiczne składa się z kilku etapów. Symulacja procesu ewolucji poprzedzona jest analizą sekwencji celu, na podstawie wyników której następuje selekcja potencjalnych szablonów i ich przyrównanie (ang. alignment) do sekwencji celu (Rys. 2). Poniżej opisane zostały kolejne etapy zmierzające do zbudowania wiarygodnego modelu białka.
) wynosi zaledwie 25 000.")
4 3.1) Analiza sekwencji 3.1.1) Identyfikacja domen Większość białek zbudowana jest z konserwowanych ewolucyjnie domen. Postuluje się, że liczba domen o odmiennej architekturze trzeciorzędowej (ang. fold) wynosi nie więcej niż kilka tysięcy, z czego dla około połowy udało się doświadczalnie rozwiązać strukturę przynajmniej jednego przedstawiciela [2]. Wykorzystanie informacji o liczbie domen, z których zbudowane jest białko, może być wskazówką dla określenia funkcji białka, jak również ma kluczowe znaczenie dla przeprowadzenia kolejnych etapów badania białka in silico. Aby zidentyfikować w badanym białku potencjalną obecność wcześniej scharakteryzowanych domen, przeprowadza się porównanie jego sekwencji z bazami motywów i profili sekwencyjnych charakterystycznych dla znanych domen (Tab. 1). W bazach domen można znaleźć informacje o dystrybucji filogenetycznej białek posiadających daną domenę, o domenach występujacych zwykle razem, zwięzłe opisy najbardziej typowych funkcji pełnionych przez domeny, odnośniki do publikacji opisujących analizy rodzin lub ich reprezentatywnych członków oraz odnośniki do innych baz danych. Domeny w bazach danych klasyfikowane są na podstawie różnych kryteriów, takich jak wzajemne podobieństwo sekwencji lub pokrewieństwo ewolucyjne. Zaleca się więc przeszukanie kilku baz danych i porównanie zaproponowanych wyników. Możliwe jest przeprowadzenie zintegrowanego przeszukiwania kilku baz danych jednocześnie, przy użyciu takich serwisów internetowych jak InterPro lub CDD (Tab. 1). Jeżeli w sekwencji celu zostanie zidentyfikowana obecność więcej niż jednej domeny, to kolejne etapy przewidywania struktury trzeciorzędowej białka powinny być początkowo przeprowadzone dla każdej z nich z osobna. Przeszukiwanie baz danych pełną sekwencją może prowadzić do wyciągnięcia błędnych wniosków o pełnej homologii dwóch niespokrewnionych białek, w których faktycznie tylko jedna z domen wykazuje wspólne pochodzenie ) Przyrównanie sekwencji Kolejnym krokiem w analizie sekwencji celu jest użycie programów (Tab. 2) do przeszukiwania baz danych i identyfikacji sekwencji podobnych do sekwencji celu (jak wspomniano wyżej, w przypadku białek wielodomenowych warto przeprowadzic taką analizę osobno dla każdej domeny). Przeszukanie bazy danych zadaną sekwencją umożliwia identyfikację fragmentów sekwencji spełniających wyznaczone przez badacza kryteria podobieństwa, świadczące o tym że sekwencja zadana i sekwencje zidentyfikowane w bazie danych posiadają region o wspólnym pochodzeniu (konserwowaną ewolucyjnie domenę lub zestaw domen). W oparciu o przyrównania sekwencji parami generowane jest przyrównanie wielosekwencyjne (ang. MSA - multiple sequence alignment), które służy do identyfikacji pozycji zmiennych i konserwowanych w całej rodzinie. Zidentyfikowane sekwencje członków rodziny można także pobrać z bazy danych i zbudować dla nich przyrównanie wielosekwencyjne innymi metodami niż poprzez porównanie fragmentów sekwencji z sekwencją zadaną. Lepsze wyniki można osiągnąć stosując programy, które generują przyrównanie wielosekwencyjne poprzez przyrównanie sekwencji każda z każdą (np. CLUSTALX, PCMA). W zależności od zastosowanego algorytmu i użytych parametrów macierzy substytucji, kar za wprowadzenie przerw itd. można uzyskać odmienne przyrównania. Dlatego też, wygenerowane przyrównanie należy porównać z wynikami zaproponowanymi przez program przeszukujący bazę danych i ewentualnie wprowadzić korektę do MSA. Dysponując wiarygodnym MSA można kontynuować przeszukiwanie bazy danych, tym

5 razem jednak wyszukując sekwencje podobne nie do pojedynczej sekwencji zadanej, ale do całej rodziny pozwala to na zidentyfikowanie sekwencji, które nagromadziły wiele mutacji i utraciły podobieństwo do najbardziej typowych członków rodziny, ale w dalszym ciągu zachowują pewne cechy wspólne dla rodziny jako całości. W szczególności można w ten sposób zidentyfikować podobieństwo między dwiema odlegle spokrewnionymi rodzinami, które razem tworzą nadrodzinę. Najbardziej popularnym narzędziem wykorzystującym powyższą strategię jest PSI- BLAST [3] (ang. Position-Specific, Iterated Basic Local Alignment Search Tool; Po wstępnym przeszukaniu bazy danych algorytmem BLAST i identyfikacji sekwencji spokrewnionych z zadaną sekwencją można zbudować profil (ang. PSSM position-specific score matrix), zawierajacy informację na temat częstości występowania (konserwacji) aminokwasów w poszczególnych pozycjach przyrównania. W kolejnych iteracjach baza danych sekwencji przeszukiwana jest przy użyciu całego profilu, który za każdym razem aktualizuje się poprzez dołączanie nowo zidentyfikowanych członków rodziny. Przeszukiwania prowadzi się do momentu, kiedy nie można zidentyfikować więcej sekwencji, które spełniałyby wyznaczone kryteria podobieństwa. Można wtedy przerwać poszukiwania lub jeżeli istnieją ku temu powody, kontynuować je z użyciem mniej rygorystycznych kryteriów. Alternatywnie, przyrównanie spokrewnionych sekwencji (MSA) może być użyte do stworzenia ukrytego modelu Markowa (ang. HMM Hidden Markov Model), który podobnie jak PSSM może być użyty do przeszukiwania baz danych i identyfikacji odlegle spokrewnionych członków rodziny [4]. Inną możliwością jest wielokrotne przeszukiwanie baz danych przy osobnym użyciu każdej z sekwencji zawartych w MSA. Strategia ta (ang. ISS intermediate sequence search) [5] jest jednak dużo bardziej kosztowna obliczeniowo. Ze względu na różnice w otrzymanych wynikach, optymalnym podejściem jest użycie kilku różnych metod i zintegrowanie ich rezultatów. Użycie PSSM do przeszukiwania baz danych pozwala niekiedy wyszukać sekwencje, których podobieństwo do celu obejmuje niewielkie obszary i niemożliwe byłoby ich zidentyfikowanie przy użyciu porównania typu sekwencja-sekwencja (ang. pairwise comparison). Z drugiej jednak strony przeszukiwanie bazy profilem może nie uwzględnić białek, które zachowały znaczące podobieństwo do kilku przedstawicieli rodziny, ale nie wykazują podobieństwa do średniej. Te lukę wypełnia właśnie zastosowanie ISS. Należy jednak pamiętać, że ISS skutkuje dużą ilością fałszywych pozytywów, czyli sekwencji, które wykazują podobieństwo do poszczególnych pojedynczych członków rodziny na skutek czystego przypadku, a nie ze względu na ich pokrewieństwo ) Edytory sekwencji Niezwykle użytecznym narzędziem służącym obróbce sekwencji są edytory takie jak np.: BioEdit ( GeneDoc ( lub DCSE ( czy SeaView ( Pozwalają one nie tylko na ręczne korygowanie przyrównań wygenerowanych automatycznie przez programy wymienione w poprzednim podrozdziale, ale umożliwiają też wizualizację różnorakich cech przyrównanych sekwencji. np. wyszczególnienie pozycji często występujących aminokwasów (Rys. 1), co może ułatwić identyfikację potencjalnych miejsc wiązania liganda lub centrum aktywnego enzymu, a skupienie się na aminokwasach hydrofobowych może pomóc w odnalezieniu elementów szczególnie istotnych dla stabilności strukturalnej białka ) Przewidywanie struktury drugorzędowej Kolejnym krokiem w analizie białka-celu jest identyfikacja elementów struktury
.")
6 drugorzędowej. Od czasu rozwiązania pierwszych struktur białek zaproponowano wiele schematów klasyfikacji elementów strukturalnych [6]. Najbardziej powszechna i najprostsza klasyfikacja wyróżnia elementy, w których kolejne aminokwasy wykazują podobną konformację: spiralnie zwinięte helisy (najczęściej występuje typ α) i rozciągnięte wstęgi (β) oraz łączące te elementy pętle. Należy podkreślić, że istnieją różne algorytmy identyfikacji helis, wstęg i pętli w strukturze białka, które nie do końca zgadzają się ze sobą, zwłaszcza jeżeli chodzi o klasyfikację aminokwasów na krańcach struktur α i β oraz elementów o konformacji rozciągniętej, które nie tworzą wiązań wodorowych z innymi elementami rozciągniętymi [7][8]. Istnieje wiele algorytmów przewidywania struktur drugorzędowych w oparciu o sekwencję białka. Najstarsze algorytmy, jak algorytm Chou i Fasmana [9], czy GOR [10] są w stanie przewidzieć, w jakiej strukturze znajdzie się dany aminokwas (α, β czy pętla) z dokładnością najwyżej 60-65% i zdecydowanie odradzamy ich używanie, mimo iż metody te są wciąż stosowane w na pozór nowoczesnych komercyjnych (zwykle bardzo drogich) programach do analizy sekwencji białek. Współczesne metody (Tab. 3) pozwalają na przewidywanie struktury drugorzędowej z dokładnością ok %, przy czym wiarygodność przewidywania konformacji heliakalnej (α) jest zazwyczaj wyższa (ok. 80%- 85%) a konformacji rozciągniętej (β) niższa (60%-70%). Najbardziej skuteczne są metody, które przeprowadzają przewidywanie struktury nie dla pojedynczych sekwencji, a dla MSA sekwencji homologicznych - zbudowanego przez badacza lub generowanego automatycznie przez stosowany algorytm. Informacja zawarta w MSA dostarcza dodatkowych informacji o konserwacji elementów hydrofobowych oraz o prawdopodobnym występowaniu pętli w regionach obejmujących delecje bądź insercje w MSA. Dodatkowo, podczas przewidywania struktury drugorzędowej warto zastosować kilka reguł. Otóż, jeśli pośród bliskich homologów znajduje się białko o znanej strukturze (rozwiązanej krystalograficznie lub przez NMR) to skopiowanie struktury drugorzędowej daje zazwyczaj lepszy wynik, niż przewidywanie jej de novo. Ponadto, przed przystąpieniem do przewidywania struktury drugorzędowej warto jest użyć MSA, z którego usunięto najbardziej rozdywergowane sekwencje. W przypadku korzystania z programu, który nie pozwala na wprowadzenie wygenerowanego przez uzytkownika MSA jako danych wejściowych, warto wykonać niezależne przewidywanie struktury drugorzędowej dla kilku członków rodziny. Otrzymanie pokrywających się wyników zwiększa pewność przewidywania. I tak jak w przypadku innych metod w miejscach dla których przewidywanie jest niejednoznaczne, warto jest porównać wyniki zaproponowane przez różne metody i zastanowić się nad przyczyną różnic (może np. w MSA wystąpiły błędy, które należy skorygować i powtórzyć przewidywanie?) ) Przewidywanie nieuporządkowania strukturalnego W niektórych białkach można zaobserwować fragmenty nieuporządkowane, które nie tworzą zdefiniowanej struktury i występują jako populacja różniących się od siebie konformacji. Często występują one w łącznikach (ang. linker) pomiędzy domenami, w długich pętlach lub N- i C-końcowych ogonach. Regiony nieustrukturalizowane występują również w elementach białek biorących udział w oddziaływaniach międzycząsteczkowych w wielu przypadkach spośród populacji możliwych konformacji tylko jedna jest zdolna do wiązania liganda, co powoduje ustrukturalizowanie danego regionu podczas formowania kompleksu międzycząsteczkowego. Przewidywanie lokalizacji potencjalnych rejonów nieustrukturalizowanych za pomocą metod bioinformatycznych (Tab. 3) może dać cenne wskazówki do dalszego przewidywaniu struktury trzeciorzędowej białka, jak i identyfikacji potencjalnych miejsc interakcji z innymi cząsteczkami.

