(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
|
|
|
- Wiktor Chmiel
- 7 lat temu
- Przeglądów:
Transkrypt
 O udzieleniu patentu europejskiego ogłoszono: 16.04.2014 Europejski Biuletyn Patentowy 2014/16 EP 2132230 B1 (13) (51) T3 Int.Cl. C07K 16/42 (2006.01) A01K 67/027 (2006.")
1 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: (97) O udzieleniu patentu europejskiego ogłoszono: Europejski Biuletyn Patentowy 2014/16 EP B1 (13) (51) T3 Int.Cl. C07K 16/42 ( ) A01K 67/027 ( ) A61P 37/06 ( ) (54) Tytuł wynalazku: Apoptotyczne przeciwciała anty-ige wiążące IgE związaną z błoną (30) Pierwszeństwo: US P (43) Zgłoszenie ogłoszono: w Europejskim Biuletynie Patentowym nr 2009/51 (45) O złożeniu tłumaczenia patentu ogłoszono: Wiadomości Urzędu Patentowego 2014/09 (73) Uprawniony z patentu: Genentech, Inc., South San Francisco, US (72) Twórca(y) wynalazku: PL/EP T3 LAWREN WU, Foster City, US MERCEDESZ BALAZS, Hayward, US HANS BRIGHTBILL, San Francisco, US ANDREW CHAN, Menlo Park, US YVONNE CHEN, San Mateo, US ANAN CHUNTHARAPAI, Colma, US MARK DENNIS, San Carlos, US TERENCE WONG, Alameda, US (74) Pełnomocnik: rzecz. pat. Dorota Rzążewska JWP RZECZNICY PATENTOWI DOROTA RZĄŻEWSKA SP. J. ul. Żelazna 28/30 Sienna Center Warszawa Uwaga: W ciągu dziewięciu miesięcy od publikacji informacji o udzieleniu patentu europejskiego, każda osoba może wnieść do Europejskiego Urzędu Patentowego sprzeciw dotyczący udzielonego patentu europejskiego. Sprzeciw wnosi się w formie uzasadnionego na piśmie oświadczenia. Uważa się go za wniesiony dopiero z chwilą wniesienia opłaty za sprzeciw (Art. 99 (1) Konwencji o udzielaniu patentów europejskich).
2 19815/14/P-RO/DR EP Opis Apoptotyczne przeciwciała anty-ige wiążące IgE związaną z błoną Powiązania z Wcześniejszymi Zgłoszeniami [0001] Wynalazek zastrzega pierwszeństwo na podstawie 35 U.S.C. 119 (e) do U.S.S.N. 60/896, 339, złożonego 22 marca Tło Wynalazku Dziedzina Wynalazku [0002] Niniejszy wynalazek dotyczy apoptotycznych przeciwciał anty-ige, kodującego je kwasu nukleinowego, ich kompozycji terapeutycznych, oraz ich zastosowania w leczeniu zaburzeń mediowanych IgE. Opis Pokrewnej Dziedziny [0003] Alergia odnosi się do niektórych chorób, w których odpowiedź immunologiczna na antygeny środowiskowe wywołuje zapalenie tkanek i zaburzenia czynności narządów. Cechy kliniczne każdej choroby alergicznej odzwierciedlają immunologiczną odpowiedź zapalną w dotkniętym narządzie lub tkance. Cechy te są na ogół niezależne od właściwości chemicznych i fizycznych antygenu. Różnorodność reakcji alergicznych wynika z udziału różnych szlaków efektorowych immunologicznych, z których każdy generuje unikalny wzorzec zapalenia. [0004] Alergia jest powszechna na całym świecie. Skłonność do specyficznych chorób różni się jednak u różnych grup wiekowych, płci i ras. Częstość występowania wrażliwości na konkretne alergeny jest określona przez skłonności genetyczne i czynniki geograficzne oraz kulturowe, które są odpowiedzialne za ekspozycję na alergen. Stan kliniczny alergii dotyka tylko niektóre osoby, które natykają się na każdy alergen. Występowanie chorób alergicznych po ekspozycji na alergen wymaga nie tylko wcześniejszego uczulenia, ale również innych czynników, które określają lokalizację reakcji do konkretnego narządu. [0005] Proces biologiczny, który poprzedza chorobę alergię po ekspozycji na alergen alergen jest, wywołuje odpowiedź immunologiczną znaną jako uczulenie lub faza uczulenia. Gdy dojdzie do uczulenia, u osobnika nie pojawiają się objawy aż do kolejnej ekspozycji na alergen. Efekt uczulania znany jest również jako pamięć immunologiczna. [0006] Jednym z głównych szlaków, indukowanych w zapaleniu, jest szlak poprzez immunoglobulinę E (IgE). IgE odgrywa ważną rolę w alergii ze względu na jej rolę jako receptorów alergenu na powierzchni komórek tucznych i bazofili. Przeciwciała IgE są przymocowane do powierzchni komórek tucznych i bazofili w części Fc cząsteczki z receptorem o wysokim powinowactwie na powierzchni komórek, zwanym FcεRI. Reakcja alergiczna jest inicjowana, gdy wielowartościowa cząsteczka alergenu wiąże się z przeciwciałami, które zajmują te receptory. Wynikiem jest mostkowanie FcεRI, który z kolei przesyła sygnał do wnętrza komórki powodując uwalnianie i aktywację mediatorów zapalenia: histaminy, leukotrienów, czynników chemotaktycznych, czynników aktywujących płytki krwi i proteinaz. Mediatory te aktywowane działają lokalnie i powodują zwiększoną
3 2- przepuszczalność naczyń, rozszerzenie naczyń krwionośnych, skurcz mięśni gładkich i wydzielanie gruczołów śluzowych. Takie zdarzenia są określane klinicznie jako faza natychmiastowa lub wczesna i występują w ciągu pierwszych minut po ekspozycji na alergen. W ciągu następnych 12 godzin dochodzi do postępującego naciekania tkanek przez komórki zapalne, postępując od neutrofilów do eozynofilów do komórek jednojądrzastych w odpowiedzi na inne mediatory chemiczne niekoniecznie całkowicie zrozumiałe. Ten okres 6-12 dni po ekspozycji na alergen jest określany jako faza późna i charakteryzuje się objawami klinicznymi zapalenia komórkowego. Biorąc pod uwagę, że reakcje fazy późnej, zwłaszcza w płucach, występują w przypadku braku reakcji fazy początkowej, nie jest nadal w pełni zrozumiałe, czy reakcja fazy późnej musi być koniecznie mediowana IgE. [0007] IgE występuje w postaci związanej z błoną i w postaci wydzielanej. Te różne postacie wydają się być wariantami splicingowymi. Dotychczasowe podejścia osiągały efekt terapeutyczny przez regulację w dół IgE biorąc za cel głównie postać wydzielaną (np. omalizumab XOLAIR ), aby zapobiegać lub rozbroić dalsze zbrojenie się układu odpornościowego. Wydzielana postać IgE jest krótszą postacią, region Fc zasadniczo kończy się w domenie CH4 (Figura 1), natomiast dłuższa postać zawiera dodatkowe reszty C- terminalne, w tym peptydy kodowane przez egzony zwane M1/M1 i M2. Chociaż niektóry opisują dwie różne postacie IgE związanego z błoną, zarówno z, jak i bez odcinka 52 aminokwasów znanego jako M1 [Batista i wsp., J. Exp. Med. 184: (1996)], Zgłaszający nie byli w stanie sprawdzić, czy wszelkie postacie związane z błoną są pozbawione odcinka M1. Konwencjonalne terapie przeciwciałami anty-ige, które wiążą się do wydzielanych postaci IgE, prowadzą do zmniejszenia jej wolnej postaci w surowicy, ale nie całkowitego stężenia w surowicy. Casale i wsp., J. Allergy Clin. Immunol. 100 (1): (1997). [0008] Chen i wsp. (2002) International Archives of Allergy and Immunology 128(4), , Chang (2006) Allergy and Astma Proceedings: The Official Journal of Regional and State Allergy Societies 27(2), S7-14, Chen i wsp. (2001) Faseb Journal 15(5), A1018 i Poggianella i wsp. (2006) Journal of Immunology 177(6), dotyczą, na ogół, odcinka M1 IgE. Chang (2006) opisuje 52 aminokwasową domenę znajduje się pomiędzy domeną CH4 i C-końcem peptydu kotwiącego w błonie IgE, określaną jako domena CepsilonmX. Niezależnie, Chen i wsp. (2002) i Chen i wsp. (2001) opisuje przeciwciała, które wiążą tę domenę, ogólnie lub w obszarze znajdującym się proksymalnie do C-końca. Niezależnie, Poggianella i wsp. (2006) donosi, że domena ta, określana jako zewnątrzkomórkowa domena proksymalna do błony (ang. extracellular membrane proximal domain, EMPD), może brać udział w procesie apoptozy. [0009] Ponadto zauważono, że w przypadku braku sygnału antygenu, receptory komórek B (np. immunoglobuliny), które są sieciowane, są skłonne do apoptozy. [0010] Niespodziewanie, Zgłaszający odkryli, że celowanie regionu N-końcowego odcinka M1 IgE za pomocą przeciwciał anty-ige możne prowadzić do indukowania apoptozy komórki B. Ponieważ potomstwo aktywowanych komórek B może prowadzić do komórek plazmatycznych, które wytwarzają i wydzielają wydzielaną postać IgE, zubożenie komórek B
4 3- produkujących IgE przez apoptozę oferuje nowatorskie podejście terapeutyczne do leczenia alergii. Streszczenie wynalazku [0011] Niniejszy wynalazek zapewnia apoptotyczne przeciwciała anty-ige, lub ich funkcjonalne fragmenty i ich zastosowanie do leczenia zaburzeń mediowanych IgE, jak zastrzeżono w zastrzeżeniach. Wynalazek dostarcza ponadto kompozycje, sposoby hamowania wytwarzania IgE i wydzielania z komórek B i sposoby specyficznego zubażania komórek B produkujących IgE i obniżanie całkowitego poziomu IgE, jak określono w załączonych zastrzeżeniach. [0012] W jednym przykładzie wykonania, wynalazek zapewnia przeciwciało anty-ige/m1, które specyficznie wiąże odcinek M1 IgE i które indukuje apoptozę w komórkach B eksprymujących IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało specyficznie zubaża komórki B produkujące IgE. W innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, przeciwciało zmniejsza zarówno całkowitą IgE w surowicy, jak i wolną IgE w surowicy. W innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało wiąże się z IgE, która jest pochodzenia ludzkiego, od małpy rezus i od małpy cynomolgus. W jeszcze innym konkretnym aspekcie, przeciwciało jest chimeryczne. W jeszcze innym konkretnym aspekcie, przeciwciało jest humanizowane. W jeszcze dalszym aspekcie, przeciwciało jest ludzkie. [0013] W innym przykładzie wykonania, wynalazek zapewnia przeciwciało anty-ige/m1, które specyficznie wiąże się z dowolnym jednym z epitopów M1 odpowiadających peptydom zidentyfikowanym na Figurze 5. W konkretnym aspekcie, przeciwciało specyficznie wiąże się z tym samym epitopem co związanym przez przeciwciało wybrane z grupy składającej się z: 47H4, 7A6, 26A11, 47H4v5, 7A6v1 i 26A11v6. W innym konkretnym aspekcie, przeciwciało wiąże się z epitopem odpowiadającym peptydowi wybranemu z grupy składającej się z: peptyd 4 (SEQ ID NO:8), peptyd 5 (SEQ ID NO:9), peptyd 7 (SEQ ID NO:11) lub peptyd 8 (SEQ ID NO:12). W jeszcze innym konkretnym aspekcie, przeciwciało wiąże się z peptydem 4 (SEQ ID NO:8), [0014] W jeszcze innym przykładzie wykonania, wynalazek zapewnia epitop M1 IgE wybrany z grupy składającej się z: peptyd 4 (SEQ ID NO:8), peptyd 5 (SEQ ID NO:9), peptyd 7 (SEQ ID NO:11) lub peptyd 8 (SEQ ID NO:12), jak określono w zastrzeżeniach. W konkretnym aspekcie, peptydem MI jest peptydem 4 (SEQ ID NO:8). [0015] W dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało anty-ige, które specyficznie wiąże się z odcinkiem M1 IgE z powinowactwem wiązania Scatcharda do ludzkiego IgE. jest równoważne z tym dla przeciwciała mysiego anty-ige/m1 47H4 lub jego humanizowanego wariantu, jak określono w zastrzeżeniach. W konkretnym aspekcie, powinowactwo jest równoważne powinowactwu wiązania 47H4. W innym konkretnym aspekcie, powinowactwo wynosi między 0.30 i 0.83 nm. W jeszcze innym konkretnym