7 3.1.5) Białka transbłonowe Około 30% białek komórkowych to białka transbłonowe. Ze względu na zakotwiczenie w błonach charakteryzują się one specyficzną budową. Ich hydrofobowe segmenty wewnątrzbłonowe przeplatane są naprzemiennie zewnątrzbłonowymi elementami hydrofilowymi. Powszechnie występują białka transbłonowe dwóch typów: zbudowane z hydrofobowych regionów α-helikalnych oraz z szeregu β-wstęg, składających się na tzw. strukturę β-baryłki (ten drugi typ białek transbłonowych jest charakterystyczny dla błony zewnętrznej bakterii gramujemnych). Dostępne są programy komputerowe wyspecjalizowane w identyfikacji obydwu typów białek błonowych (Tab. 4). Niektóre z nich oprócz identyfikacji transbłonowych elementów struktury drugorzędowej i łączących je pętli przewidują także ich topologię względem błony, umożliwiając identyfikację fragmentów zorientowanych do wewnątrz i na zewnątrz komórki. 3.2) Metody przewidywania struktury trzeciorzędowej poprzez identyfikację szablonu strukturalnego Przewidywanie struktury białka metodą modelowania homologicznego opiera się na wykorzystaniu szablonu homologicznego białka o znanej strukturze. Często jednak analiza sekwencji, nawet z użyciem profili czy ukrytych modeli Markowa nie jest w stanie doprowadzić do identyfikacji homologicznego białka o strukturze rozwiązanej doświadczalnie. Nie oznacza to jednak, że taki homolog nie istnieje, a jedynie, że nie można go wykryć poprzez porównywanie samych sekwencji. Teoretycznie liczba możliwych konformacji białka jest nieskończona, jednak w świecie białek występuje ograniczona liczba sposobów zwinięcia białka, którą szacuje się na kilka tysięcy [2] [11] [12] [13]. Obserwacja, iż białka mogą przyjmować podobne zwinięcie (ang. fold) pomimo niewielkiego, bądź wręcz braku podobieństwa sekwencyjnego, stała się podstawą rozwoju metod rozpoznających architekturę przestrzenną białka (ang. fold recognition) i ograniczających przewidywanie struktury trzeciorzędowej białka do odgadnięcia, czy któraś ze znanych struktur innych białek jest podobna do rzeczywistej (nieznanej podczas przewidywania) struktury analizowanego celu oraz czy te znane struktury mogłyby być użyte jako szablony do modelowania. Programy służące rozpoznawaniu architektury białka składają się zwykle z następujących modułów: 1) bazy danych struktur, 2) metody umożliwiającej porównanie sekwencji celu do sekwencji białek zawartych w bazie danych, 3) algorytmu obliczającego optymalne przyrównanie sekwencji celu i szablonu, 4) metody szacującej istotność i poprawność przyrównania i jego ocenę statystyczną [14]. Zasadniczo istnieją dwie strategie porównywania sekwencji celu z sekwencjami białek z bazy danych - wykorzystujące informacje o strukturze trzeciorzędowej szablonu lub pomijające ją ) Metody sekwencyjne Metody sekwencyjne (Tab. 5) opierają się jedynie na podobieństwie sekwencyjnym celu i szablonu, nie uwzględniając informacji o strukturze szablonu. Dokonują jedynie przyrównania sekwencji celu i sekwencji szablonu, zazwyczaj wykorzystując informacje o sekwencjach homologicznych i de facto porównując nie same sekwencje, a całe profile, lub ukryte modele Markowa. Czasami używa się meta-profili, które oprócz informacji o konserwacji sekwencji aminokwasowej, w porównywanych rodzinach, zawierają przewidywaną strukturę drugorzędową, przewidywane preferencje poszczególnych aminokwasów w sekwencji względem stopnia solwatacji itp. Informacja ta pochodzi z analizy sekwencji, a nie z danych doświadczalnych (np. pomimo, że dla potencjalnych szablonów porównywanych z sekwencją celu struktura drugorzędowa jest znana, to tutaj jest ona przewidywana de novo ). Metody te dają najlepsze rezultaty w przypadku stosunkowo
.")
8 bliskiego podobieństwa sekwencyjnego pomiędzy celem a szablonem ) Metody przewlekania (ang. threading ) Metody przewlekania (Tab. 5) w swojej funkcji oceniającej prawdopodobieństwo, że dana struktura może być dobrym szablonem, zawierają oszacowanie kompatybilności sekwencji celu z doświadczalnie określoną strukturą. Ortodoksyjne metody przewlekania używają potencjałów fizyko-chemicznych, aby obliczyć energię oddziaływania aminokwasów celu gdy badana sekwencja dopasowana jest optymalnie do rusztowania jakie stanowi struktura szablonu. Praktyka wykazuje jednak, że takie metody są stosunkowo mało skuteczne, prawdopodobnie na skutek niedoskonałości oceny energii oddziaływań. Najlepsze spośród współczesnych metod przewlekania łączą w swoich funkcjach oceny dopasowania zarówno podobieństwo sekwencyjne celu i szablonu (lub raczej odpowiadających im profili), jak i podobieństwo przewidywanej struktury celu z doświadczalnie określoną strukturą szablonu (pokrywanie się elementów struktury drugorzędowej, usytuowanie na powierzchi białka aminokwasów przewidywanych jako uwodnione itp.). Metody służące rozpoznawaniu architektury białka są w stanie poprawnie zidentyfikować odpowiedni szablon do modelowania w ponad 40% przypadków, gdy sekwencja celu nie wykazuje statystycznie istotnego podobieństwa do żadnego białka o znanej strukturze. Słabym punktem wszystkich tych metod (zarówno opartych o same sekwencje jak i o sekwencje i struktury) jest brak skutecznej funkcji oceniającej przyrównania sekwencji celu i szablonu. Często zdarza się, że białko o strukturze podobnej do prawdziwej struktury białka-celu znajduje się wśród np. dziesięciu najlepiej ocenionych potencjalnych szablonów, jednak jego ocena jest zbliżona do oceny dziewięciu innych przewidywań, które są fałszywymi pozytywami ) Budowa modelu - korygowanie przyrównania sekwencji celu do szablonu Jak wspomniano, model białka generowany jest przez program symulujący ewolucję celu z szablonu na podstawie zaproponowanego przyrównania ich sekwencji. Niewątpliwie najważniejszym etapem jest więc wybór szablonu oraz poprawne przyrównanie jego sekwencji z sekwencją celu. Te dwa czynniki w głównej mierze determinują poprawność modelu. Model oparty na błędnym szablonie bądź na niepoprawnym przyrównaniu praktycznie zawsze będzie błędny. Korekta przyrównania następować powinna w oparciu o 1) dane literaturowe (jak np. identyfikacja wzajemnie odpowiadających sobie aminokwasów tworzących miejsce wiązania podobne w obu białkach mimo ich ogólnego braku podobieństwa sekwencyjnego), 2) sprawdzenie, czy wprowadzone delecje i insercje znajdują się w obrębach pętli, w których to rejonach zmiany są dużo bardziej dynamiczne niż w zazwyczaj wysoko konserwowanej strukturze rdzenia, 3) ocenę modelu pod względem występowania cech charakterystycznych dla dobrze zwiniętych i upakowanych białek Najbardziej popularnymi wśród wolno dostepnych programów do modelowania homologicznego (symulującymi ewolucję sekwencji celu w oparciu o strukturę szablonu) są MODELLER ( [15] i SWISS-MODEL ( [16]. Podstawowe różnice pomiędzy tymi programami to zastosowanie różnych algorytmów do budowy modelu oraz różne procedury wykorzystywane do minimizacji energii. W oparciu o przyrównanie sekwencyjne SWISS-MODEL ustala regiony konserwowane, w których konformacja łańcucha głównego nie zmieni się lub zmieni niewiele i po prostu kopiuje ich koordynaty. Taki niepełny model używany jest jako rusztowanie do wymodelowania insercji i delecji poprzez wstawienie z bazy danych takich fragmentów struktury, których końce mają podobną odległość, co końce rusztowania i których sekwencja najbardziej przypomina sekwencję modelowanego odcinka. Łańcuchy boczne aminokwasów są dobudowywane w oparciu o

9 konformację łańcuców w szablonie. Jeżeli aminokwasy są identyczne lub posiadają identyczne fragmenty to koordynaty ich atomów są kopiowane z szablonu do celu, natomiast dla aminokwasów, które uległy substytucji, łańcuch boczny wybierany jest z listy konformacji naczęściej występujących w białkach i wstawiany tak, by spowodować jak najmniej konfliktów sterycznych z atomami innych aminokwasów. Tak powstały model poddawany jest minimizacji energii z użyciem pola siłowego GROMOS 96 ( MODELLER nie kopiuje w sposób jawny koordynatów przestrzennych szablonu, natomiast ustala odległości i kąty pomiędzy atomami szablonu i następnie przenosi je jako więzy przestrzenne (ang. restrains) na odpowiadające im atomy homologicznych aminokwasów celu. Dodatkowe więzy mają na celu zapewnienie białko-podobności modelu, tj. opisują idealne wartości długości wiązań i kątów pomiędzy atomami. Dodatkowo użytkownik może dodać własne więzy pochodzące z analizy doświadczalnej (np. wyniki mutagenezy lub sieciowania) lub bioinformatycznej (np. przewidywanie struktury drugorzędowej dla regionu, który nie ma homologicznego odpowiednika w szablonie). Model budowany jest tak, aby zminimalizować naruszenie wszystkich więzów. W końcowym etapie budowy modelu przeprowadzana jest minimalizacja energii w polu siłowym CHARMM22 ( aby zapewnić poprawną stereochemię i korzystne oddziaływania pomiędzy grupami funkcyjnymi. Obydwa te programy umożliwiają modelowanie struktury celu w oparciu o pojedynczy szablon, jak i o cały zestaw homologicznych szablonów (np. odpowiadających różnym domenom lub podjednostkom w multimerze). Oba programy dostarczają zwykle modeli o porónywalnej jakości, a wybór jednego z nich jest dyktowany przede wszystkim przez doświadczenie badacza. Z doświadczenia autorów SWISS-MODEL lepiej nadaje się do modelowania białek o wysokim podobieństwie sekwencji, zwłaszcza w oparciu o jeden szablon i gdy liczba insercji i delecji w sekwencji celu jest niewielka, natomiast MODELLER lepiej sprawdza się w modelowaniu odległych homologów oraz gdy konieczne jest równoczesne zastosowanie wielu szablonów strukturalnych. Jak przy innych analizach bioinformatycznych, zalecanym podejściem jest zastosowanie obu w.w. metod (a także innych, jeżeli użytkownik ma do nich dostęp), a następnie porównanie wyników i w szczególności analiza różnic, która może pomóc w identyfikacji różnych artefaktów procesu modelowania charakterystycznych dla poszczególnych programów ) Ocena modelu Dostępnych jest wiele metod służących ocenie poprawności struktury białka, z których większość powstała z myślą o analizie modeli krystalograficznych. Można je jednak stosować również do weryfikacji modeli teoretycznych (Tab. 6). Metody te analizują takie cechy strukturalne jak geometria, stereochemia, czy też kompatybilność charakteru fizykochemicznego danego aminokwasu z kontekstem strukturalnym w jakim został umieszczony (np. czy hydrofobowe grupy boczne otoczone są innymi grupami hydrofobowymi i czy naładowane grupy boczne są eksponowane na zewnątrz białka lub sparowane z grupami bocznymi o przeciwnym ładunku). Różnice w ocenie modeli różnymi metodami wynikają z czułości i przypisywania różnych wag poszczególnym komponentom funkcji oceny. Należy podkreślić, że najczęściej spotykane funkcje oceny służą określeniu, czy analizowany model jest białko-podobny, czy też wykazuje nietypowe cechy, ale nie są w stanie obiektywnie stwierdzić, czy jest on poprawny, czy błędny bez odniesienie do danych doświadczalnych o wysokiej rozdzielczości (pochodzących z badań krystalograficznych lub NMR). Ponadto analiza struktur białek o wysokiej rozdzielczości wskazuje, że nietypowe cechy strukturalne można napotkać stosunkowo często i że są one najczęściej związane ze
.")
10 specyficzną funkcją pełnioną przez białko. Wynika to z faktu, że w procesie ewolucji pozytywnej selekcji podlega nie tylko stabilność strukturalna białka, ale też jego zdolność do interakcji z innymi cząsteczkami, które to cechy często bywają wzajemnie niekompatybilne. Dlatego interpretacji wyników oceny modelu struktury białka należy dokonywać w kontekście jego funkcji biologicznej (o ile jest ona znana). Na przykład wiele miejsc wiązania ligandów (np. jonów metalu) tworzonych jest w białkach przez ujemnie naładowane grupy boczne znajdujące się w otoczeniu hydrofobowym. Identyfikacja takich niekompatybilnych konstelacji grup bocznych w modelu strukturalnym może sugerować istnienie miejsca ważnego dla funkcji (zwłaszcza, jeżeli reszta modelu oceniona jest jako białko-podobna ), choć może także oznaczać, że podczas budowy modelu wystąpił błąd (np. ze względu na nieprawidłowe przyrównanie celu do szablonu) i aminokwasy są przesunięte względem faktycznej pozycji. W takich sytuacjach należy koniecznie porównać ocenę białkopodobności modelu z oceną szablonu, rozważyć konserwację kontrowersyjnych aminokwasów w obrębie rodziny, oraz przeanalizować możliwości wygenerowania alternatywnych modeli, opartych o zmienione przyrównania sekwencji lub korzystających z innych szablonów. Należy podkreślić, że ocena poprawności modelu teoretycznego wyłącznie pod względem stereochemii (np. popularne w badaniach krystalograficznych badanie wykresu Ramachandrana) ma zwykle niewielki sens dla modeli teoretycznych wygenerowanych metodami modelowania homologicznego. Parametry stereochemiczne, wartości kątów i długości wiązań w modelach homologicznych są najczęściej bardzo podobne do ich odpowiedników w użytych szablonach strukturalnych i nie odzwierciedlają dobrze stopnia poprawności samego modelu. Np. można łatwo wygenerować zupełnie błędny model struktury białka wykazujący doskonałą stereochemię (np. przez błąd w przyrównaniu sekwencji celu do szablonu), jak i model bardzo bliski strukturze natywnej, w którym długości i kąty wiązań będą dalekie od idealnych (np. z powodu użycia kilku szablonów w których homologiczne aminokwasy miały odmienną konformację). Autorzy niniejszego rozdziału gorąco polecają użycie metod oceniających właściwości znacznie trudniejsze do podrobienia w błędnych modelach, zwłaszcza. kompatybilność aminokwasów z otoczeniem (np. VERIFY3D, PROSAII, lub ANOLEA) czy upakowanie atomów w rdzeniu białka (SOESA). Ostatnio zaproponowano specjalną metodę PROQ [17] służącą do oszacowania jakości modeli teoretycznych. W przeciwieństwie do wyżej wymienionych metod opracowanych oryginalnie dla struktur krystalicznych i dopiero później zaadaptowanych przez modelarzy teoretycznych, celem PROQ nie jest identyfikacja struktur nienatywnych (w odróżnieniu od idealnej struktury natywnej), a odróżnienie modeli poprawnych (choć w dalszym ciągu niekoniecznie idealnych) od zupełnie błędnych. Od wcześniej omawianych algorytmów PROQ różni się jednak tym, że ocenia jakość na poziomie całego modelu a nie jego poszczególnych elementów (atomów czy aminokwasów) i nie pozwala na wyróżnienie fragmentów porawnych i tych, które należałoby poprawić. Jak wspomniano wcześniej, zalecanym podejściem jest użycie kilku alternatywnych metod oceny struktury modelu i porónanie ich wyników. Autorzy chcieliby uczulić czytelników na fakt, iż ze względu na łatwość wygenerowania modeli zupełnie błędnych, przy jakimkolwiek użyciu modelu teoretycznego należy KONIECZNIE podawać ocenę jego poprawności. Modele nie ocenione za pomocą opisanych tu metod (lub innych, sprawdzających podobne parmetry), są praktycznie bezwartościowe. 4) Od sekwencji do modelu poprzez jedno kliknięcie myszy Teoretyczne modele białek, które otrzymały pozytywną ocenę w testach poprawności, mogą służyć zazwyczaj jako przybliżona i z założenia obarczona drobnymi błędami, ale w
 tworzonych jest w białkach przez ujemnie naładowane grupy boczne znajdujące się w otoczeniu hydrofobowym.")
11 miarę wiarygodna mapa, która może naprowadzić na ślad aminokwasów ważnych dla funkcji białka. Biorąc jednak pod uwagę mnogość dostępnych metod przewidywania różnych cech strukturalnych oraz konieczność konfrontacji ich wyników, wygenerowanie wiarygodnego modelu struktury białka nie jest zadaniem łatwym. Zastymulowało to rozwój programów, najczęściej zaimplementowanych jako serwisy internetowe, zwanych Meta-serwerami. Jako dane wejściowe przyjmują one od użytkownika sekwencję celu, przesyłają ją następnie do innych serwisów analizujących sekwencje białek, gromadzą ich wyniki i przedstawiają podsumowanie rezultatów. Meta-serwery najczęściej prezentują przewidywania dotyczące potencjalnej przynależności do rodziny białek, przewidywanej struktury drugorzędowej, rozpoznania architektury, a niekiedy generują również wstępne modele. Jako przykład omówiony zostanie Meta-serwer GeneSilico [18] ( Użytkownik może nakazać analizę przygotowanego przez siebie przyrównania wielosekwencyjnego (MSA) lub pojedynczej sekwencji. W przypadku pojedynczej sekwencji MSA zostanie wygenerowany automatycznie. Dodatkowo użytkownik decyduje o pozostawieniu lub usunięciu z przyrównania regionów, w których występują przerwy. Powoduje to ograniczenie przewidywania do regionów, które wykazują największe podobieństwo do struktury szablonu. Meta-serwer GeneSilico oferuje przewidywanie obecności domen, struktury drugorzędowej, występowania elementów transbłonowych oraz prezentuje wyniki identyfikacji potencjalnych szablonów proponowane przez różne metody, służące rozpoznawaniu architektury białka. Wyniki przedstawiane są w postaci przyrównań sekwencji celu i proponowanych szablonów. Każde z przyrównań posiada dodatkowo ocenę oszacowaną przez algorytm generujący wynik. Na podstawie rezultatów threadingu generowane jest przewidywanie konsensusowe z użyciem algorytmów Pcons2 i 5. Nie proponują one dodatkowego szablonu, czy przyrównania, a jedynie wybierają 10 potencjalnie najlepszych przyrównań spośród zaproponowanych przez inne metody. Użytkownik serwera ma również możliwość otrzymania wstępnych modeli struktury trzeciorzędowej celu, zbudowanych automatycznie na podstawie zaproponowanych przyrównań. Modele te są często niekompletne i mogą zawierać znaczące błędy, nawet jeżeli zostały zbudowane na bazie poprawnie wybranego szablonu, jednak stanowią użyteczny punkt wyjściowy dla dalszej analizy i budowy ostatecznego modelu. 5) Modele hybrydowe Większość prezentowanych wyżej metod, w założeniu, buduje wstępny model struktury celu w oparciu o jeden szablon. Nakłada to ograniczenia na nowopowstający model przyjmuje on architekturę podobną do architektury struktur rozwiązanych eksperymentalnie, mimo, że natywna struktura celu może wykazywać unikalne cechy bądź stanowić hybrydę cech występujących u kilku spokrewnionych białek o znanej strukturze. Ponadto metody FR umożliwiają zbudowanie poprawnych modeli jedynie dla takich sekwencji, które posiadają potencjalne szablony w bazie danych rozwiązanych struktur. Ograniczenia te stały się bodźcem do tworzenia metod konstruujących modele hybrydowe, zapoczątkowanych przez pionierski program 3D-SHOTGUN opracowany przez Daniela Fischera [19]. W metodzie tej punktem wyjścia jest zbiór wyników wygenerowanych przez metody rozpoznające architekturę białka. Spośród nich wybierane są przyrównania celszablon, które zostały ocenione przez poszczególne metody jako najlepsze. Dla nich generowane są prowizoryczne modele składające się jedynie z koordynatów atomów węgli Cα tych aminokwasów szablonu, którym udało się przyporządkować jakiś aminokwas pochodzący z sekwencji celu. Algorytm 3D-SHOTGUN identyfikuje podzbiory modeli wykazujące wzajemne podobieństwo strukturalne oraz, przy zastosowaniu programu MaxSub [20], buduje przyrównanie przestrzenne wszystkich wyselekcjonowanych struktur. Tworzenie