5 4- aspekcie, powinowactwo jest równoważne powinowactwu wiązania 47H4v5. W innym konkretnym aspekcie, powinowactwo wynosi około 1.5 nm. [0016] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało anty- IgE/M1 zawierające HVE łańcucha ciężkiego i łańcucha lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało ponadto zawiera regiony zmienne ciężkich i lekkich łańcuchów sekwencji przeciwciał pojawiających się na każdej z Figur 6A-6F. W innym konkretnym aspekcie, przeciwciało zawiera pełnej długości ciężkie i lekkie łańcuchy przeciwciał pojawiających się na każdej z Figur 6A-6F. W jeszcze innym konkretnym aspekcie, ciężkie i lekkie łańcuchy sekwencji przeciwciał pojawiają się na dowolnej z Figur 6A-6F. W innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało jest afukozylowane. [0017] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia kompozycję zawierającą przeciwciało anty-ige/m1 zawierające HVE łańcucha ciężkiego i łańcucha lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F w połączeniu z co najmniej jednym farmaceutycznie dopuszczalnym nośnikiem, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało jest afukozylowane. [0018] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia kompozycję zawierającą przeciwciało anty-ige/m1 zawierające HVE łańcucha ciężkiego i łańcucha lekkiego przeciwciała występującego na dowolnej z Figur 8-13 z jednym lub większą liczbą leków wybranych z grupy składającej się z: przeciwciało anty-ige, lek przeciwhistaminowy, lek rozszerzający oskrzela, glikokortykoid, NSAID, antagonista TNF, antagonista integryny, środek immunosupresyjny, antagonista IL-4, antagonista IL-13, podwójny antagonista IL- 4/IL-13, DMARD, przeciwciało, które wiąże się z markerem powierzchniowym komórki B i przeciwciało BAFF, jak określono w zastrzeżeniach. W konkretnym aspekcie, kompozycja zawiera ponadto co najmniej jeden farmaceutycznie dopuszczalny nośnik. [0019] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia wyizolowany kwas nukleinowy kodujący HVR łańcucha ciężkiego przeciwciała anty-ige/m1 pojawiający się na każdej z Figur 6A-6F, jak określono w zastrzeżeniach. W konkretnym aspekcie, wyizolowany kwas nukleinowy dalej obejmuje kwas nukleinowy kodujący HVR łańcucha lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W innym konkretnym aspekcie, przeciwciało jest chimeryczne. W jeszcze innym konkretnym aspekcie, przeciwciało jest humanizowane. W dalszym aspekcie, przeciwciało jest ludzkie. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało jest afukozylowane. W jeszcze dalszym aspekcie, kwas nukleinowy obejmuje ponadto wektor odpowiedni do ekspresji kwasu nukleinowego, jak określony w zastrzeżeniach. W jeszcze innym konkretnym
6 5- aspekcie, wektor obejmuje ponadto komórki gospodarza odpowiednie do ekspresji kwasu nukleinowego, jak określono w zastrzeżeniach. W jeszcze innym konkretnym aspekcie, komórka gospodarza jest komórką eukariotyczną lub komórką prokariotyczną. W jeszcze innym konkretnym aspekcie, eukariotyczna komórka jest komórką ssaka taką, jak komórka jajnika chomika chińskiego (ang. Chinese Hamster Ovary, CHO). [0020] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia proces wytwarzania przeciwciała anty-ige/m1, lub jego funkcjonalnego fragmentu, które specyficznie wiąże się z odcinkiem M1 IgE, obejmujący hodowanie komórki gospodarza zawierającej kwas nukleinowy kodujący przeciwciało lub jego fragment w postaci odpowiedniej do ekspresji, w warunkach odpowiednich do wytworzenia takiego przeciwciała lub fragmentu, i odzyskanie przeciwciała lub jego fragmentu, jak określono w zastrzeżeniach. [0021] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia artykuł przemysłowy obejmujący pojemnik zawierający kompozycję ujawnioną w niniejszym opisie i ulotkę wskazującą zastosowanie do leczenia zaburzenia mediowanego IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, artykuł przemysłowy jest fiolką. W innym konkretnym aspekcie, artykuł przemysłowy jest ampułkostrzykawką. W jeszcze innym konkretnym aspekcie, ampułkostrzykawka jest ponadto umieszczona na urządzeniu do wstrzykiwania. W innym konkretnym aspekcie, urządzeniem do wstrzykiwania jest autoiniektor. [0022] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie specyficznego zubażania komórek B produkujących IgE obejmującym podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało zawiera HVR łańcucha ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W innym konkretnym aspekcie, sposób zmniejsza całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, sposób zmniejsza zarówno wolną IgE w surowicy, jak i całkowitą IgE w surowicy. W innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC. [0023] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie leczenia zaburzenia mediowanego IgE obejmującym podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało specyficznie zubaża komórki B produkujące IgE. W innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą i wolną IgE. W innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało zawiera HVR łańcucha
7 6- ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 vl-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC. W jeszcze innym konkretnym aspekcie, zaburzenie mediowane IgE jest wybrane z grupy składającej się z: alergiczny nieżyt nosa, astma (np. astma alergiczna i astma niealergiczna), atopowe zapalenie skóry, alergiczna gastroenteropatia, nadwrażliwość (np. anafilaksja, pokrzywka, alergie pokarmowe itd.), alergiczna aspergiloza oskrzelowopłucna, choroby pasożytnicze, śródmiąższowe zapalenie pęcherza moczowego, zespół hiper-ige, ataksja-teleangiektazja, zespół Wiskotta-Aldricha, limfoplazja atymiczna (ang. athymic lymphoplasia), szpiczak IgE i reakcja przeszczep przeciwko gospodarzowi. W jeszcze innym jeszcze bardziej konkretnym aspekcie, zaburzenie mediowane IgE jest alergią pokarmową, anafilaksją, kontaktowym zapaleniem skóry i plamicą alergiczną. [0024] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie leczenia zaburzenia mediowanego IgE obejmującym podawanie kompozycji zawierającej terapeutycznie skuteczną ilość przeciwciała anty- IgE/M1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE w kombinacji z terapeutycznie skuteczną ilością co najmniej jednego leku wybranego z grupy składającej się z: przeciwciało anty-ige, lek przeciwhistaminowy, lek rozszerzający oskrzela, glikokortykoid, NSAID, lek zmniejszający przekrwienie, lek przeciwkaszlowy, lek przeciwbólowy, antagonista TNF, antagonista integryny, środek immunosupresyjny, antagonista IL-4, antagonista IL-13, podwójny antagonista IL-4/IL-13, DMARD, przeciwciało, które wiąże się z markerem powierzchniowym komórki B i antagonista BAFF, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało specyficznie zubaża komórki B produkujące IgE. W innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą i wolną IgE. W innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało zawiera HVR łańcucha ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC. [0025] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie leczenia zaburzenia mediowanego IgE obejmujący łączny schemat leczenia podawania terapeutycznie skutecznej ilości przeciwciała anty- IgE/M1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE, przed, równocześnie z lub po podaniu znanego sposobu leczenia zaburzeń alergicznych, jak określono w zastrzeżeniach. W konkretnym aspekcie, kombinacja obejmuje podawanie przeciwciała anty-ige, leku przeciwhistaminowego, leku rozszerzającego oskrzela, glikokortykoidu, niesteroidowego leku przeciwzapalnego, immunosupresanta, antagonisty IL-4, antagonisty IL-13, podwójnego antagonisty IL-4/IL-13,
8 7- leku zmniejszającego przekrwienie, leku przeciwkaszlowego lub leku przeciwbólowego. W innym konkretnym aspekcie, przeciwciało anty-ige/m1 podaje się w kombinacji ze schematem leczenia odczulania alergenu. W konkretnym aspekcie, przeciwciało specyficznie zubaża komórki B produkujące IgE. W innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, przeciwciało zmniejsza całkowitą i wolną IgE. W innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało zawiera HVR łańcucha ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC. [0026] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie zapobiegania produkcji IgE indukowanej alergenem, obejmującym podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało zawiera HVR łańcucha ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W innym konkretnym aspekcie, sposób specyficznie zubaża komórki B produkujące IgE. W jeszcze innym konkretnym aspekcie, sposób zmniejsza całkowite IgE w surowicy. W kolejnym innym specyficznym aspekcie, sposób zmniejsza zarówno wolną IgE w surowicy, jak i całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC. [0027] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia przeciwciało według wynalazku do zastosowania w sposobie zmniejszania produkcji IgE indukowanej alergenem, obejmującym podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1, które specyficznie wiąże się z odcinkiem M1 IgE i indukuje apoptozę w komórkach B eksprymujących IgE, jak określono w zastrzeżeniach. W konkretnym aspekcie, przeciwciało zawiera HVR łańcucha ciężkiego i lekkiego przeciwciała występującego na dowolnej z Figur 6A-6F. W innym konkretnym aspekcie, sposób specyficznie zubaża komórki B produkujące IgE. W jeszcze innym konkretnym aspekcie, sposób zmniejsza całkowitą IgE w surowicy. W innym konkretnym aspekcie, sposób zmniejsza zarówno wolną IgE w surowicy, jak i całkowitą IgE w surowicy. W jeszcze innym konkretnym aspekcie, IgE w surowicy jest specyficzna dla alergenu. W jeszcze innym konkretnym aspekcie, przeciwciało jest wybrane z grupy składającej się z: 26A11, 26A11 v1-16, 7A6, 7A6v1, 47H4, 47H4v1-6. W jeszcze innym konkretnym aspekcie, przeciwciałem jest 47H4v5. W jeszcze innym konkretnym aspekcie, przeciwciało wykazuje aktywność ADCC.