12 modeli hybrydowych dla każdego podzbioru struktur podobnych odbywa się poprzez wybór elementów struktury drugorzędowej występujących najczęściej w zbiorze modeli. Fragmenty te są następnie łączone ze sobą, na zasadzie prostych transformacji geometrycznych. Powstałe w ten sposób modele hybrydowe są oceniane poprzez wyznaczenie stopnia podobieństwa każdego z nich do całego zbioru modeli. Ostatecznie generowana jest seria modeli, które mogą stanowić hybrydy struktur o zupełnie różnych architekturach. Jednak metoda ta nie przynosi w pełni satysfakcjonujących rezultatów. To co jest w niej cechą konkurencyjną w stosunku do innych metod, czyli pośrednie wykorzystanie wielu szablonów i alternatywnych przyrównań, jest również jej piętą Achillesową. Modele wygenerowane przez 3D- SHOTGUN posiadają często duże przerwy pomiędzy węglami Cα, które powinny ze sobą sąsiadować, jak i przestrzenne zbitki koordynatów, z których nie można w prosty sposób odtworzyć białkopodobnej struktury pełnoatomowej. Tym niemniej, jeżeli weźmie się pod uwagę odległości pomiędzy przewidywaną i rzeczywistą pozycją poszczególnych węgli Cα, to poszatkowane modele wygenerowane metodą 3D-SHOTGUN okazują się zazwyczaj bardziej podobne do struktury rzeczywistej niż białkopodobne modele wygenerowane przez oryginalne metody dostarczające danych metodzie hybrydowej [21] [22]. Mimo iż interpretacja funkcjonalna tych fragmentów modelu, które naruszają prawa fizyki rządzące zarówno zwijaniem się struktury białka, jak i wykonywaniem jego funkcji w komórce jest wysoce problematyczna, modele 3D-SHOTGUN mogą stanowić użyteczny punkt wyjścia do prób rozwiązania konfliktów sterycznych i wygenerowania modeli poddających się interpretacji. Innym przykładem metody hybrydowej jest zaproponowany przez Paula Bates'a i wspólpracowników [23], algorytm genetyczny pozwalający na rekombinację modeli pełnoaatomowych zbudowanych w oparciu o odmienne szablony i przyrównania ( Selekcja modeli wyjściowych odbywa się przy użyciu tzw. funkcji dopasowania (ang. fitness), w której skład wchodzi ocena energii oddziaływania poszczególnych aminokwasów ze sobą i energia solwatacji grup bocznych. Miejsca rekombinacji modeli wyznaczane są losowo w obrębach pętli, łączących ze sobą elementy struktur drugorzędowych. Modele pochodne tworzone są poprzez łączenie fragmentów modeli wyjściowych, czemu towarzyszy naprawianie geometrii pętli, jeśli była ona zaburzona przez rekombinacje. W algorytmie genetycznym Batesa, oprócz rekombinacji, zaimplementowany został także proces mutacji polegający na nałożeniu na siebie dwóch struktur i uśrednieniu ich koordynat kartezjańskich. Po każdej rundzie rekombinacji usuwane są z populacji te modele, które wykazują ewidentne zaburzenia budowy (więcej niż jedno złamanie łańcucha głównego oraz więcej niż 4% nieplanarnych wiązań peptydowych). Unika się w ten sposób tworzenia zbitek koordynat i innych nieprawdopodobnych konformacji często obecnych w modelach generowanych przez 3D-SHOTGUN. Podstawowym ograniczeniem metod 3D-SHOTGUN i algorytmu genetycznego Batesa jest fakt, że dobre rozwiązania można znaleźć tylko wtedy, gdy w początkowym zbiorze przyrównań sekwencji cel-szablon znajduje się przynajmniej jedno, chociaż częściowo poprawne rozwiązanie. Jeżeli dany element struktury drugorzędowej budowanego modelu jest błędnie przewidziany we wszystkich elementach populacji wyjściowej, to w końcowym modelu również pojawi się błędne rozwiązanie. Autorzy poniższego rozdziału opracowali własną metodę budowania modeli hybrydowych, która pozwala na ominięcie powyższego ograniczenia. Podejście w stylu potwora FRankensteina (ang. Frankenstein s Monster approach ) [24] polega na konstrukcji modelu spełniającego kryteria białkopodobności, a pochodzącego ze zbioru wstępnych (i w znacznej mierze niebiałkopodobnych modeli generowanych na podstawie przyrównań zaproponowanych przez metody FR. W modelach wyjściowych identyfikuje się białkopodobne fragmenty (np. przy użyciu programu VERIFY3D [25]) a następnie

13 przyrównuje się je ze sobą. Potwór FRankensteina konstruowany jest w pierwszym rzędzie z fragmentów najczęściej występujących we wszystkich modelach (niezależnie od oceny) a następnie jest uzupełniany fragmentami ocenionymi jako najbardziej białkopodobne. Tak skonstruowany potwór przypomina modele generowane przez 3D-SHOTGUN, tj. może być niekompletny i wykazywać konflikty steryczne na styku fragmentów. Struktura potwora służy jednak nie jako fizyczny model struktury białka, a jako referencja do wyznaczenia nowego przyrównania pomiędzy sekwencją celu a zbiorem szablonów, na podstawie którego generowany jest model drugiej generacji, tym razem w zamierzeniu białkopodobny. W modelu tym ocenia się kompatybilność sekwencji aminokwasowej z otoczeniem dla wszystkich elementów struktury drugorzędowej. Regiony o nisko ocenionej kompatybilności ( niebiałkowe ) traktuje sie jako niedopasowane, chyba że użytkownik nakaże ich specjalne traktowanie (w ten sposób można uwzględnić np. dane doświadczalne wskazujące na centrum aktywne, które może wykazywać cechy uznane za niebiałkowe ). Dla każdego regionu uznanego za niedopasowany podejmuje się próbę poprawienia lokalnego dopasowania sekwencji do struktury poprzez wygenerowanie alternatywnych przyrównań, w których sekwencję systematycznie przesuwa się w stronę obydwu końców danego elementu struktury drugorzędowej. Dla każdego przyrównania budowany jest nowy model, który podlega ocenie. Następnie przeprowadza się rekombinację fragmentów o najlepszej ocenie i tworzy potwora drugiej generacji i całą procedurę modelownia powtarza się aż do otrzymania modelu o oczekiwanej jakości lub zatrzymuje, w razie gdy kolejne próby poprawy dopasowania na poziomie przyrównania sekwencji nie są w stanie poprawić oceny modelu na poziomie struktury trzeciorzędowej. Jeżeli w modelu wygenerowanym na tym etapie brakuje elementów na N- i C- końcach lub występują duże insercje, dla których nie udaje się wygenerować białkopodobnych struktur to mogą być one ewentualnie dodane de novo (patrz niżej). Ostateczny model otrzymuje się po zastosowaniu procedury granicznej minimizacji energetycznej, mającej na celu usunięcie ewentualnych zawad sterycznych oraz optymizację długości i kątów wiązań. Modele wygenerowane za pomocą powyższej procedury są prawie zawsze białkopodobne. Metoda potwora Frankensteina ta najlepiej działa dla przypadków, w których serwery FR wiarygodnie przewidują konkretną architekturę, nie ma jedynie zgody co do preferowanego przyrównania sekwencyjnego. Tym niemniej, jeżeli wśród przyrównań sekwencyjnych stanowiących punkt wyjścia znajduje się tylko jedno poprawne przewidywanie architekturze trzeciorzędowej, to nadal istnieje szansa, że wygenerowany zostanie model o poprawnie wymodelowanym rdzeniu strukturalnym, w którym wiele błędów występujących w oryginalnym przyrównaniu zostanie naprawionych. W przypadkach kiedy nie można zidentyfikować właściwego szablonu, potwora Frankensteina nie da się ożywić i jedyną możliwością pozostaje zastosowanie metod de novo. 6) Modelowanie struktury białka de novo Przykładem metody de novo budującej modele z fragmentów jest ROSETTA [26] [27] [28], rozwijana przez zespół kierowany przez Davida Bakera. Metoda ta łączy w sobie pewne cechy modelowania homologicznego i symulacji zwijania łańcucha polipeptydowego. Modele budowane są z krótkich, 9- i 3-aminokwasowych fragmentów znanych struktur, tworzących bibliotekę możliwych konformacji. W przeciwieństwie do tradycyjnych metod modelowania z użyciem szablonów, danemu regionowi sekwencji nie jest przyporządkowywana na stałe jedna konformacja. ROSETTA przeprowadza symulację, w trakcie której 9- lub 3-aminokwasowe odcinki sekwencji celu przyjmują różne konformacje w oparciu o model probabilistyczny opisujący związki między konformacją i sekwencją fragmentów. Lista konformacji dopuszczalnych dla wszystkich odcinków sekwencji ustalana jest na początku symulacji na podstawie lokalnego podobieństwa odcinków sekwencji celu i

14 przewidywanej struktury drugorzędowej do sekwencji i obserwowanej konformacji fragmentów tworzących bibliotekę. Kolejna różnica pomiędzy ROSETTĄ i poprzednio wymienionymi programami polega na tym, że fragmenty do budowy modelu pobierane są z niespokrewnionych struktur, które mogą wykazywać globalnie odmienną architekturę. Ostateczny model generowany jest przez ROSETTĘ w oparciu o ocenę energii oraz/lub przez identyfikację globalnych konformacji, które najczęściej powtarzały się w całej symulacji. ROSETTA jest obecnie jednym z niewielu programów, które potrafią niekiedy wygenerować model o zgrubnie prawidłowej strukturze bez odniesienia do żadnego specyficznego szablonu (czyli de novo ), co umożliwia próby przewidywania struktury trzeciorzędowej dla białek, które nie posiadają homologa o znanej strukturze. Tym niemniej, należy podkreślić, że otrzymanie prawidłowego modelu de novo jest nadal raczej rzadkością niż regułą. Możliwości programu ROSETTA zostały wykorzystane w meta-serwerze ROBETTA [29] [30], który umożliwia konstruowanie modelu białka częściowo w oparciu o szablon, a częściowo de novo. ROBETTA automatycznie dzieli sekwencję celu na regiony, które można wymodelować w oparciu o szablon i na takie, które nie wykazują globalnego podobieństwa do żadnej ze znanych struktur. Część białka jest modelowana poprzez tradycyjną homologię, natomiast pozostała część jest zwijana poprzez wstawianie 9 i 3- aminokwasowych fragmentów. Metoda ta wydaje się być szczególnie obiecująca w przypadku potrzeby wymodelowania struktury białek, które w stosunku do dostępnych szablonów wykazują obecność sporych insercji i/lub wydłużonych końców, których nie da się wymodelować innymi metodami niż de novo. Niestety, ze względu na duży koszt obliczeniowy związany z idealnym włączaniem elementów modelowanych de novo do łańcucha modelowanego homologiczne oraz z upakowaniem przestrzennym tych elementów wzajemnie wobec siebie i wobec reszty białka, obecna wersja meta-serwera ROBETTA często generuje modele z nieciągłościami i zbitkami koordynat, podobnymi do tych otrzymywanych z metody 3D-SHOTGUN. Wprowadza to konieczność naprawiania i udokładniania modeli wynikowych przez zastosowanie innych metod. 7) Perspektywy Metody przewidywania struktury trzeciorzędowej białek są rozwijane przez grupy bioinformatyków na całym świecie, a efekty jakie można uzyskać przy ich zastosowaniu zdają się być coraz lepsze (CASP- I choć jakość modeli teoretycznych zwykle odbiega od jakości struktur rozwiązanych dzięki krystalografii, to jednak mogą być one pomocne w interpretacji danych doświadczalnych i np. służyć do wyliczenia rzeczywistej struktury metodą podstawienia molekularnego [31] (MR, ang. Molecular Replacement). W niektórych przypadkach może umożliwić to rozwiązanie struktury białka bezpośrednio z danych dyfrakcyjnych, dzięki czemu czasochłonny i kosztowny etap uzyskiwania pochodnych ciężkoatomowych analizowanego białka może stać się zbędny. Wykazano, że w niektórych przypadkach udaje się rozwiązać strukturę białkacelu dzięki użyciu modeli stworzonych na bazie szablonów, których podobieństwo sekwencyjne do sekwencji celu jest niższe niż 30%, nawet jeżeli dla struktur samych szablonów procedura MR zawodziła [32]. Kluczowe w takich sytuacji jest uzyskanie poprawnego przyrównania sekwencji celu do szablonu i identyfikacja regionów niepewnych oraz nieustrukturalizowanych. Modele białek można również, z dużym powodzeniem, stosować do przewidywania ich funkcji. Przy obecnych dysproporcjach w wiedzy na temat sekwencji i struktur trzeciorzędowych tego typu analizy są często jedynym sposobem, aby uzyskać odpowiedź na najważniejsze pytania biologii dotyczące funkcji poznanych białek oraz przebiegu procesów, w których one uczestniczą. Jedną z możliwych analiz tego typu jest rozpatrywanie
, co")
15 właściwości powierzchni modelu, np. przy zastosowaniu mapowania powierzchni białek na obiekty sferyczne i porównywaniu tych sfer. W takim wypadku nieuniknione niedokładności w budowie modelu nie mają znaczenia dla wyników eksperymentu [33], a w literaturze można znaleźć bardzo wiele przykładów kiedy wyniki tego typu przewidywań teoretycznych pokryły się z wynikami uzyskanymi poźniej drogą doświadczalną [34]. Jedna z najintensywniej rozwijanych gałęzi bioinformatyki jest bezpośrednio powiązana z przemyslem farmaceutycznym i dotyczy projektowania leków. Aktualny stopień zaawansowania metod przewidywania struktur trzeciorzędowych pozwala na tworzenie na tyle dokładnych modeli białek, np. białek bakteryjnych, że możliwym jest poznanie, drogą czysto teoretyczną, cząsteczek molekularnych z jakimi one oddziaływują. Powyższe przykłady dowodzą jak biologia teoretyczna może w wielu wypadkach wychodzić naprzeciw biologii doświadczalnej, także tej, która ma swoje praktyczne zastosowanie w dziedzinach niezwykle ważnych dla ludzkości. Autorzy są przekonani, że przewidywanie struktury przestrzennej białka stanie się wkrótce elementarną częścią większości eksperymentów białkowych, ułatwiając nie tylko fazę projektową eksperymentu, ale często również interpretację wyników eksperymentalnych. Podziękowania Autorzy chcieliby gorąco podziękować wszystkim twórcom metod bioinformatycznych, które są nieodpłatnie udostępniane społeczności akademickiej. Dziękujemy także pozostałym członkom naszego Zespołu za owocne dyskusje i za wkład w rozwój nowych metod badawczych. Nasze badania naukowe możliwe są dzięki wsparciu finansowemu zapewnionemu przez granty i stypendia z KBN, NIH, EMBO, HHMI, EU 6FP, FNP i BIF. Literatura [1] Anfinsen CB (1973) Principles that govern the folding of protein chains. Science, 181: [2] Chothia C (1992) Proteins. One thousand families for the molecular biologist. Nature, 357(6379):543-4 [3] Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25: [4] Karplus K, Barrett C, Hughey R (1998). Hidden Markov models for detecting remote homologies. Bioinformatics,14: [5] Park J, Teichmann SA, Hubbard T, Chothia C (1997) Intermediate sequences increase the detection of homology between sequences. J Mol Biol., 273: [6] Rost B Review: protein secondary structure prediction continues to rise. J Struct Biol, 134: [7] Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22: [8] Frishman D, Argos P (1995) Knowledge-based protein secondary structure assignment. Proteins, 23: [9] Chou PY, Fasman GD (1974) Prediction of protein conformation. Biochemistry, 13: [10] Garnier J, Osguthorpe DJ, Robson B (1978) Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol., 120:
![teoretycznych pokryły się z wynikami uzyskanymi poźniej drogą doświadczalną [34].](/docs-images/40/3554325/images/page_15.jpg "Jedna z najintensywniej rozwijanych gałęzi bioinformatyki jest bezpośrednio powiązana z przemyslem farmaceutycznym i dotyczy projektowania leków.")
16 [11] Gerstein M, Levitt M (1997) A structural census of the current population of protein sequences. Proc Natl Acad Sci U S A., 94: [12] Wolf YI, Grishin NV, Koonin EV (2000) Estimating the number of protein folds and families from complete genome data. J Mol Biol., 299: [13] Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature, 420: [14] Fischer D, Elofsson A, Rice D, Eisenberg D (1996) Assessing the performance of fold recognition methods by means of a comprehensive benchmark. Pac. Symp. Biocomput, [15] Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234: , [16] Peitsch MC (1996) ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans. 24: [17] Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L (2004) The importance of alignment accuracy for molecular replacement. Acta Crystallogr D Biol Crystallogr. 60: [18] Kurowski MA, Bujnicki JM (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res, 31: [19] Fischer D (2003) 3D-SHOTGUN: A novel, cooperative, fold-recognition meta-predictor. Proteins: Structure, Function and Genetics 51: [20] Siew N, Elofsson A, Rychlewski L, Fischer D (2000) MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics, 16: [21] Sasson I, Fischer D (2003) Modeling three-dimensional protein structures for CASP5 using the 3D-SHOTGUN meta-predictors. Proteins, 53 Suppl 6: [22] Fischer D (2003) 3DS3 and 3DS5 3D-SHOTGUN meta-predictors in CAFASP3. Proteins, 53 Suppl 6: [23] Bates PA, Kelley LA, MacCallum RM, Sternberg MJ (2001) Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D- PSSM. Proteins; Suppl 5: [24] Kosiński J, Cymerman IA, Feder M, Kurowski MA, Sasin JM, Bujnicki JM A "FRankenstein's monster" approach to comparative modeling: merging the finest fragments of Fold-Recognition models and iterative model refinement aided by 3D structure evaluation. Proteins 53 Suppl 6: [25] Eisenberg D, Luthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol, 277: [26] Bradley P, Chivian D, Meiler J, Misura KM, Rohl CA, Schief WR, Wedemeyer WJ, Schueler-Furman O, Murphy P, Schonbrun J, Strauss CE, Baker D (2003) Rosetta predictions in CASP5: successes, failures, and prospects for complete automation. Proteins;53 Suppl 6: [27] Bonneau R, Tsai J, Ruczinski I, Chivian D, Rohl C, Strauss CE, Baker D (2001) Rosetta in CASP4: progress in ab initio protein structure prediction. Proteins, Suppl 5: [28] Rohl CA, Strauss CE, Misura KM, Baker D (2004) Protein structure prediction using Rosetta. Methods Enzymol.; 383: [29] Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res; 32(Web Server issue): [30] Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D (2003) Automated prediction of CASP-5 structures using the Robetta server. Proteins, 53 Suppl, 6: [31] Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L (2004) The importance of
![[13] Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature, 420: 218-23.](/docs-images/40/3554325/images/page_16.jpg "[14] Fischer D, Elofsson A, Rice D, Eisenberg D (1996) Assessing the performance of fold recognition methods by means of a comprehensive benchmark. Pac. Symp. Biocomput, 300-18.")
17 alignment accuracy for molecular replacement. Acta Crystallogr D Biol Crystallogr. 60: [32] Claude JB, Suhre K, Notredame C, Claviere JM, Abergel C (2004) CaspR: a web server for automated molecular replacement using homology modeling. Nucleic Acids Research,32, (Web Server issue): [33] Pawlowski K, Godzik A (2001) Surface Map Comparison: Studying Function Diversity of Homologous Proteins. J Mol Biol; 309: [34] Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC (2001) A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J Biol Chem, 276:
![Nucleic Acids Research,32, (Web Server issue): 606-609 [33] Pawlowski K, Godzik A (2001) Surface Map Comparison: Studying Function Diversity of Homologous](/docs-images/40/3554325/images/page_17.jpg "Proteins. J Mol Biol; 309:793-806.")
18 a) NBB ADTKEVLEAREAYFKSLGGSMKAMTGVAK---AFDAEAAKVEAAKLEKI--LATDVAPLFPAGTSSTDLP 1A7V QT--DVIAQRKAILKQMGEATKPIAAMLKGEAKFDQA---VVQKSLAAIADDSKKLPALFPADSK--TGG 1GQA ADAEHVVEARKGYFSLVALEFGPLAAMAKGEMPYDAAAAKAHASDLVTL--TKYDPSDLYAPGTSADDVK 1NBB 1A7V 1GQA GQTEAKAAIWANMDDFGAKGKAMHEAGGAVIAAANAGDG-AAFGAALQKLGGTCKACHDDYREED-- -DTAALPKIWEDKAKFDDLFAKLAAAATAAQGT---IKDEASLKANIGGVLGNCKSCHDDFRAKKS- G-TAAKAAIWQDADGFQAKGMAFFEAVAALEPAA--GAGQKELAAAVGKVGGTCKSCHDDFRVKR- b) 1NBB 1A7V 1GQA Rysunek 1 Porównanie dywergencji sekwencji i struktur na podstawie białek należacych do rodziny cytochromów c. A) Przyrównanie sekwencji aminokwasowych cytochromów c pochodzących z następujących organizmów: 1NBB - Rhodobacter capsulatus, 1A7V - Rhodopseudomonas palustris, 1GQA - Rhodobacter sphaeroides. B) Przyrównanie rozwiązanych krystalograficznie struktur w.w. białek. Białka homologiczne mimo utraty podobieństwa sekwencji zachowują ewidentne podobieństwo struktury trzeciorzędowej.