9 8- [0028] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia kompozycję użyteczną do dowolnego z opisanych poprzednio sposobów, jak określono w zastrzeżeniach. [0029] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia zastosowanie kompozycji do dowolnego z poprzednio opisanych sposobów, jak określono w zastrzeżeniach. [0030] W jeszcze dalszym przykładzie wykonania, wynalazek dostarcza mysie hybrydomy zdeponowane w ATCC 21 marca 2007 z oznaczeniem wybranym z grupy składającej się z: 7A6.18, IC , 47G4.6.2, 47H , 42H4.6.9, 42A , 26A11.6.5, 51D , 45C1.6.14, 26B , 28E W konkretnym aspekcie, wynalazek zapewnia przeciwciało wydzielane przez zdeponowane hybrydomy. [0031] W jeszcze dalszym przykładzie wykonania, wynalazek zapewnia zwierzę transgeniczne, które eksprymuje ludzki odcinek M1 IgE, jak określono w zastrzeżeniach. Krótki Opis Rysunków [0032] Figury 1A-B przedstawiają dopasowanie wybranych regionów stałych łańcuchów IgE człowieka (SEQ ID NO:1, mapy rezus (SEQ ID NO:2) i małpy cynomolgous (SEQ ID NO:3). Pokazane są przybliżone lokalizacje CH2, CH3, CH4, M1, domen transbłonowych i wewnątrzkomórkowych. Figury 2A-2C są wykresami Scatcharda FACS specyficzności różnych przeciwciał anty-m1. Figura 2A-1 do 2A-6 pokazuje wiązanie względem krótkiej postaci IgE (bez M1 ), a Figury 2B-1 do 2B-6 pokazują wiązanie względem długiej postaci (z M1 ). Figury 2C-1 do 2C-6 pokazują wiązanie przeciwko IgE wyeksprymowanej przez linię komórkową U266. Zacieniona krzywa pokazuje intensywność fluorescencji kontroli Ab, natomiast krzywa niezacieniona pokazuje względną fluorescencję badanego przeciwciała. Figury 2D-F pokazują specyficzności wiązania mysich przeciwciał anty-ige/m1 47H4, 26A11 i 7A6. Figura 2D pokazuje, że 47H4 wiąże się do ludzkiego, rezus i cyno IgE/M1, ale nie do IgE bez M1. Figura 2E pokazuje, że 47H4 wiązał się do U266, natomiast 26A11 i 7A6 nie. Figura 2F pokazuje, że 47H4 i 7A6 wiążą się do M1 rezus i cyno, natomiast 26A11 wiąże tylko rezus. Figury 2G-1 pokazują specyficzności wiązania przeciwciał humanizowanych anty-ige/m1 47H4v5, 2611v6 i 7A6v1. Figura 2G pokazuje, że wszystkie trzy humanizowane warianty 47H4v5, 26A11v6 i 7A6v1 są specyficzne dla IgE-M1, ale nie IgE bez M1. Figura 2H pokazuje, że warianty 47H4v5 i 26A11v6 (ale nie 7A6v1) wiążą U266. Figura 2I pokazuje, że 47H4v5 i 7A6v1 wiążą rezus i cyno M1, natomiast 26a11v6 wiąże wyłącznie rezus M1. Figury 3A-L są wykresami FACS pokazującymi względne powinowactwa powinowactw wiązania różnych przeciwciał anty-m1 za pomocą seryjnych rozcieńczeń podanych na Figurze 3M. Figura 3N pokazuje względne powinowactwa
10 9- dla każdego przeciwciała. Figura 3O podsumowuje powinowactwa mysich przeciwciał 47H4, 26A11 i 7A6 mierzonych na podstawie analizy Scatcharda wobec ludzkiego, rezus i cyno M1. O ile nie wskazano inaczej, podane liczby są uśrednionymi średnimi. Figura 3P podsumowuje powinowactwa podanych wariantów humanizowanych przeciwciał 47H4 i 26A11 mierzonych na podstawie analizy Scatcharda wobec ludzkiego, rezus i cyno M1. Figury 4A-D pokazuje badanie względnego wiązania/blokowania przeciwciał anty- M1. Figury 4A-1 do 4A-20 są wykresami FACS pokazującymi przeciwciała, które blokowały lub częściowo blokowały wiązanie. Figura 4B jest wykresem dwuwymiarowym przedstawiającym względną zdolność do blokowania wiązania innych przeciwciał (częściowo lub całkowicie stosując stosunek molowy 1:1). Figura 4C jest wykresem dwuwymiarowym, który bardziej skupia się na konkretnie wskazanych przeciwciałach, w których badania blokowania powtórzono ze stosunkiem molowym 10:1. Figura 4D jest schematem, który pokazuje grupy wynikające z badań wiązania/blokowania epitopów. Figury 5A-C pokazują badania wiązania epitopów przeprowadzone za pomocą 47H4, 7A6 i 26A11. Figura 5A przedstawia odpowiednio odcinek M1, w tym sąsiednie reszty N-końcowe i C-końcowe (SEQ ID NO:3), i peptydy 1-15 M1, (SEQ ID NO:5-19), wykorzystane do określenia epitopu wiążącego. Figury 5B i 5D przedstawiają, że macierzyste mysie przeciwciała 47H4 wiąże peptyd 4, 7A6 wiąże peptyd 4 i 5 i 26A11 wiąże peptydy 7 i 8. Figury 5C i 5E przedstawiają, że humanizowane warianty 47H4v5 wiąże peptyd 4, natomiast 7A67v1 wiąże peptydy 4 i 5, a 26A11v6 wiąże peptydy 7 i 8, utrzymując tym samym specyficzność epitopu macierzystych mysich przeciwciał. Figury 6A-F przedstawiają sekwencje zmiennych łańcuchów lekkich i łańcuchów ciężkich mysiego przeciwciała 26A11, 7A6 i 47H4 i ich różnych humanizowanych wariantów. Pozycje są numerowane według Kabata oraz regiony hiperzmienne, które były szczepione zmiennej sieci konsensusowej, (Kappa I dla lekkiego, podgrupa III dla ciężkiego łańcucha) są w kwadracie. Figura 6A przedstawia, w stosunku do ludzkiego łańcucha lekkiego kappa I (SEQ ID NO:20), zmienny łańcuch lekki 26A11 (SEQ ID NO:21) i humanizowane warianty 1,4 (SEQ ID NO:22), warianty 2,5 (SEQ ID NO:23), warianty 3,6 (SEQ ID NO:24), warianty 13,15 (SEQ ID NO:25) i warianty 14,16 (SEQ ID NO:26). Figura 6B przedstawia, w stosunku do ludzkiego łańcucha lekkiego kappa I (SEQ ID NO:20), zmienny łańcuch lekki 7A6 (SEQ ID NO:27) i wariant humanizowany I (SEQ ID NO:28). Figura 6C przedstawia, w stosunku do ludzkiego łańcucha lekkiego kappa I (SEQ ID NO:20), zmienny łańcuch lekki 47H4 (SEQ ID NO:29) i humanizowane warianty 1,3 (SEQ ID NO:30) i warianty 2, 4-6 (SEQ ID NO:31). Figura 6D przedstawia, w stosunku do ludzkiego III łańcucha ciężkiego (SEQ ID NO:32), zmienny łańcuch ciężki 26A11 (SEQ ID NO:33) i humanizowane warianty 1-3, 13, 14 (SEQ ID NO:34) i warianty 4-6, 15, 16 (SEQ ID NO:35). Figura 6E przedstawia, w
11 10- stosunku do ludzkiego łańcucha ciężkiego (SEQ ID NO:32), zmienny łańcuch ciężki 7A6 (SEQ ID NO:36) i wariant humanizowany 1 (SEQ ID NO:37). Figura 6F przedstawia, w stosunku do ludzkiego III łańcucha ciężkiego (SEQ ID NO:32), zmienny łańcuch ciężki 47H4 (SEQ ID NO:38), i humanizowane warianty 1,2 (SEQ ID NO:39), warianty 3-4 (SEQ ID NO:40), wariant 5 (SEQ ID NO:41) i wariant 6 (SEQ ID NO:42). Figury 7A-G przedstawiają aktywność apoptotyczną macierzystych przeciwciał anty- M1 w komórkach Daudi transfekowanych IgE-M1. Figura 7A jest wykresem FACS pokazującym, że w istocie IgM jest eksprymowana na poziomie wyższym niż IgE. Figura 7B przedstawia odpowiednio wpływ sieciowania przeciwciał anty-igm [F(ab ) 2 ] i zastosowanie kamptotecyny do indukowania apoptozy. Komórki barwiące się pozytywne dla aneksyny, ale negatywne dla PI umierają, podczas gdy komórki wybarwione pozytywnie dla zarówno aneksyny i PI są martwe. Figura 7C jest graficznym przedstawieniem całkowitej obserwowanej apoptozy, w którym jasny słupek pokazuje komórki anneksyna-(+) i PI-(-), natomiast ciemne słupki pokazują anneksyna-(+) i PI-(+). Figury 7D-7G są graficznym przedstawieniem apoptozy indukowanej anty-m1 przy stężeniach 25, 10, 1, 0.1, 0.01 i µg/ml. Figura 7D przedstawia wyniki zastosowania przeciwciał M1 grupy epitopów A1, w tym 7A6, 47H4, 47G4, 42A5, 42H4 (wraz z kontrolami MAE-11 i GP120) bez sieciowania. Figura 7E przedstawia wyniki zastosowania przeciwciał M1 grupy epitopów A2, w tym C11, 26A11, 51D2, 45C1) i grupy epitopów B/C, w tym 26B11 i 28E9 bez sieciowania. Figura 7F pokazuje grupę epitopów A1 z sieciowaniem, a Figura 7G pokazuje grupy epitopów A2 i B/C z sieciowaniem. Figury 8A-B przedstawia aktywność apoptotyczną humanizowanych wariantów anty- M1 w komórkach Daudi transfekowanych IgE-M1 traktowanych różnymi humanizowanymi wariantami przeciwciała anty-m1 przy stężeniach 25, 10, 1, 0.1, 0.01 i µg/ml. Figura 8A przedstawia, że humanizowane warianty 47H4v5, 26A11v6 i 7A6v1 indukują apoptozę w szeregu 30-40% przy większych stężeniach (tj µg/ml warianty 47H4 i 26A11, 1, 10 i 25 µg wariant 7A6). Figura 8B pokazuje aktywność apoptotyczną przeciwciała tego samego na komórkach Daudi transfekowanych IgE-M1, które są traktowane w obecności sieciującego przeciwciała F(ab )2 koziego anty-ludzka IgG. Wszystkie przeciwciała indukowały maksymalne poziomy apoptotyczne 70-90%, z pewnym spadkiem aktywności apoptotycznej w wysokich stężeniach (np. 47H4 -v1, - v2, 26A11 -v1, -v14). Figura 8C pokazuje, że zarówno typ dziki, jak i afukozylowany 47H4v5 były w stanie indukować apoptozę na podobnym poziomie. Figury 9A-B1-2 przedstawia zdolność mysich przeciwciał anty-m1 do wywołania przepływu wapnia w komórkach Daudi-IgE/M1. Figura 9A jest kontrolą przedstawiającą wpływ anty-igm i MAE11, natomiast Figura 9B1-B2 pokazują wpływ stosowania wskazanych przeciwciał mysich anty-m1.