19 Rysunek 2 Etapy modelowania homologicznego.
20 Tabela 1 Programy służące przeszukiwaniu baz domen białkowych: Program URL ( Programy służące przeszukiwaniu baz domen białkowych PFAM SMART smart.embl-heidelberg.de/ TIGRFAMs PRODOME prodes.toulouse.inra.fr/prodom/2002.1/html/home.php PROSITE us.expasy.org/prosite/ SBASE hydra.icgeb.trieste.it/~kristian/sbase/ BLOCKS bioinfo.weizmann.ac.il/blocks/ COGs CDD INTERPRO CDART PRINTS Tabela 2 Programy służące obliczaniu przyrównań wielosekwencyjnych Program URL ( Programy służące obliczaniu przyrównań wielosekwencyjnych ClustalW TCoffee igs-server.cnrs-mrs.fr/tcoffee/tcoffee_cgi/index.cgi Macaw ftp://ncbi.nlm.nih.gov/pub/macaw/ PCMA ftp://iole.swmed.edu/pub/pcma/ Tabela 3 Programy służące przewidywaniu struktury drugorzędowej białek Program URL ( Serwisy służące przewidywaniu elementów struktury α/β/pętla PSIPRED bioinf.cs.ucl.ac.uk/psipred/ SSPRO PHD cubic.bioc.columbia.edu/predictprotein/ PROF PRED2ARY APSSP2 PREDATOR bioweb.pasteur.fr/seqanal/interfaces/predator-simple.html NNSSP bioweb.pasteur.fr/seqanal/interfaces/nnssp-simple.html HMMSTR NPREDICT Przewidywanie innych typów struktur drugorzędowych TURNS COILS Serwisy służące przewidywaniu rejonów nieustrukturalizowanych NORSP cubic.bioc.columbia.edu/services/norsp/ GLOBPLOT globplot.embl.de/ PONDR Meta-serwery integrujące przewidywania generowane przez inne metody JPRED NPS@ npsa-pbil.ibcp.fr META-PP cubic.bioc.columbia.edu/meta/ Serwis prezentujący ocenę wiarygodności przewidywań struktury drugorzędowej EVA cubic.bioc.columbia.edu/eva/doc/intro_sec.html

Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania? Dlaczego? Struktura krystaliczną czy NMR (to samo białko, ta sama rozdzielczość)? Strukturę
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania? Dlaczego? Struktura krystaliczną czy NMR (to samo białko, ta sama rozdzielczość)? Strukturę
Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Modelowanie homologiczne
 Modelowanie homologiczne Struktura trzeciorzędowa ułatwia planowanie eksperymentów oraz interpretację otrzymanych wyników Struktura trzeciorzędowa Hemoglobiny - na 226 białek z tej rodziny zawsze grupa
Modelowanie homologiczne Struktura trzeciorzędowa ułatwia planowanie eksperymentów oraz interpretację otrzymanych wyników Struktura trzeciorzędowa Hemoglobiny - na 226 białek z tej rodziny zawsze grupa
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład VI Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład VI Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Modelowanie białek ab initio / de novo
 Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) Algorytmy i Struktury Danych PIŁA
 PAWLICKI Piotr (55567) Algorytmy i Struktury Danych PIŁA") OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) 16.01.2003 Algorytmy i Struktury Danych PIŁA ALGORYTMY ZACHŁANNE czas [ms] Porównanie Algorytmów Rozwiązyjących problem TSP 100 000 000 000,000 10 000 000
OSTASZEWSKI Paweł (55566) PAWLICKI Piotr (55567) 16.01.2003 Algorytmy i Struktury Danych PIŁA ALGORYTMY ZACHŁANNE czas [ms] Porównanie Algorytmów Rozwiązyjących problem TSP 100 000 000 000,000 10 000 000
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Modelowanie białek ab initio / de novo
 Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Lingwistyczny system definicyjny wykorzystujący korpusy tekstów oraz zasoby internetowe.
 Lingwistyczny system definicyjny wykorzystujący korpusy tekstów oraz zasoby internetowe. Autor: Mariusz Sasko Promotor: dr Adrian Horzyk Plan prezentacji 1. Wstęp 2. Cele pracy 3. Rozwiązanie 3.1. Robot
Lingwistyczny system definicyjny wykorzystujący korpusy tekstów oraz zasoby internetowe. Autor: Mariusz Sasko Promotor: dr Adrian Horzyk Plan prezentacji 1. Wstęp 2. Cele pracy 3. Rozwiązanie 3.1. Robot
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
166 Wstęp do statystyki matematycznej
 166 Wstęp do statystyki matematycznej Etap trzeci realizacji procesu analizy danych statystycznych w zasadzie powinien rozwiązać nasz zasadniczy problem związany z identyfikacją cechy populacji generalnej
166 Wstęp do statystyki matematycznej Etap trzeci realizacji procesu analizy danych statystycznych w zasadzie powinien rozwiązać nasz zasadniczy problem związany z identyfikacją cechy populacji generalnej
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Aproksymacja funkcji a regresja symboliczna
 Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA. Marta Szachniuk
 Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Kombinatoryczna analiza widm 2D-NOESY w spektroskopii Magnetycznego Rezonansu Jądrowego cząsteczek RNA Marta Szachniuk Plan prezentacji Wprowadzenie do tematyki badań Teoretyczny model problemu Złożoność
Modelowanie białek ab initio / de novo
 Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Modelowanie białek ab initio / de novo Słowniczek de novo - od początku, na nowo ab initio - od początku Słowniczek de novo - kategoria metod przewidywania struktury, w których nie używa się wzorców homologicznych
Testowanie hipotez statystycznych. Wnioskowanie statystyczne
 Testowanie hipotez statystycznych Wnioskowanie statystyczne Hipoteza statystyczna to dowolne przypuszczenie co do rozkładu populacji generalnej (jego postaci funkcyjnej lub wartości parametrów). Hipotezy
Testowanie hipotez statystycznych Wnioskowanie statystyczne Hipoteza statystyczna to dowolne przypuszczenie co do rozkładu populacji generalnej (jego postaci funkcyjnej lub wartości parametrów). Hipotezy
Badanie długości czynników sieciujących metodami symulacji komputerowych
 Badanie długości czynników sieciujących metodami symulacji komputerowych Agnieszka Obarska-Kosińska Prof. dr hab. Bogdan Lesyng Promotorzy: Dr hab. Janusz Bujnicki Zakład Biofizyki, Instytut Fizyki Doświadczalnej,
Badanie długości czynników sieciujących metodami symulacji komputerowych Agnieszka Obarska-Kosińska Prof. dr hab. Bogdan Lesyng Promotorzy: Dr hab. Janusz Bujnicki Zakład Biofizyki, Instytut Fizyki Doświadczalnej,
OPTYMALIZACJA HARMONOGRAMOWANIA MONTAŻU SAMOCHODÓW Z ZASTOSOWANIEM PROGRAMOWANIA W LOGICE Z OGRANICZENIAMI
 Autoreferat do rozprawy doktorskiej OPTYMALIZACJA HARMONOGRAMOWANIA MONTAŻU SAMOCHODÓW Z ZASTOSOWANIEM PROGRAMOWANIA W LOGICE Z OGRANICZENIAMI Michał Mazur Gliwice 2016 1 2 Montaż samochodów na linii w
Autoreferat do rozprawy doktorskiej OPTYMALIZACJA HARMONOGRAMOWANIA MONTAŻU SAMOCHODÓW Z ZASTOSOWANIEM PROGRAMOWANIA W LOGICE Z OGRANICZENIAMI Michał Mazur Gliwice 2016 1 2 Montaż samochodów na linii w
Bioinformatyka wykład 10
 Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Hierarchiczna analiza skupień
 Hierarchiczna analiza skupień Cel analizy Analiza skupień ma na celu wykrycie w zbiorze obserwacji klastrów, czyli rozłącznych podzbiorów obserwacji, wewnątrz których obserwacje są sobie w jakimś określonym
Hierarchiczna analiza skupień Cel analizy Analiza skupień ma na celu wykrycie w zbiorze obserwacji klastrów, czyli rozłącznych podzbiorów obserwacji, wewnątrz których obserwacje są sobie w jakimś określonym
RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych
 RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych Joanna Wiśniewska Promotor: dr inż. P. Łukasiak Spis treści 1. Zakres pracy magisterskiej 2. Struktura białka 3. Struktura kwasów nukleionowych
RMSD - Ocena jakości wybranych molekularnych struktur przestrzennych Joanna Wiśniewska Promotor: dr inż. P. Łukasiak Spis treści 1. Zakres pracy magisterskiej 2. Struktura białka 3. Struktura kwasów nukleionowych
Automatyczne tworzenie trójwymiarowego planu pomieszczenia z zastosowaniem metod stereowizyjnych
 Automatyczne tworzenie trójwymiarowego planu pomieszczenia z zastosowaniem metod stereowizyjnych autor: Robert Drab opiekun naukowy: dr inż. Paweł Rotter 1. Wstęp Zagadnienie generowania trójwymiarowego
Automatyczne tworzenie trójwymiarowego planu pomieszczenia z zastosowaniem metod stereowizyjnych autor: Robert Drab opiekun naukowy: dr inż. Paweł Rotter 1. Wstęp Zagadnienie generowania trójwymiarowego
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Opis efektów kształcenia dla programu kształcenia (kierunkowe efekty kształcenia) WIEDZA. rozumie cywilizacyjne znaczenie matematyki i jej zastosowań
 WIEDZA. rozumie cywilizacyjne znaczenie matematyki i jej zastosowań") TABELA ODNIESIEŃ EFEKTÓW KSZTAŁCENIA OKREŚLONYCH DLA PROGRAMU KSZTAŁCENIA DO EFEKTÓW KSZTAŁCENIA OKREŚLONYCH DLA OBSZARU KSZTAŁCENIA I PROFILU STUDIÓW PROGRAM KSZTAŁCENIA: POZIOM KSZTAŁCENIA: PROFIL KSZTAŁCENIA:
TABELA ODNIESIEŃ EFEKTÓW KSZTAŁCENIA OKREŚLONYCH DLA PROGRAMU KSZTAŁCENIA DO EFEKTÓW KSZTAŁCENIA OKREŚLONYCH DLA OBSZARU KSZTAŁCENIA I PROFILU STUDIÓW PROGRAM KSZTAŁCENIA: POZIOM KSZTAŁCENIA: PROFIL KSZTAŁCENIA:
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Modelowanie jako sposób opisu rzeczywistości. Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka
 Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
Spis treści. Przedmowa... XI. Rozdział 1. Pomiar: jednostki miar... 1. Rozdział 2. Pomiar: liczby i obliczenia liczbowe... 16
 Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
Bioinformatyka wykład 8, 27.XI.2012
 Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Dokowanie molekularne. Karol Kamel Uniwersytet Warszawski
 molekularne Wstęp Dokowanie metoda modelowania molekularnego, pozwalająca na znalezienie położenia (i konformacji) liganda w miejscu wiążącym receptora. Informacja ta pozwala na ocenę energii swobodnej
molekularne Wstęp Dokowanie metoda modelowania molekularnego, pozwalająca na znalezienie położenia (i konformacji) liganda w miejscu wiążącym receptora. Informacja ta pozwala na ocenę energii swobodnej
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
w analizie wyników badań eksperymentalnych, w problemach modelowania zjawisk fizycznych, w analizie obserwacji statystycznych.
 Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(), zwaną funkcją aproksymującą
Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(), zwaną funkcją aproksymującą
Weryfikacja hipotez statystycznych. KG (CC) Statystyka 26 V / 1
 Statystyka 26 V / 1") Weryfikacja hipotez statystycznych KG (CC) Statystyka 26 V 2009 1 / 1 Sformułowanie problemu Weryfikacja hipotez statystycznych jest drugą (po estymacji) metodą uogólniania wyników uzyskanych w próbie
Weryfikacja hipotez statystycznych KG (CC) Statystyka 26 V 2009 1 / 1 Sformułowanie problemu Weryfikacja hipotez statystycznych jest drugą (po estymacji) metodą uogólniania wyników uzyskanych w próbie
Bioinformatyka wykład 9
 Bioinformatyka wykład 9 14.XII.21 białkowa bioinformatyka strukturalna krzysztof_pawlowski@sggw.pl 211-1-17 1 Plan wykładu struktury białek dlaczego? struktury białek geometria i fizyka modyfikacje kowalencyjne
Bioinformatyka wykład 9 14.XII.21 białkowa bioinformatyka strukturalna krzysztof_pawlowski@sggw.pl 211-1-17 1 Plan wykładu struktury białek dlaczego? struktury białek geometria i fizyka modyfikacje kowalencyjne
Wprowadzenie do analizy korelacji i regresji
 Statystyka dla jakości produktów i usług Six sigma i inne strategie Wprowadzenie do analizy korelacji i regresji StatSoft Polska Wybrane zagadnienia analizy korelacji Przy analizie zjawisk i procesów stanowiących
Statystyka dla jakości produktów i usług Six sigma i inne strategie Wprowadzenie do analizy korelacji i regresji StatSoft Polska Wybrane zagadnienia analizy korelacji Przy analizie zjawisk i procesów stanowiących
na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das
 na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das wykonała: Marta Szynczewska bioinformatyka Uniwersytet Jagielloński Struktura I-rzędowa
na podstawie artykułu: Modeling Complex RNA Tertiary Folds with Rosetta Clarence Yu Cheng, Fang-Chieh Chou, Rhiju Das wykonała: Marta Szynczewska bioinformatyka Uniwersytet Jagielloński Struktura I-rzędowa
Wykład 4: Wnioskowanie statystyczne. Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA
 Wykład 4: Wnioskowanie statystyczne Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA Idea wnioskowania statystycznego Celem analizy statystycznej nie jest zwykle tylko
Wykład 4: Wnioskowanie statystyczne Podstawowe informacje oraz implementacja przykładowego testu w programie STATISTICA Idea wnioskowania statystycznego Celem analizy statystycznej nie jest zwykle tylko
Systemy uczące się Lab 4
 Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW
 PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
Elementy modelowania matematycznego
 Elementy modelowania matematycznego Modelowanie algorytmów klasyfikujących. Podejście probabilistyczne. Naiwny klasyfikator bayesowski. Modelowanie danych metodą najbliższych sąsiadów. Jakub Wróblewski
Elementy modelowania matematycznego Modelowanie algorytmów klasyfikujących. Podejście probabilistyczne. Naiwny klasyfikator bayesowski. Modelowanie danych metodą najbliższych sąsiadów. Jakub Wróblewski
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
Wzorcowe efekty kształcenia dla kierunku studiów biotechnologia studia pierwszego stopnia profil ogólnoakademicki
 Załącznik nr 2 do Uchwały Rady Wydziału Biochemii, Biofizyki i Biotechnologii UJ z dnia 19 czerwca 2018 r. w sprawie programu i planu studiów na kierunku BIOTECHNOLOGIA na poziomie studiów pierwszego stopnia
Załącznik nr 2 do Uchwały Rady Wydziału Biochemii, Biofizyki i Biotechnologii UJ z dnia 19 czerwca 2018 r. w sprawie programu i planu studiów na kierunku BIOTECHNOLOGIA na poziomie studiów pierwszego stopnia
MultiSETTER: web server for multiple RNA structure comparison. Sandra Sobierajska Uniwersytet Jagielloński
 MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
Projektowanie oprogramowania cd. Projektowanie oprogramowania cd. 1/34
 Projektowanie oprogramowania cd. Projektowanie oprogramowania cd. 1/34 Projektowanie oprogramowania cd. 2/34 Modelowanie CRC Modelowanie CRC (class-responsibility-collaborator) Metoda identyfikowania poszczególnych
Projektowanie oprogramowania cd. Projektowanie oprogramowania cd. 1/34 Projektowanie oprogramowania cd. 2/34 Modelowanie CRC Modelowanie CRC (class-responsibility-collaborator) Metoda identyfikowania poszczególnych
Algorytm Genetyczny. zastosowanie do procesów rozmieszczenia stacji raportujących w sieciach komórkowych
 Algorytm Genetyczny zastosowanie do procesów rozmieszczenia stacji raportujących w sieciach komórkowych Dlaczego Algorytmy Inspirowane Naturą? Rozwój nowych technologii: złożone problemy obliczeniowe w
Algorytm Genetyczny zastosowanie do procesów rozmieszczenia stacji raportujących w sieciach komórkowych Dlaczego Algorytmy Inspirowane Naturą? Rozwój nowych technologii: złożone problemy obliczeniowe w
Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
 Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Testowanie hipotez statystycznych
 9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Program MC. Obliczyć radialną funkcję korelacji. Zrobić jej wykres. Odczytać z wykresu wartość radialnej funkcji korelacji w punkcie r=
 Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
Program MC Napisać program symulujący twarde kule w zespole kanonicznym. Dla N > 100 twardych kul. Gęstość liczbowa 0.1 < N/V < 0.4. Zrobić obliczenia dla 2,3 różnych wartości gęstości. Obliczyć radialną
Generator testów Bioinformatyka wer / 0 Strona: 1
 Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Statystyki: miary opisujące rozkład! np. : średnia, frakcja (procent), odchylenie standardowe, wariancja, mediana itd.
, odchylenie standardowe, wariancja, mediana itd.") Wnioskowanie statystyczne obejmujące metody pozwalające na uogólnianie wyników z próby na nieznane wartości parametrów oraz szacowanie błędów tego uogólnienia. Przewidujemy nieznaną wartości parametru
Wnioskowanie statystyczne obejmujące metody pozwalające na uogólnianie wyników z próby na nieznane wartości parametrów oraz szacowanie błędów tego uogólnienia. Przewidujemy nieznaną wartości parametru
Diagramy ERD. Model struktury danych jest najczęściej tworzony z wykorzystaniem diagramów pojęciowych (konceptualnych). Najpopularniejszym
. Najpopularniejszym") Diagramy ERD. Model struktury danych jest najczęściej tworzony z wykorzystaniem diagramów pojęciowych (konceptualnych). Najpopularniejszym konceptualnym modelem danych jest tzw. model związków encji (ERM
Diagramy ERD. Model struktury danych jest najczęściej tworzony z wykorzystaniem diagramów pojęciowych (konceptualnych). Najpopularniejszym konceptualnym modelem danych jest tzw. model związków encji (ERM
Statystyki: miary opisujące rozkład! np. : średnia, frakcja (procent), odchylenie standardowe, wariancja, mediana itd.
, odchylenie standardowe, wariancja, mediana itd.") Wnioskowanie statystyczne obejmujące metody pozwalające na uogólnianie wyników z próby na nieznane wartości parametrów oraz szacowanie błędów tego uogólnienia. Przewidujemy nieznaną wartości parametru
Wnioskowanie statystyczne obejmujące metody pozwalające na uogólnianie wyników z próby na nieznane wartości parametrów oraz szacowanie błędów tego uogólnienia. Przewidujemy nieznaną wartości parametru
METODY ROZWIĄZYWANIA RÓWNAŃ NIELINIOWYCH
 METODY ROZWIĄZYWANIA RÓWNAŃ NIELINIOWYCH Jednym z zastosowań metod numerycznych jest wyznaczenie pierwiastka lub pierwiastków równania nieliniowego. W tym celu stosuje się szereg metod obliczeniowych np:
METODY ROZWIĄZYWANIA RÓWNAŃ NIELINIOWYCH Jednym z zastosowań metod numerycznych jest wyznaczenie pierwiastka lub pierwiastków równania nieliniowego. W tym celu stosuje się szereg metod obliczeniowych np:
Komputerowe wspomaganie projektowanie leków
 Komputerowe wspomaganie projektowanie leków wykład V Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Komputerowe wspomaganie projektowanie leków wykład V Prof. dr hab. Sławomir Filipek Grupa BIOmodelowania Uniwersytet Warszawski, Wydział Chemii oraz Centrum Nauk Biologiczno-Chemicznych Cent-III www.biomodellab.eu
Generator testów bioinformatyka wer / Strona: 1
 Przedmiot: wyklad monograficzny Nazwa testu: bioinformatyka wer. 1.0.6 Nr testu 10469906 Klasa: 5 IBOS Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Aminokwas jest to związek organiczny zawierający A) grupę
Przedmiot: wyklad monograficzny Nazwa testu: bioinformatyka wer. 1.0.6 Nr testu 10469906 Klasa: 5 IBOS Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Aminokwas jest to związek organiczny zawierający A) grupę
Mechatronika i inteligentne systemy produkcyjne. Modelowanie systemów mechatronicznych Platformy przetwarzania danych
 Mechatronika i inteligentne systemy produkcyjne Modelowanie systemów mechatronicznych Platformy przetwarzania danych 1 Sterowanie procesem oparte na jego modelu u 1 (t) System rzeczywisty x(t) y(t) Tworzenie
Mechatronika i inteligentne systemy produkcyjne Modelowanie systemów mechatronicznych Platformy przetwarzania danych 1 Sterowanie procesem oparte na jego modelu u 1 (t) System rzeczywisty x(t) y(t) Tworzenie
Sterowanie wielkością zamówienia w Excelu - cz. 3
 Sterowanie wielkością zamówienia w Excelu - cz. 3 21.06.2005 r. 4. Planowanie eksperymentów symulacyjnych Podczas tego etapu ważne jest określenie typu rozkładu badanej charakterystyki. Dzięki tej informacji
Sterowanie wielkością zamówienia w Excelu - cz. 3 21.06.2005 r. 4. Planowanie eksperymentów symulacyjnych Podczas tego etapu ważne jest określenie typu rozkładu badanej charakterystyki. Dzięki tej informacji
Sterowanie procesem i jego zdolność. Zbigniew Wiśniewski
 Sterowanie procesem i jego zdolność Zbigniew Wiśniewski Wybór cech do kart kontrolnych Zaleca się aby w pierwszej kolejności były brane pod uwagę cechy dotyczące funkcjonowania wyrobu lub świadczenia usługi