12 11- Figura 10 wykazuje zdolność humanizowanego przeciwciała anty-ige/m1 47H4 v5 wariantu wt i afukozylowanego do indukcji ADCC. Natomiast warianty wt i afukozylowany indukowały podobną maksymalną cytotoksyczność, przy czym wariant afukozylowany ( AF ) wykazywał silniejsze działanie niż postać typu dzikiego (EC50 AF 0.83 nm, EC50 wt 6.6 nm). Figura 11A=11F są sekwencjami pełnej długości ciężkich i lekkich łańcuchów mysich przeciwciał anty-ige/m1. Regiony zmienne zaznaczono kursywą, natomiast HVR (regiony hiperzmienne) są podkreślone. Figura 11A przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 7A6 (SEQ ID NO: 43 i 44). Figura 11B przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 47H4 (SEQ ID NOS: 45 i 46). Figura 11C przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 26A11 (SEQ ID NO: 47 i 48). Figura 11D przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 45C1 (SEQ ID NOS: 49 i 50). Figura 11E przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 28E9 (SEQ ID NO: 51 i 52). Figura 11F przedstawia odpowiednio ciężkie i lekkie łańcuchy mysiego przeciwciała 1C11 (SEQ ID NOS: 53 i 54). Figury 12A-I przedstawia zdolność mysich przeciwciał anty-ige/m1 do hamowania wytwarzania IgE w surowicy i komórek plazmatycznych produkujących IgE w atopowym modelu hu-scid. Figura 12A jest graficznym przedstawieniem projektu doświadczalnego. Figury 12B-C przedstawiają, że leczenie mysimi przeciwciałami anty-ige/m1 zmniejszało poziomy IgE o 65-84%. Figury 12D-E przedstawiają, że zmniejszenie komórek produkujących IgE in vivo wyniosło 19-69%. Figury 12F-G przedstawiają, że poziomy innych immunoglobulin (np. IgG1-4, IgA, IgM) były względnie niezmienione. Figury 12H-I przedstawiają, że nie obserwowano zmniejszenia całkowitej liczby komórek plazmatycznych śledziony. Figury 13A-H pokazują wpływ wariantu humanizowanego 47H4v5 na poziomy immunoglobulin w atopowym modelu hu-scid. Figura 13A jest graficznym przedstawieniem projektu doświadczalnego. Figury 13B-D przedstawiają, że IgE w surowicy zmniejszyła się o 79%, i że komórki plazmatyczne produkujące IgE uległy zmniejszeniu o 75%. Figury 13E-F pokazuje brak obniżenia stężenia innych immunoglobulin surowicy. Figury 13G-H pokazuje brak obniżenia całkowitych poziomów komórek plazmatycznych, wykazując w ten sposób, że 47H4 specyficznie zmniejsza komórki plazmatyczne produkujące IgE, które stanowią bardzo niewielką część całkowitej ilości komórek plazmatycznych. Figury 14A-D ilustrują wytwarzanie myszy knock-in hum1. Figura 14A pokazuje lokalizację egzonu M1 na locus myszy IgE. Figura 14B przedstawia schemat rekombinacji w locus myszy IgE powodujący utworzenie docelowego allelu. Figura 14C pokazuje PCR genotypowania pasma 668 pb u myszy wt i pasma 457 bp w knock-in M1. Figura 14D przedstawia Southern Blot, w którym 7.4 kb fragment HindIII u myszy wt, staje się 3 kb fragmentem w knockin hu-m1, a 14.1 kb fragment
13 12- BamHI staje się 18.1 fragmentem w alleli knockin hu-m1. Pokazano zarówno myszy typu dzikiego, jak i heterozygotyczne. Figury 15A-I przedstawiają zdolność przeciwciał anty-ige/m1 do zapobiegania wytwarzaniu przeciwciał IgE w pierwotnej odpowiedzi immunologicznej. Figura 15A jest schematem pokazującym oś czasu dla projektu doświadczalnego, w tym podawania przeciwciał TNP/OVA i anty-ige/m1. Figura 15B jest wykres antygenowo specyficznych poziomów IgE w czasie i pokazuje, że, podczas gdy antygenowo specyficzna IgE u zwierząt kontrolnych (tj. gp120) osiągnęła wartość szczytową między 8 i 14 dniem, anty-ige/m1 zapobiegła jakiemukolwiek wzrostowi, i zmierzone poziomy antygenowo specyficznej IgE nie różniły się istotnie od myszy nieimmunizowanych. Figury 15C-D pokazuje, że leczenie anty-ige/m1 zapobiegało wzrostowi antygenowo specyficznej IgE w surowicy w odpowiednio w dniach 8 i 14, i nie było statystycznie różne od myszy nieimmunizowanych (Figura 15E). Figury 15F-I przedstawiają, że poziomy antygenowo specyficznej IgG1 nie zostały znacząco zmienione przez anty-ige/m1 przez 28 dni doświadczenia (oprócz niewielkiej różnicy w dniu 14). Figury 16A-K przedstawiają zdolność przeciwciał anty-ige/m1 do zapobiegania powstawaniu antygenowo specyficznej IgE w pamięci lub wtórnej odpowiedzi immunologicznej. Figura 16A jest schematem przedstawiającym osie czasu wtórnej dawki przypominającej TNP-OVA i podanie przeciwciał anty-ige/m1, które podano najpierw w dniu 28. Figura 16B jest wykresem poziomów antygenowo specyficznej IgE w czasie i przedstawia, że wtórna odpowiedź na dawkę przypominającą TNP- OVA w dniu 28 jest szybsza, osiągając maksimum po 4 dniach, a nie 8-9 dniach w pierwotnej odpowiedzi. Figury 16C-D przedstawiają, że antygenowo specyficzne poziomy IgE u zwierząt leczonych anty-ige/m1 były znacznie zmniejszone w porównaniu z kontrolą dla izotypu, 59-65% na dzień 32 i 90-93%% na dzień 35. Figura 16E przedstawia, że na dzień 42 (12 dni początkowego podawania przeciwciała anty-ige), poziomy antygenowo specyficznej IgE zmniejszyły się do poziomu statystycznie nieróżniącego się od nieleczonych myszy kontrolnych. Figury 16F-H przedstawiają, że pomiędzy dniami 28 i 49, podawanie anty-ige/m1 zmniejszyło poziomy IgE w surowicy o 74-84%, i średni poziom dzienny antygenowo specyficznej IgE został również zmniejszony o 74-83%. Figury 16J-K przedstawiają, że poziomy antygenowo specyficznej IgG1 nie zostały znacząco zmienione przez anty-ige/m1. Figury 17A-D ilustrują zdolność przeciwciał anty-ige/m1 do profilaktycznego zmniejszenia produkcji IgE w odpowiedzi na zakażenie Nippostrongylus brasiliensis ( NB ). Figura 17A jest schematem pokazującym projekt doświadczenia. Zwierzęta były leczone trzy razy w tygodniu, począwszy od dnia 0 do dnia 21. Figura 17B przedstawia poziomy IgE w czasie o odpowiedzi na zakażenie NB z przeciwciałami anty-ige/m1 i kontrolnym. Figury 17C-D przedstawiają, że w dniu 15, zwierzęta
14 13- leczone anty-ige/m1 miały zmniejszone poziomy IgE w surowicy nie istotne statystycznie od myszy niezakażonych. Figury 18A-I ilustrują zdolność przeciwciał anty-ige/m1 do leczenia terapeutycznego szczytowej odpowiedzi IgE na Nippostrongylus brasiliensis ( NB ), Figura 18A jest schematem pokazującym projekt doświadczenia. Zwierzęta leczono trzy razy w tygodniu, pomiędzy dniami 11 a 21. Figura 18B przedstawia poziomy IgE w czasie w odpowiedzi na infekcję NB z przeciwciałami anty-tgeim1 i kontrolnymi. Figura 18C-D przedstawiają, że anty-ige/m1 zmniejszało poziomy IgE w surowicy o 82-89% w ciągu czterech dni leczenia. Figura 18E pokazuje, że na dzień 21 poziomy IgE u zwierząt leczonych anty-ige/m1 o 97-98%, i osiągnęły poziom nie istotny statystycznie w stosunku do niezakażonej grupy kontrolnej. Figury 18F-G wykazują, że komórki plazmatyczne produkujące IgE (oznaczone ilościowo przez Elispot) w węzłach chłonnych i śledzionie uległy zmniejszeniu o odpowiednio 88-94% i 57-66%. Figury 18H-I pokazują, że całkowite komórki plazmatyczne (CD138+) zarówno w węzłach chłonnych, jak i śledzionie wzrosły we wszystkich grupach traktowania w stosunku do myszy niezakażonych, i że leczenie anty-ige/m1 nie zmieniło znacznie całkowitej liczby komórek plazmatycznych w obu narządach. Wyniki te wykazują zdolność przeciwciał anty-ige/m1 do zmniejszenia stężenia IgE w surowicy przez zubożenie komórek produkujących IgE in vivo. Figury 19A-G ilustrują zdolność przeciwciał anty-ige/m1 do leczenia terapeutycznego odpowiedzi IgE występującej pod koniec cyklu zakażenia Nippostrongylus brasiliensis ( NB ). Figura 19A jest schematem pokazującym projekt doświadczenia, w którym zwierzęta leczono trzy razy w tygodniu, począwszy od dnia 40. Figura 19B pokazuje, że szczyt produkcji IgE wystąpił około dnia 15 i, że wszystkie przeciwciała anty-ige/m1 zmniejszały IgE w surowicy. Figura 19C-D pokazuje, że przeciwciało anty-ige/m1 wykazuje znaczne zmniejszenie zarówno odpowiednio bezwzględnych i znormalizowanych poziomów IgE, w stosunku do rozpoczęcia leczenia. Figury 19E-G pokazują, że leczenie anty-ige/m1 istotnie zmniejszyły poziomy IgE w surowicy, w porównaniu do kontroli izotypowej migg1 anty-gp120 między dniami 48 a 55. Szczegółowy Opis Korzystnego Przykładu Wykonania Ogólne techniki [0033] Realizacja praktyczna niniejszego wynalazku będzie wykorzystywać, o ile nie wskazano inaczej, konwencjonalne techniki biologii molekularnej (w tym techniki rekombinacji), mikrobiologii, biologii komórkowej, biochemii i immunologii, które są w zakresie biegłości w tej dziedzinie. Takie techniki są w pełni wyjaśnione w piśmiennictwie, np., Molecular Cloning: A Laboratory Manual, wydanie drugie (Sambrook i wsp., 1989); Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Animals Cell Culture (R.I. Freshney, ed., 1987); Methods in Enzymology (Academic Press, Inc.);. Current Protocols in Molecular Biology (F.M. Ausubel i wsp., eds 1987, i okresowe aktualizacje); PCR: The Polymerase
15 14- Chain Reaction, (Mullis i wsp., ed., 1994); A Practical Guide to Molecular Cloning (Perbal Bernard V., 1988); Phage Display: A Laboratory Manual (Barbas i wsp., 2001). Rozwój i aktywacja limfocytów [0034] Dwoma głównymi rodzajami limfocytów u ludzi są T (pochodzące z grasicy) i B (pochodzące ze szpiku kostnego). Komórki te pochodzą z krwiotwórczych komórek macierzystych w szpiku kostnym i wątrobie płodowej, które są zaangażowane w ścieżkę rozwoju limfoidalnego. Potomstwo tych komórek macierzystych podąża rozbieżnymi ścieżkami dojrzewania do limfocytów B lub T. Rozwój ludzkich limfocytów B odbywa się w pełni w szpiku kostnym. Komórki T, z drugiej strony, powstają z niedojrzałych prekursorów, które opuszczają szpik i przechodzą przez krwiobieg do grasicy, gdzie proliferują i różnicują się w dojrzałe limfocyty T. [0035] Dojrzałe limfocyty, które powstają w grasicy i szpiku kostnym są w spoczynku, lub w stanie spoczynku, to znaczy, że są mitotycznie nieaktywne. Po rozproszeniu do krwiobiegu, te naiwne lub dziewicze limfocyty, podróżują do różnych wtórnych lub obwodowych narządów limfatycznych takich, jak śledziona, węzły chłonne lub migdałki. Większość dziewiczych limfocytów ma z natury krótki czas życia i umierają kilka dni po opuszczeniu szpiku lub grasicy. Jednakże, jeśli taka komórka odbiera sygnały, które wskazują na obecność antygenu, mogą aktywować się i przechodzić kolejne rundy podziału komórkowego. Niektóre z powstałych komórek potomnych powracają następnie do stanu spoczynku, aby stać się limfocytami pamięci komórkami B i T, które są w istocie przygotowane do następnego spotkania ze stymulującym alergenem. Inne potomstwo aktywowanych limfocytów dziewiczych jest komórkami efektorowymi, które przetrwają tylko kilka dni, ale przeprowadzają konkretne działania obronne. [0036] Aktywacja limfocytów odnosi się do uporządkowanej serii zdarzeń, za pośrednictwem których spoczynkowe limfocyty ulegają przemianie, gdy są stymulowane do podziału i dają potomstwo, przy czym niektóre stają się komórkami efektorowymi. Pełna odpowiedź obejmuje zarówno indukowanie proliferacji (mitogenezę) i ekspresję funkcji immunologicznych. Limfocyty stają się aktywowane, gdy specyficzne ligandy wiążą się do receptorów na ich powierzchni. Ligandy te różnią się dla komórek T i komórek B, ale wynikowe wewnątrzkomórkowe mechanizmy fizjologiczne są podobne. [0037] Chociaż same obce antygeny mogą wywoływać aktywację limfocytów, zwłaszcza duże polimeryczne antygeny sieciujące immunoglobuliny na powierzchni komórek B lub innych glikoprotein na komórkach T. Jednakże większość antygenów nie jest polimeryczna i nawet gdy wiążą się bezpośrednio do komórek B w dużych ilościach, nie doprowadzają do aktywacji. Komórki B aktywowane są tymi samymi wspólnymi antygenami, gdy są kostymulowane przez prawie aktywowane limfocyty T pomocnicze. Takie pobudzenie może nastąpić ze strony limfokin wydzielanych przez komórki T, ale jest przekazywane najbardziej efektywnie poprzez bezpośredni kontakt komórki B z białkami powierzchniowymi komórek T, które oddziałują z niektórymi receptorami powierzchniowymi komórki B do wytwarzania sygnału wtórnego.
16 15- Komórki B [0038] Charakterystyczną cechą komórek B jest zdolność do syntezy immunoglobulin. Immunoglobuliny (Ig) stanowią bardzo różnorodną rodzinę białek składającą się z pokrewnych rodzajów polipeptydów zwanych łańcuchami ciężkimi i łańcuchami lekkimi. Każda Ig wiąże się specyficznie z dużym powinowactwem z własnym specyficznym antygenem. Dojrzałe limfocyty B mogą eksprymować immunoglobuliny w dwóch różnych formach, z których każdy służy unikalnymi funkcjami. Immunoglobuliny limfocytów B w stanie spoczynku (dziewiczych lub pamięci) są eksprymowane na powierzchni komórki, gdzie działają głównie jako receptor na błonie dla konkretnych antygenów. Natomiast, komórki efektorowe komórek B (komórki plazmatyczne) wydzielają immunoglobuliny do otaczającego środowiska. Takie wydzielane immunoglobuliny zachowują zdolność do rozpoznawania i wiązania (vis a vis postaci związanej z błoną na spoczynkowych komórkach B), i są zwykle określane jako przeciwciała. [0039] Gdy aktywowany limfocyt B ulega podziałowi, niektóre z ich potomstwa stają się komórkami B pamięci, podczas gdy pozostała część różnicuje się w komórki plazmatyczne. Jako że komórki plazmatyczne mają stosunkowo krótką żywotność, chyba, że nowe komórki plazmatyczne są produkowane, populacja szybko gaśnie i immunoglobuliny nie są dłużej wydzielane. W rezultacie, aktywacja limfocytów B zazwyczaj prowadzi do uzyskania przejściowej fali proliferacji, a następnie wybuchu, który zwiększa wydzielanie przeciwciał, a następnie ustępuje w ciągu kilku dni lub kilku tygodni. Komórki B są komórkami typu pierwotnego zaangażowanymi w odporność humoralną, lub efekt ochronny mediowany przez płyny tkankowe. Ponieważ przeciwciała anty-ige/m1 wynalazku rzeczywiście zubażają komórki B, w tym komórki B pamięci, można je stosować do resetowania pamięci. Zatem skutkiem tego może być to, że składnik B-komórkowy, który napędza reakcję alergiczną u osoby, może być stłumiony, jeśli nie usunięty. Komórki T [0040] Limfocyty T nie wykazują ekspresji immunoglobulin, lecz zamiast tego wykrywają obecność substancji obcych za pomocą białek powierzchniowych, zwanych receptorami komórek T. Receptory te rozpoznają antygeny albo przez bezpośredni kontakt, albo poprzez wpływanie na aktywność innych komórek immunologicznych. Wraz z makrofagami, komórki T są komórkami typu pierwotnego zaangażowanymi w odporność komórkową. [0041] W odróżnieniu od komórek B, komórki T mogą wykryć substancje obce tylko w określonych sytuacjach. W szczególności, limfocyty T rozpoznają obce białko wyłącznie wtedy, gdy są najpierw cięte na małe peptydy, a następnie prezentowane na powierzchni drugiej komórki gospodarza, nazywanej komórką prezentującą antygen (APC). Wiele typów komórek gospodarza może prezentować antygeny w pewnych warunkach, lecz pewne typy są szczególnie przystosowane do tego celu i są szczególnie ważne dla kontrolowania aktywności komórek T i obejmują one makrofagi i inne komórki B. Prezentacja antygenu, zależy po części od konkretnych białek, zwanych białkami głównego układu zgodności tkankowej (MHC), na powierzchni komórek prezentujących. Zatem, w celu pobudzenia odporności
17 16- komórkowej, obce peptydy muszą być prezentowane na komórkach T, w połączeniu z peptydami MHC, a połączenie to musi być rozpoznawane przez receptor komórek T. [0042] Istnieją dwa istotne podzbiory komórek T: limfocyty T cytotoksyczne (komórki T c lub CTL) i komórki T pomocnicze (T H ), które mogą być w przybliżeniu określone na podstawie ekspresji na powierzchni komórki markera CD8 i CD4. Komórki Tc odgrywają ważną rolę w obronie wirusowej i mogą zabijać wirusy bezpośrednio poprzez rozpoznanie pewnych peptydów wirusowych wyrażanych na powierzchni komórek. Komórki T H promują proliferację, dojrzewanie i funkcję immunologiczną innych typów komórek, np. wydzielanie limfokin w celu kontroli działalności komórek B, makrofagów i komórek T cytotoksycznych. Zarówno limfocyty T dziewicze i pamięci zazwyczaj pozostają w stanie spoczynku i w tym stanie nie wykazują znaczącej aktywności cytotoksycznej lub pomocnika. Po aktywacji komórki te przechodzą kilka rund podziału mitotycznego produkując komórki potomne. Niektóre z tych komórek potomnych powracają w stan spoczynku jako komórki pamięci, ale inne stają się komórkami efektorowymi, które aktywnie eksprymują pomocników aktywności cytotoksycznej. Te komórki potomne przypominają ich rodziców: komórki CD4+ mogą wyłącznie produkować potomstwo CD4+, natomiast komórki CD8+ mogą dać wyłącznie potomstwo CD8+. Efektorowe komórki T eksprymują markery powierzchniowe komórek, które nie są eksprymowane w spoczywających komórkach T takie, jak CD25, CD28, CD29, CD40L, receptory transferyny i białka MHC klasy II. Gdy bodziec aktywujący zostanie wycofany, aktywność cytotoksyczna lub pomocnika stopniowo opada w ciągu kilku dni, ponieważ komórki efektorowe umierają lub powracają do stanu spoczynkowego. [0043] Podobnie do aktywacji komórek B, odpowiedzi limfocytów T na większość antygenów wymagają dwóch typów równoczesnych bodźców. Pierwszym z nich jest antygen, który, jeśli prezentowany odpowiednio przez białka MHC na komórce prezentującej antygen, może być rozpoznawany i wiązany przez receptory komórek T. Choć ten kompleks antygen- MHC wysyła sygnał do wnętrza komórki, jest to zwykle niewystarczające, aby doprowadzić do aktywacji komórek T. Pełna aktywacja taka, jak występująca w przypadku komórek T pomocniczych, wymaga kostymulacji innymi specyficznymi ligandami zwanymi kostymulatorami, które ulegają ekspresji na powierzchni komórki prezentującej antygen. Aktywacja cytotoksycznych komórek T, z drugiej strony na ogół wymaga IL-2, cytokiny wydzielanej przez aktywowane komórki pomocnicze T. Odpowiedź Immunologiczna [0044] Trzy podstawowe właściwości funkcjonalne systemu immunologicznego ssaków odróżniające go od innych obron organizmu obejmują: (1) specyficzność - zdolność do rozpoznawania i odpowiadania lub nie odpowiadania samodzielnie wśród ogromnej liczby cząsteczek docelowych, (2) dyskryminacja - umiejętność określenia siebie od nie-siebie tak, aby pokojowo współistnieć z wszystkimi niezliczonymi białkami i innymi materiałami organicznymi, a jednocześnie energicznie reagować na obcy materiał, który jest wprowadzany do organizmu, i (3) pamięć - zdolność do formowania przez doświadczenie takie, że kolejne spotkania z konkretnym obcym patogenem sprowokuje szybszą i energiczną odpowiedź, niż ta, która miała miejsce przy początkowym spotkaniu. Ponieważ antagoniści
18 17- IgE według wynalazku wywołują apoptozę komórek B przenoszących IgE, oczekuje się, że złagodzą lub nawet wymażą pamięć immunologiczną na poszczególne antygeny. Oczekuje się, że będzie to szczególnie korzystne, gdy silna odpowiedź immunologiczna na alergeny jest patologiczna, jak to ma miejsce w przypadku chorób atopowych. [0045] Dziewicze limfocyty są stale uwalniane z pierwotnych narządów limfoidalnych do obwodu, z których każdy zawiera receptory powierzchniowe, które pozwalają na wiązanie antygenu. Wiązanie antygenu w komórkach B jest mediowane przez immunoglobuliny związane z powierzchnią, natomiast w komórkach T jest mediowane przez receptory komórek T. Gdy dziewicze limfocyty nie są aktywowane, umierają w ciągu kilku dni po wejściu do obwodu. Te, które są aktywowane, przeżywają i rozmnażają się, uzyskując komórki potomne, które następnie mogą ulegać dalszym cyklom aktywacji i proliferacji. Szybkość i intensywność odpowiedzi na dany antygen jest określona głównie przez selekcję klonów: im większa populacja komórek potomnych lub klonów specyficznych dla danego antygenu, tym większa liczba komórek, które mogą rozpoznawać i uczestniczą w odpowiedzi immunologicznej. Każda odpowiedź immunologiczna jest złożoną i misternie regulowaną sekwencją wydarzeń z udziałem kilku typów komórek. Jest ona wyzwalany, gdy immunogen wchodzi do ciała i napotyka wyspecjalizowaną klasę komórek zwanych komórki prezentujące antygen (APC). Te APC wychwytują małą ilość immunogenu i prezentują go w postaci, która może być uznana za antygenowo specyficzne limfocyty pomocnicze T. Te komórki pomocnicze T wtedy stają się aktywne i promują z kolei aktywację innych klas limfocytów takich, jak komórki B lub cytotoksyczne komórki T. Aktywowane limfocyty następnie proliferują i realizują swoje specyficzne funkcje efektorowe. Na każdym etapie tego procesu, limfocyty i komórki APC komunikują się ze sobą za pomocą bezpośredniego kontaktu lub przez wydzielanie cytokin regulacyjnych. [0046] Egzogenne antygeny, które są wychwytywane przez APC przechodzą szereg zmian zwanych przetwarzaniem antygenu. Takie przetwarzanie, zwłaszcza immunogenów białkowych polega na denaturacji i częściowym trawieniu proteolitycznym, tak, że immunogen rozszczepia się do krótkich peptydów. Ograniczona liczba powstałych peptydów następnie asocjuje niekowalencyjnie z białkami MHC klasy II i są transportowane do powierzchni APC, proces znany jako prezentacja antygenu. Limfocyt pomocniczy T CD4+, który pozostaje w bezpośrednim kontakcie z APC, może stać się aktywowany, ale uczyni to wyłącznie, gdy wyeksprymował białko receptora komórek T, które może rozpoznawać i wiązać się z konkretnym kompleksem peptyd-mhc prezentowanym na APC. [0047] Komórki pomocnicze T (T H ) są głównymi koordynatorami odpowiedzi immunologicznej, ponieważ są one potrzebne do aktywacji pozostałych dwóch limfatycznych komórek efektorowych: cytotoksycznych komórek T (Tc) i komórek plazmatycznych wydzielających przeciwciała. Aktywacja T H następuje wcześnie w odpowiedzi immunologicznej i wymaga co najmniej dwóch sygnałów. Jeden sygnał jest zapewniony przez wiązanie receptora antygenu komórki T z antygenowym kompleksem peptyd-mhc na powierzchni APC, który jest przesyłany poprzez kompleks białka CD3, natomiast uważa się, że drugi, ko-stymulujący sygnał przez APC wynika z wiązania odrębnego białka