Sterowanie procesem i jego zdolność Zbigniew Wiśniewski Wybór cech do kart kontrolnych Zaleca się aby w pierwszej kolejności były brane pod uwagę cechy dotyczące funkcjonowania wyrobu lub świadczenia usługi
Zadania ze statystyki, cz.6
 Zadania ze statystyki, cz.6 Zad.1 Proszę wskazać, jaką część pola pod krzywą normalną wyznaczają wartości Z rozkładu dystrybuanty rozkładu normalnego: - Z > 1,25 - Z > 2,23 - Z < -1,23 - Z > -1,16 - Z
Zadania ze statystyki, cz.6 Zad.1 Proszę wskazać, jaką część pola pod krzywą normalną wyznaczają wartości Z rozkładu dystrybuanty rozkładu normalnego: - Z > 1,25 - Z > 2,23 - Z < -1,23 - Z > -1,16 - Z
METODY CHEMOMETRYCZNE W IDENTYFIKACJI ŹRÓDEŁ POCHODZENIA
 METODY CHEMOMETRYCZNE W IDENTYFIKACJI ŹRÓDEŁ POCHODZENIA AMFETAMINY Waldemar S. Krawczyk Centralne Laboratorium Kryminalistyczne Komendy Głównej Policji, Warszawa (praca obroniona na Wydziale Chemii Uniwersytetu
METODY CHEMOMETRYCZNE W IDENTYFIKACJI ŹRÓDEŁ POCHODZENIA AMFETAMINY Waldemar S. Krawczyk Centralne Laboratorium Kryminalistyczne Komendy Głównej Policji, Warszawa (praca obroniona na Wydziale Chemii Uniwersytetu
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii
 Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Analiza i projektowanie oprogramowania. Analiza i projektowanie oprogramowania 1/32
 Analiza i projektowanie oprogramowania Analiza i projektowanie oprogramowania 1/32 Analiza i projektowanie oprogramowania 2/32 Cel analizy Celem fazy określania wymagań jest udzielenie odpowiedzi na pytanie:
Analiza i projektowanie oprogramowania Analiza i projektowanie oprogramowania 1/32 Analiza i projektowanie oprogramowania 2/32 Cel analizy Celem fazy określania wymagań jest udzielenie odpowiedzi na pytanie:
Komputerowe wspomaganie projektowania leków
 Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Komputerowe wspomaganie projektowania leków MECHANIKA MOLEKULARNA I KWANTOWA W MM korzysta się z równań wynikających z praw fizyki klasycznej i stosuje się je do jader atomów z pominięciem elektronów,
Modelowanie motywów łańcuchami Markowa wyższego rzędu
 Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
Modelowanie motywów łańcuchami Markowa wyższego rzędu Uniwersytet Warszawski Wydział Matematyki, Informatyki i Mechaniki 23 października 2008 roku Plan prezentacji 1 Źródła 2 Motywy i ich znaczenie Łańcuchy
Opis postępowania przy eksportowaniu geometrii z systemu Unigraphics NX do pakietu PANUKL (ver. A)
") 1 Opis postępowania przy eksportowaniu geometrii z systemu Unigraphics NX do pakietu PANUKL (ver. A) Przedstawiony poniżej schemat przygotowania geometrii w systemie Unigraphics NX na potrzeby programu
1 Opis postępowania przy eksportowaniu geometrii z systemu Unigraphics NX do pakietu PANUKL (ver. A) Przedstawiony poniżej schemat przygotowania geometrii w systemie Unigraphics NX na potrzeby programu
Testowanie hipotez statystycznych
 Agenda Instytut Matematyki Politechniki Łódzkiej 2 stycznia 2012 Agenda Agenda 1 Wprowadzenie Agenda 2 Hipoteza oraz błędy I i II rodzaju Hipoteza alternatywna Statystyka testowa Zbiór krytyczny Poziom
Agenda Instytut Matematyki Politechniki Łódzkiej 2 stycznia 2012 Agenda Agenda 1 Wprowadzenie Agenda 2 Hipoteza oraz błędy I i II rodzaju Hipoteza alternatywna Statystyka testowa Zbiór krytyczny Poziom
Optymalizacja ciągła
 Optymalizacja ciągła 5. Metoda stochastycznego spadku wzdłuż gradientu Wojciech Kotłowski Instytut Informatyki PP http://www.cs.put.poznan.pl/wkotlowski/ 04.04.2019 1 / 20 Wprowadzenie Minimalizacja różniczkowalnej
Optymalizacja ciągła 5. Metoda stochastycznego spadku wzdłuż gradientu Wojciech Kotłowski Instytut Informatyki PP http://www.cs.put.poznan.pl/wkotlowski/ 04.04.2019 1 / 20 Wprowadzenie Minimalizacja różniczkowalnej
Wnioskowanie bayesowskie
 Wnioskowanie bayesowskie W podejściu klasycznym wnioskowanie statystyczne oparte jest wyłącznie na podstawie pobranej próby losowej. Możemy np. estymować punktowo lub przedziałowo nieznane parametry rozkładów,
Wnioskowanie bayesowskie W podejściu klasycznym wnioskowanie statystyczne oparte jest wyłącznie na podstawie pobranej próby losowej. Możemy np. estymować punktowo lub przedziałowo nieznane parametry rozkładów,
mgr inż. Magdalena Deckert Poznań, r. Metody przyrostowego uczenia się ze strumieni danych.
 mgr inż. Magdalena Deckert Poznań, 30.11.2010r. Metody przyrostowego uczenia się ze strumieni danych. Plan prezentacji Wstęp Concept drift i typy zmian Algorytmy przyrostowego uczenia się ze strumieni
mgr inż. Magdalena Deckert Poznań, 30.11.2010r. Metody przyrostowego uczenia się ze strumieni danych. Plan prezentacji Wstęp Concept drift i typy zmian Algorytmy przyrostowego uczenia się ze strumieni
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych
 Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
Proteomika: umożliwia badanie zestawu wszystkich lub prawie wszystkich białek komórkowych Zalety w porównaniu z analizą trankryptomu: analiza transkryptomu komórki identyfikacja mrna nie musi jeszcze oznaczać
Wprowadzenie do PKI. 1. Wstęp. 2. Kryptografia symetryczna. 3. Kryptografia asymetryczna
 1. Wstęp Wprowadzenie do PKI Infrastruktura klucza publicznego (ang. PKI - Public Key Infrastructure) to termin dzisiaj powszechnie spotykany. Pod tym pojęciem kryje się standard X.509 opracowany przez
1. Wstęp Wprowadzenie do PKI Infrastruktura klucza publicznego (ang. PKI - Public Key Infrastructure) to termin dzisiaj powszechnie spotykany. Pod tym pojęciem kryje się standard X.509 opracowany przez
Priorytetyzacja przypadków testowych za pomocą macierzy
 Priorytetyzacja przypadków testowych za pomocą macierzy W niniejszym artykule przedstawiony został problem przyporządkowania priorytetów do przypadków testowych przed rozpoczęciem testów oprogramowania.
Priorytetyzacja przypadków testowych za pomocą macierzy W niniejszym artykule przedstawiony został problem przyporządkowania priorytetów do przypadków testowych przed rozpoczęciem testów oprogramowania.
Programowanie i techniki algorytmiczne
 Temat 2. Programowanie i techniki algorytmiczne Realizacja podstawy programowej 1) wyjaśnia pojęcie algorytmu, podaje odpowiednie przykłady algorytmów rozwiązywania różnych 2) formułuje ścisły opis prostej
Temat 2. Programowanie i techniki algorytmiczne Realizacja podstawy programowej 1) wyjaśnia pojęcie algorytmu, podaje odpowiednie przykłady algorytmów rozwiązywania różnych 2) formułuje ścisły opis prostej
Zagadnienia: wprowadzenie podstawowe pojęcia. Doświadczalnictwo. Anna Rajfura
 Zagadnienia: wprowadzenie podstawowe pojęcia Doświadczalnictwo 1 Termin doświadczalnictwo Doświadczalnictwo planowanie doświadczeń oraz analiza danych doświadczalnych z użyciem metod statystycznych. Doświadczalnictwo
Zagadnienia: wprowadzenie podstawowe pojęcia Doświadczalnictwo 1 Termin doświadczalnictwo Doświadczalnictwo planowanie doświadczeń oraz analiza danych doświadczalnych z użyciem metod statystycznych. Doświadczalnictwo
Opis zakładanych efektów kształcenia na studiach podyplomowych WIEDZA
 Opis zakładanych efektów kształcenia na studiach podyplomowych Nazwa studiów: BIOSTATYSTYKA PRAKTYCZNE ASPEKTY STATYSTYKI W BADANIACH MEDYCZNYCH Typ studiów: doskonalące Symbol Efekty kształcenia dla studiów
Opis zakładanych efektów kształcenia na studiach podyplomowych Nazwa studiów: BIOSTATYSTYKA PRAKTYCZNE ASPEKTY STATYSTYKI W BADANIACH MEDYCZNYCH Typ studiów: doskonalące Symbol Efekty kształcenia dla studiów
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
STATYSTYKA MATEMATYCZNA
 STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery
 http://xqtav.sourceforge.net XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery dr hab. Jerzy Tyszkiewicz dr Andrzej Kierzek mgr Jacek Sroka Grzegorz Kaczor praca mgr pod
http://xqtav.sourceforge.net XQTav - reprezentacja diagramów przepływu prac w formacie SCUFL przy pomocy XQuery dr hab. Jerzy Tyszkiewicz dr Andrzej Kierzek mgr Jacek Sroka Grzegorz Kaczor praca mgr pod
Spacery losowe generowanie realizacji procesu losowego
 Spacery losowe generowanie realizacji procesu losowego Michał Krzemiński Streszczenie Omówimy metodę generowania trajektorii spacerów losowych (błądzenia losowego), tj. szczególnych procesów Markowa z
Spacery losowe generowanie realizacji procesu losowego Michał Krzemiński Streszczenie Omówimy metodę generowania trajektorii spacerów losowych (błądzenia losowego), tj. szczególnych procesów Markowa z
Algorytm genetyczny (genetic algorithm)-
-") Optymalizacja W praktyce inżynierskiej często zachodzi potrzeba znalezienia parametrów, dla których system/urządzenie będzie działać w sposób optymalny. Klasyczne podejście do optymalizacji: sformułowanie
Optymalizacja W praktyce inżynierskiej często zachodzi potrzeba znalezienia parametrów, dla których system/urządzenie będzie działać w sposób optymalny. Klasyczne podejście do optymalizacji: sformułowanie
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
1. Symulacje komputerowe Idea symulacji Przykład. 2. Metody próbkowania Jackknife Bootstrap. 3. Łańcuchy Markova. 4. Próbkowanie Gibbsa
 BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
Recenzja pracy doktorskiej mgr Tomasza Świsłockiego pt. Wpływ oddziaływań dipolowych na własności spinorowego kondensatu rubidowego
 Prof. dr hab. Jan Mostowski Instytut Fizyki PAN Warszawa Warszawa, 15 listopada 2010 r. Recenzja pracy doktorskiej mgr Tomasza Świsłockiego pt. Wpływ oddziaływań dipolowych na własności spinorowego kondensatu
Prof. dr hab. Jan Mostowski Instytut Fizyki PAN Warszawa Warszawa, 15 listopada 2010 r. Recenzja pracy doktorskiej mgr Tomasza Świsłockiego pt. Wpływ oddziaływań dipolowych na własności spinorowego kondensatu
Generator testów 1.3.1 Bioinformatyka_zdalne wer. 1.0.13 / 0 Strona: 1
 Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...