19 18- transmitującego sygnał na powierzchni komórek T ze specyficznym ligandem na APC. Jedną ze znanych takich interakcji jest białko komórek T CD28 i rodzina białek powierzchniowych APC zwana B7. Inne pary białek powierzchniowych mogą również mediować ko-stymulację. [0048] Wspólnie, te dwa sygnały indukują komórki T pomocnicze do rozpoczęcia wydzielania cytokiny znanej jako interleukina-2 (IL-2) a także do rozpoczyna ekspresji specyficznych receptorów IL-2 o wysokim powinowactwie na powierzchni. IL-2 jest bardzo silnym czynnikiem mitogennym dla limfocytów T i jest niezbędny dla proliferacji odpowiedzi aktywowanych komórek T. Działanie IL-2 na komórki, z których jest wydzielana - zjawisko znane jako efekt autokrynny. Stwierdzono ponadto, że nawet jeśli komórki T nie otrzymały obu sygnałów, nie będą proliferowały, jeśli jego własne receptory IL-2 na powierzchni są zablokowane. IL-2 może także działać na komórki, w bezpośrednim sąsiedztwie, w tak zwanym mechanizmie parakrynnym. Efekt ten jest szczególnie ważny dla aktywacji komórek Tc, które na ogół nie produkują wystarczająco IL-2 do stymulowania własnej proliferacji. Oprócz IL-2, aktywowane komórki T H wydzielają inne cytokiny i promują wzrost, różnicowanie i funkcje komórki B, makrofagi i inne komórki. [0049] Kontakt pomiędzy i APC oraz antygenowo specyficzną komórką T H również ma wpływ na APC - jednym z najważniejszych z nich jest uwalnianie IL-1. Uważa się, że ta cytokina działa w sposób autokrynny w celu zwiększenia ekspresji powierzchniowych białek MHC klasy II i różnych cząsteczek adhezyjnych wzmacniając w ten sposób wiązanie komórki T H i zwiększając prezentację antygenu. W tym samym czasie, IL-1 działa w sposób parakrynny na komórki TH w celu promowania wydzielania IL-2 i ekspresji receptorów IL-2. [0050] Podczas aktywacji komórek T H w sposób opisany uprzednio, niektóre komórki B mogły również zajmować immunogen przez ich receptory antygenów, które są związane z błoną postaciami przeciwciał, które będą później wydzielane. W odróżnieniu od komórek T, komórki B rozpoznają immunogen w postaci wolnej, nieprzetworzonej. Specyficzne wiązanie antygenu stanowi jeden rodzaj sygnału, który może prowadzić do aktywacji komórek B. Drugi typ jest zapewniony przez aktywowane komórki T H, które eksprymują białka ułatwiające aktywację komórki B poprzez wiązanie z receptorami nieimmunoglobuliny na jego powierzchni. Te sygnały pochodzące z T H, które działają w każdej komórce B niezależnie od jego specyficzności antygenowej, są określane jako czynniki pomocnicze. Te czynniki pomocnicze obejmują IL-2, IL-4 i IL-6. Jednakże pomoc jest bardziej skutecznie osiągana poprzez kontakt komórka-komórka, co umożliwia białkom na powierzchni komórek T bezpośredni kontakt z tymi na komórki B. Najbardziej skuteczna postać pomocy mediowanej kontaktem występuje, gdy białko zwane ligandem CD40 (CD40L), które jest eksprymowane na komórkach TH dopiero po tym, jak staną się aktywne, wiąże się z białkiem zwanym CD40 na komórkach B. W procesie zwanym aktywacją przypadkową (ang. bystander activation), kontakt z aktywowaną komórką B może być nawet wystarczający do aktywacji spoczynkowych komórek B, nawet jeśli jej immunoglobuliny powierzchniowe nie są zaangażowane w antygen. [0051] Limfocyty Tc działają w celu wyeliminowania komórek, w których zachodzi ekspresja obcych antygenów na swojej powierzchni takich, jak komórki gospodarza zainfekowane
20 19- wirusem. Większość komórek Tc eksprymuje CD8 zamiast CD4 i tym samym rozpoznaje antygeny w połączeniu z białkami MHC klasy I, a nie białkami MHC klasy II. Gdy komórka somatyczna jest zakażona wirusem, niektóre immunogenne białka wirusowe mogą być poddane przetworzeniu w komórce i powstałe peptydy mogą wówczas pojawić się w postaci kompleksów powierzchniowych z cząsteczkami MHC klasy I. Te kompleksy peptyd-mhc mogą następnie być rozpoznawane przez receptor komórek T antygenowo specyficznego klonu, zapewniając jeden z dwóch sygnałów niezbędnych do aktywacji komórek Tc. Ten pierwszy sygnał sam indukuje receptory IL-2 o wysokim powinowactwie na komórce Tc. Drugi sygnał jest dostarczany przez IL-2 wydzielaną z aktywowanego w pobliżu limfocytu T H. Po otrzymaniu obu sygnałów aktywowane komórki Tc nabierają aktywności cytotoksycznej, umożliwiając zabicie komórki, do której jest ona związana, jak również inne komórki zawierające te same kompleksy peptyd-mhc klasy I. W niektórych przypadkach, występuje zabijanie, ponieważ Tc uwalnia specyficzne toksyny do komórki docelowej; w innych Tc indukuje komórki docelowe do popełnienia samobójstwa przez apoptozę. Aktywne komórki Tc również proliferują, powodując powstanie dodatkowych komórek Tc o tej samej specyficzności antygenowej. IgE/Komórka Tuczna/Szlak Mediatorowy. [0052] Przeciwciała IgE są przymocowane do powierzchni komórek tucznych i bazofili w części Fc cząsteczki z receptorem o wysokim powinowactwie na powierzchni komórek, zwanym FcεRI. Reakcja alergiczna jest inicjowana, gdy wielowartościowa cząsteczka alergenu wiąże się z przeciwciałami, które zajmują te receptory. Wynikiem jest mostkowanie FcεRI, który z kolei przesyła sygnał do wnętrza komórki powodując uwalnianie i aktywację mediatorów zapalenia: histaminy, leukotrienów, czynników chemotaktycznych, czynników aktywujących płytki krwi i proteinaz. Mediatory te aktywowane działają lokalnie i powodują zwiększoną przepuszczalność naczyń, rozszerzenie naczyń krwionośnych, skurcz mięśni gładkich i wydzielanie gruczołów śluzowych. Takie zdarzenia są określane klinicznie jako faza natychmiastowa lub wczesna, i występują w ciągu pierwszych minut po ekspozycji na alergen. W ciągu następnych 12 godzin dochodzi do progresywnego naciekania tkanek komórek zapalnych, wychodząc z neutrofilów do eozynofilów do komórek jednojądrzastych w odpowiedzi na inne mediatory chemiczne niekoniecznie całkowicie zrozumiałe. Ten okres 6-12 dni po ekspozycji na alergen jest określany jako faza późna i charakteryzuje się objawami klinicznymi zapalenia komórkowego. Biorąc pod uwagę, że reakcje fazy późnej, zwłaszcza w płucach, występują w przypadku braku reakcji fazy początkowej, całkowicie zrozumiałe nadal nie jest, czy reakcja fazy późnej musi być koniecznie mediowana IgE. [0053] Mechanizm ten jest przede wszystkim odpowiedzialny za anafilaksję, pokrzywkę i choroby atopowe takie, jak alergiczny nieżyt nosa, astma alergiczna, atopowe zapalenie skóry i alergiczna gastroenteropatia. Szlak Efektor Limfocytów T/Limfokina [0054] Niektóre choroby alergiczne są mediowane przez reakcję alergenu z efektorowymi limfocytami T uczulonymi na specyficzny alergen z wcześniejszej ekspozycji. Gdy alergen
21 20- napotyka komórki T CD4+, zostają one aktywowane do wytworzenia limfokiny, co skutkuje przez okres od kilku dni do akumulacją nacieku komórek jednojądrzastych. I. Definicje [0055] Alergen lub immunogen jest dowolną cząsteczką, która może wywołać odpowiedź immunologiczną. W stosowanym tu kontekście, określenie obejmuje albo samą cząsteczkę antygenową, albo jej źródło takie, jak ziarno pyłku, naskórek zwierząt, jad owadów lub produkt spożywczy. To kontrastuje z określeniem antygen, który odnosi się do cząsteczki mogącej być specyficznie rozpoznawaną przez immunoglobulinę lub receptor limfocytów T. Każda obca substancja zdolna do indukowania odpowiedzi immunologicznej jest potencjalnym alergenem. Znanych jest wiele różnych środków chemicznych pochodzenia naturalnego i syntetycznego, które są alergenne. Złożone naturalne organiczne środki chemiczne, zwłaszcza białka, mogą powodować alergię mediowaną przeciwciałami, natomiast proste związki organiczne, chemikalia nieorganiczne i metale korzystniej powodują alergię zależną od komórek T. W pewnych przypadkach, sam alergen może odpowiadać więcej niż jednemu rodzajowi alergii. Ekspozycja na alergen nastąpić przez inhalację, wstrzykiwanie, wstrzykiwanie lub skórę. [0056] Określenie przeciwciało obejmuje przeciwciała monoklonalne (w tym przeciwciała o pełnej długości, które mają region Fc immunoglobuliny), kompozycje przeciwciała o poliepitopowej specyficzności, przeciwciała wielospecyficzne (np. bispecyficzne przeciwciała, diaciała i cząsteczki jednołańcuchowe, a także fragmenty przeciwciał (np. Fab, F(ab ) 2, i Fv). Określenie immunoglobulina (Ig) jest tu używane zamiennie z przeciwciałem. [0057] Podstawowa 4-łańcuchowa jednostka przeciwciała jest heterotetrameryczną glikoproteiną złożoną z dwóch identycznych łańcuchów lekkich (L) oraz dwóch identycznych łańcuchów ciężkich (H). Przeciwciało IgM składa się z 5 podstawowych jednostek heterotetramerowych wzdłuż dodatkowego polipeptydu zwanego łańcuchem J i zawiera 10 miejsc wiązania antygenu, natomiast przeciwciała klasy IgA zawierają od 2-5 podstawowych 4-łańcuchowych jednostek, które mogą polimeryzować tworząc poliwalentne skupiska w połączeniu z łańcuchem J. W przypadku IgG, 4-łańcuchowa jednostka ma na ogół około daltonów. Każdy łańcuch L jest połączony z łańcuchem H jednym kowalencyjnym wiązaniem dwusiarczkowym, natomiast dwa łańcuchy H są połączone ze sobą za pomocą jednego lub więcej wiązań dwusiarczkowych, w zależności od izotypu łańcucha H. Każdy łańcuch H i L ma także rozmieszczone w regularnych odstępach wewnątrzłańcuchowe mostki dwusiarczkowe. Każdy łańcuch H ma na N-końcu, domenę zmienną (V H ), a następnie trzy domeny stałe (C H ) dla każdego z łańcuchów α i γ i cztery domeny C H dla izotypów μ i ε. Każdy łańcuch L ma na N-końcu, domenę zmienną (V L ), a następnie domenę stałą na swoim drugim końcu. V L jest dopasowane z V H, a C L jest dopasowane do pierwszej domeny stałej łańcucha ciężkiego (C H 1). Uważa się, że poszczególne reszty aminokwasowe tworzą łącznik pomiędzy domenami zmiennymi łańcucha lekkiego i łańcucha ciężkiego. Parowanie V H i V L tworzy pojedyncze miejsce wiązania antygenu. Dla struktury i właściwości różnych klas przeciwciał, zobacz np. Basic and Clinical Immunology, 8. wydanie, Daniel P. Sties, Abba I.
22 21- Terr i Tristram G. Parsolw (eds), Appleton & Lange, Norwalk, CT, 1994, strona 71 i Rozdział 6. [0058] Łańcuch L z jakichkolwiek gatunków kręgowców może być przypisany do jednego z dwóch wyraźnie odrębnych typów, nazywanych kappa i lambda, na podstawie sekwencji aminokwasowych ich domen stałych. W zależności od sekwencji aminokwasowej domeny stałej ich łańcuchów ciężkich (CH), immunoglobuliny można przypisać do różnych klas lub izotypów. Istnieje pięć klas immunoglobulin: IgA, IgD, IgE, IgG i IgM, mające ciężkie łańcuchy oznaczone odpowiednio α, δ, ε, γ i µ. Klasy γ oraz α dzielą się na podgrupy w oparciu o względnie drobne różnice w sekwencji i funkcji CH, np. ludzie eksprymują następujące podklasy: IgG1, IgG2, IgG3, IgG4, IgA1 i IgA2. [0059] Wyizolowane przeciwciało jest takim, które zidentyfikowano, oddzielono i/lub odzyskano ze składnika jego środowiska (np. naturalnego lub rekombinacyjnego). Korzystnie wyizolowany polipeptyd jest wolny od asocjacji z wszystkimi innymi elementami z jego środowiska. Zanieczyszczającymi składnikami jego środowiska, takimi, jak te wynikające z rekombinowanych komórek transfekowanych, są materiały, które zwykle przeszkadzają w badaniu, zastosowaniu diagnostycznym lub zastosowaniu terapeutycznym przeciwciała i mogą obejmować enzymy, hormony i inne białkowe lub niebiałkowe substancje rozpuszczone. W korzystnych wykonaniach, polipeptyd będzie oczyszczony: (1) w stopniu większym niż 95% wagowych przeciwciała, jak określono, na przykład, metodą Lowry ego, a w niektórych przykładach wykonania, w stopniu większym niż 99% wagowych; (1) w stopniu wystarczającym do uzyskania co najmniej 15 reszt N-terminalnej lub wewnętrznej sekwencji aminokwasowej za pomocą wirującego sekwenatora kubkowego, lub (3) do homogenności przez SDS-PAGE w warunkach nieredukujących lub warunkach redukujących przy użyciu błękitu Coomassie lub, korzystnie, barwienia srebrem. Izolowane przeciwciało obejmuje przeciwciało in situ w komórkach rekombinowanych, ponieważ co najmniej jeden składnik naturalnego otoczenia przeciwciała nie będzie obecny. Zazwyczaj, jednakże, izolowany polipeptyd lub przeciwciało będzie przygotowane przez przynajmniej jeden etap oczyszczania. [0060] Region zmienny lub domena zmienna przeciwciała odnosi się do domen aminokońcowych łańcucha ciężkiego lub lekkiego przeciwciała. Każda z domen zmiennych łańcucha ciężkiego i łańcucha lekkiego może być określana jako odpowiednio VH lub VL. Domeny te są na ogół najbardziej zmiennymi częściami przeciwciała (w stosunku do innych przeciwciał o tej samej klasie) i zawierają miejsca wiązania antygenu. [0061] Określenie zmienny odnosi się do faktu, że pewne odcinki domen zmiennych różnią się sekwencją pomiędzy przeciwciałami. Domena V pośredniczy w wiązaniu antygenu i definiuje specyficzność określonego przeciwciała do jego określonego antygenu. Jednakże, zmienność nie jest równomiernie rozłożona w całym zakresie domen zmiennych. Zamiast tego jest ona skoncentrowana w trzech segmentach, zwanych regionami hiperzmiennymi (HVR) zarówno w domenie zmiennej łańcucha ciężkiego, jak i łańcucha lekkiego. Bardziej konserwatywne części domen zmiennych nazywane są regionami zrębowymi (FR). Każda z domen zmiennych natywnych łańcuchów ciężkich i lekkich zawiera cztery regiony FR, w
23 22- większości przyjmujące konfigurację arkusza P, połączone trzema, HVR, które tworzą pętle łączące, a w niektórych przypadkach stanowią część struktury beta-kartki. HVR w każdym łańcuchu są utrzymywane w bliskim sąsiedztwie przez regiony FR i wraz z regionami HVR z drugiego łańcucha przyczyniają się do utworzenia miejsca wiążącego antygen przeciwciał (patrz Kabat i wsp., Sequences of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, MD (1991)). Domeny stałe nie są bezpośrednio zaangażowane w wiązanie przeciwciała z antygenem, ale wykazują różne funkcje efektorowe takie, jak udział przeciwciała w toksyczności komórkowej zależnej od przeciwciał. [0062] Określenie przeciwciało monoklonalne w znaczeniu stosowanym w niniejszym dokumencie odnosi się do przeciwciała otrzymanego z populacji zasadniczo jednorodnych przeciwciał, tj. poszczególne przeciwciała tworzące populację są identyczne, z wyjątkiem możliwych naturalnie występujących mutacji i/lub modyfikacji po translacyjnych (np. izomeryzacje, amidowania), które mogą być obecne w niewielkich ilościach. Przeciwciała monoklonalne są wysoce specyficzne, skierowane przeciw pojedynczym miejscom antygenowym. W przeciwieństwie do preparatów przeciwciał poliklonalnych, które zazwyczaj zawierają różne przeciwciała skierowane przeciwko różnym determinantom (epitopom), każde przeciwciało monoklonalne jest skierowane przeciwko pojedynczej determinancie na antygenie. W uzupełnieniu do ich specyficzności, przeciwciała monoklonalne są korzystne, ponieważ są syntetyzowane przez hodowlę hybrydom, niezanieczyszczone przez inne immunoglobuliny. Modyfikator monoklonalne wskazuje na charakter przeciwciała, jako uzyskanego z zasadniczo homogenicznej populacji przeciwciał i nie należy tego interpretować jako wymóg wytwarzania tego przeciwciała jakąś określoną metodą. Na przykład, przeciwciała monoklonalne stosowane zgodnie z niniejszym wynalazkiem mogą być wykonane za pomocą różnych technik, włączając, na przykład, metodę hybrydomy (np. Kohler i Milstein., Nature, 256: (1975); Hongo i wsp., Hybridoma, 14 (3): (1995), Harlow i wsp., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling i wsp., in: Monoclonal Antibodies and T-Cell Hybridomas (Elsevier, N.Y., 1981)), metody rekombinacji DNA (patrz np. Patent US Nr 4,816,567), technologie prezentacji fagowej (patrz np. Clackson i wsp., Nature, 352: (1991); Marks i wsp., J. Mol. Biol. 222: (1992); Sidhu i wsp., J. Mol. Biol. 338(2): (2004); Lee i wsp., J. Mol. Biol. 340(5): (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101 (34): (2004); i Lee i wsp., J. Immunol. Methods 284(1-2): (2004), i technologie wytwarzania przeciwciał ludzkich lub podobnych do ludzkich u zwierząt, które mają części lub wszystkie ludzkie loci immunoglobulin lub sekwencje genów kodujących ludzkie immunoglobuliny (patrz np. WO 1998/24893; WO 1996/34096; WO 1996/33735; WO 1991/10741; Jakobovits i wsp., Proc. Natl. Acad. Sci. USA 90: 2551 (1993); Jakobovits i wsp., Nature 362: (1993); Bruggemann i wsp., Year in Immunol. 7:33 (1993); Patenty US Nr 5,545,807; 5,545,806; 5,569,825; 5,625,126; 5,633,425; i 5,661,016; Marks i wsp., Biotechnology 10: (1992); Lonberg i wsp., Nature 368: (1994); Morrison, Nature 368: (1994); Fishwild i wsp., Nature Biotechnol. 14: (1996); Neuberger, Nature Biotechnol. 14: 826 (1996); i Lonberg i Huszar, Intern. Rev. Immunol. 13: (1995).
24 23- [0063] Określenie nagie przeciwciało odnosi się do przeciwciała, które nie jest skoniugowane z ugrupowaniem cytotoksycznym lub radioaktywnym. [0064] Określenia przeciwciało pełnej długości, nienaruszone przeciwciało lub pełne przeciwciało są używane zamiennie w odniesieniu do przeciwciała w zasadniczo nienaruszonej postaci, w przeciwieństwie do fragmentu przeciwciała. W szczególności całe przeciwciała obejmują te z ciężkich i lekkich łańcuchów w tym region Fc. Domeny stałe mogą być domenami stałymi o natywnych sekwencjach (np. ludzkie domeny stałe o natywnych sekwencjach) lub sekwencjami aminokwasów jej wariantów. W niektórych przypadkach, nienaruszone przeciwciało może posiadać jedną lub więcej funkcji efektorowych. [0065] Fragment przeciwciała obejmuje część nienaruszonego przeciwciała, korzystnie region wiążący antygen i/lub region zmienny nienaruszonego przeciwciała. Przykłady fragmentów przeciwciał obejmują fragmenty Fab, Fab, F(ab ) 2 i Fv; diaciała; przeciwciała liniowe (patrz Patent US 5,641,870, Przykład 2; Zapata i wsp., Protein Eng. 8(10): [1995]); cząsteczki przeciwciał jednołańcuchowych i wieloswoiste przeciwciała utworzone z fragmentów przeciwciał. [0066] Trawienie przeciwciał papainą wytwarza dwa identyczne fragmenty wiążące antygen, zwane fragmentami Fab, oraz resztkowy fragment Fc, oznaczenie odzwierciedlające zdolność do łatwej krystalizacji. Fragment Fab składa się z całego łańcucha L wraz z domeną regionu zmiennego łańcucha H (V H ), i pierwszą domeną stałą jednego łańcucha ciężkiego (C H 1). Każdy fragment Fab jest jednowartościowy w odniesieniu do wiązania antygenu, tj. ma pojedyncze miejsce wiązania antygenu. Traktowanie pepsyną przeciwciała daje pojedynczy duży fragment F(ab ) 2, który z grubsza odpowiada fragmentowi dwóm połączonym wiązaniem dwusiarczkowym fragmentów Fab o różnej aktywności wiązania antygenu i nadal jest zdolny do krzyżowego wiązania antygenów. Fragmenty Fab różnią się od fragmentów Fab przez posiadanie kilku dodatkowych reszt na końcu karboksylowym domeny C H 1 zawierającym jedną lub więcej cystein z regionu zawiasowego przeciwciała. Fab -SH jest oznaczeniem w niniejszym opisie dla Fab, w którym reszta (reszty) cysteiny domen stałych posiadają wolną grupę tiolową. Fragmenty przeciwciała F(ab ) 2 pierwotnie zostały wyprodukowane jako pary fragmentów Fab, które mają między nimi cysteiny zawiasowe. Inne chemiczne połączenia fragmentów przeciwciał są także znane. [0067] Fragment Fc obejmuje części karboksylowe obu łańcuchów H, spojonych ze sobą mostkami dwusiarczkowymi. Funkcje efektorowe przeciwciał są determinowane przez sekwencje w regionie Fc, w regionie, który jest również rozpoznawany przez receptory Fc (FcR), znajdujące się na określonych typach komórek. [0068] Fv jest minimalnym fragmentem przeciwciała, który zawiera pełne miejsce rozpoznawania i miejsce wiązania antygenu. Ten fragment składa się z dimeru jednej domeny regionu zmiennego łańcucha ciężkiego i jednej domeny zmiennej łańcucha lekkiego w ścisłym, niekowalencyjnym połączeniu. Z fałdowania tych dwóch domen pochodzi sześć pętli hiperzmiennych (po 3 pętle z każdego łańcucha H i L), które sprawiają, że reszty
25 24- aminokwasowe przyczyniają się do wiązania antygenu i nadają przeciwciału specyficzność wiązania przeciwciała. Jednakże, nawet pojedyncza domena zmienna (lub połowa Fv zawierająca tylko trzy HVR specyficzne dla antygenu) ma zdolność rozpoznawania i wiązania antygenu, choć z niższym powinowactwem niż całe miejsce wiązania. [0069] Jednołańcuchowe Fv skracane także jako sfv lub scfv to fragmenty przeciwciał, które zawierają domeny VH i VL połączone w pojedynczy łańcuch polipeptydowy. Korzystnie, polipeptyd sfv zawiera ponadto polipeptydowy łącznik pomiędzy domenami V H i V L, który umożliwia tworzenie przez sfv pożądanej struktury do wiązania antygenu. Dla przeglądu sfv, patrz Pluckthun w The Pharmacology of Monoclonal Antibodies, tom 113, Rosenburg i Moore eds., Springer-Verlag, New York, pp (1994). [0070] Funkcjonalne fragmenty przeciwciał według wynalazku obejmują część nienaruszonego przeciwciała ogólnie zawierającą region wiązania antygenu lub region zmienny nienaruszonego przeciwciała lub region F przeciwciała, który zachowuje lub ma zmodyfikowaną zdolność wiązania FcR. Przykłady fragmentów przeciwciał obejmują przeciwciała liniowe, cząsteczki przeciwciał jednołańcuchowych i wielospecyficzne przeciwciała utworzone z fragmentów przeciwciał. [0071] Określenie diaciała odnosi się do małych fragmentów przeciwciał, wytworzonych przez konstruowanie fragmentów sfv (patrz poprzedzający akapit) z krótkimi łącznikami (około 5-10 reszt) pomiędzy domenami V H i V L tak, że między łańcuchami, ale nie w obrębie łańcucha uzyskuje się parowanie domen V, uzyskując w ten sposób fragment dwuwartościowy, tj. fragment mający dwa miejsca wiązania antygenu. Bispecyficzne diaciała są heterodimerami dwóch skrzyżowanych fragmentów sfv, w których domeny V H i V L dwóch przeciwciał są obecne na różnych łańcuchach polipeptydowych. Diaciała opisano dokładniej w, na przykład, EP 404,097; WO 93/11161; Hollinger i wsp., Proc. Natl. Acad. Sci. USA 90: (1993). [0072] Tutaj przeciwciała monoklonalne specyficznie obejmują przeciwciała chimeryczne (immunoglobuliny), w których część łańcucha ciężkiego i/lub lekkiego jest identyczna z lub homologiczna do odpowiednimi sekwencjami w przeciwciałach pochodzących od określonego gatunku lub należących do określonej klasy lub podklasy przeciwciał, natomiast pozostały łańcuch (pozostałe łańcuchy) są identyczna z lub homologiczna z odpowiednimi sekwencjami w przeciwciałach pochodzących od innego gatunku lub należących do innej klasy lub podklasy przeciwciał, jak również fragmentami takich przeciwciał, o ile wykazują one pożądaną aktywność biologiczną (Patent US Nr 4,816,567; Morrison i wsp., Proc. Natl. Acad. Sci. USA, 81: (1984)). Przeciwciała chimeryczne będące tu przedmiotem zainteresowania obejmują przeciwciała PRIMATIZED, przy czym region wiązania antygenu przeciwciała pochodzi z przeciwciała wytworzonego przez np. immunizowanie makaków antygenem. Stosowane tu określenie humanizowane przeciwciało jest stosowane jako podzestaw definicji chimerycznych przeciwciał.
26 25- [0073] Humanizowanymi postaciami przeciwciał nie-ludzkich (np. mysich) są przeciwciała chimeryczne, zawierające minimalną sekwencję pochodzącą z nie-ludzkiej immunoglobuliny. W jednym przykładzie wykonania, humanizowane przeciwciało jest ludzką immunoglobuliną (przeciwciało biorcy), w której reszty HVR biorcy są zastąpione przez reszty z HVR z innego gatunku niż człowiek (przeciwciało dawcy) takiego, jak mysz, szczur, królik lub naczelnego niebędącego człowiekiem, o pożądanej specyficzności, powinowactwie i/lub pojemności. W niektórych przypadkach, reszty FR z immunoglobuliny ludzkiej zastępuje się odpowiadającymi resztami gatunku innego niż człowiek. Ponadto, humanizowane przeciwciała mogą zawierać reszty, które nie występują ani w przeciwciele biorcy, ani w przeciwciele dawcy. Te modyfikacje mogą być dokonane w celu dalszego udoskonalenia właściwości przeciwciała takich, jak powinowactwo wiązania. Na ogół, humanizowane przeciwciało będzie zawierać zasadniczo wszystkie z co najmniej jednej, a typowo dwóch, domen zmiennych, w których wszystkie lub zasadniczo wszystkie hiperzmienne pętle odpowiadają tym z nie-ludzkiej sekwencji immunoglobulin, a wszystkie lub zasadniczo wszystkie regiony FR są tymi o sekwencji immunoglobuliny ludzkiej, chociaż regiony FR mogą zawierać jeden lub więcej pojedynczych podstawień reszt FR, które poprawiają działanie przeciwciała, np. wykazujących powinowactwo wiązania, izomeryzację, immunogenność, itd. Liczba tych podstawień aminokwasowych w FR wynosi zwykle nie więcej niż 6 w łańcuchu H i w łańcuchu L, nie więcej niż 3. Humanizowane przeciwciało opcjonalnie będzie również zawierać co najmniej część regionu stałego immunoglobuliny (Fc), zwykle tego z ludzkiej immunoglobuliny. W celu uzyskania dalszych szczegółów, patrz, np., Jones i wsp., Nature 321: (1986); Riechmann i wsp., Nature 332: (1988); i Presta, Curr. Op. Struct. Biol. 2: (1992). Patrz także na przykład Vaswani i Hamilton, Ann. Allergy, Astma & Immunol. 1: (1998); Harris, Biochem. Soc. Transactions 23: (1995); Hurle i Gross, Curr. Op. Biotech. 5: (1994); i Pat. US Nr 6,982,321 i 7,087,409. [0074] Ludzkie przeciwciało to takie, które posiada sekwencję aminokwasową odpowiadającą przeciwciału wytwarzanemu przez organizm człowieka i/lub zostało wykonane przy użyciu dowolnej techniki wytwarzania ludzkich przeciwciał, jak ujawniono w niniejszym opisie. Ta definicja ludzkiego przeciwciała wyklucza w szczególności humanizowane przeciwciała zawierające reszty wiążące antygen pochodzące od gatunków innych niż człowiek. Ludzkie przeciwciała mogą być wytwarzane przy użyciu różnych technik znanych w dziedzinie, w tym bibliotek prezentacji fagowej. Hoogenboom i Winter, J. Mol. Biol., 227:381 (1991); Marks i wsp., J. Mol. Biol., 222:581 (1991). Dostępne również do wytwarzania ludzkich przeciwciał monoklonalnych są metody opisane w Cole i wsp., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985); Boerner i wsp., J. Immunol., 147(1):86-95 (1991). Patrz także van Dijk i van de Winkel, Curro Opin. Pharmacol., 5: (2001). Przeciwciała ludzkie można wytworzyć przy podawaniu antygenu zwierzęciu transgenicznemu, które zostało zmodyfikowane w celu wytworzenia takich przeciwciał w odpowiedzi na działanie antygenu, ale których endogenne loci zostały wyłączone, np. immunizowane ksenomyszy (patrz np. US Pat. Nry 6,075,181 i 6,150,584 dla technologii XENOMOUSE ). Patrz również na przykład Li i wsp., Proc. Natl. Acad. Sci.
27 26- USA, 103: (2006) dla ludzkich przeciwciał wytwarzanych przez technologię hybrydomy z ludzkimi komórkami B. [0075] Określenie region hiperzmienny, HVR lub HV stosowane w niniejszym kontekście, odnosi się do regionów domeny zmiennej przeciwciała, które są hiperzmienne pod względem sekwencji i/lub które tworzą strukturalne pętle. Ogólnie, przeciwciała zawierają sześć HVR; trzy w VH (H1, H2, H3) i trzy w VL (L1, L2, L3). W natywnych przeciwciał, H3 i L3 prezentują najwięcej różnorodności z sześciu HVR, a w szczególności uważa się, że H3 odgrywa wyjątkową rolę w nadawaniu znakomitej specyficzności przeciwciał. Np. Xu i wsp. Immnunity 13:37-45 (2000); Johnson i Wu w Methods in Molecular Biology 248:1-25 (Lo, ed., Human Press, Totowa, NJ, 2003)). Rzeczywiście, naturalnie występujące przeciwciała wielbłądowatych, składające się tylko z łańcucha ciężkiego są funkcjonalne i trwałe w nieobecności łańcucha lekkiego. Patrz, na przykład, Hamers-Casterman i wsp., Nature 363: (1993) i Sheriff i wsp., Nature Struck. Biol. 3: (1996). [0076] Wiele delineacji HVR jest w użyciu i są tu ujęte. HVR, które są regionami determinującymi komplementarność (CDR) Kabata są oparte o zmienność sekwencji i są używane najczęściej (Kabat i wsp., supra). Chothia odwołuje się do lokalizacji pętli strukturalnych (Chothia i Lesk J. Mol. Biol. 196: (1987)). HVR AbM stanowią kompromis pomiędzy CDR według Kabata i pętlami strukturalnymi Chothia, i są stosowane przez oprogramowanie do modelowania przeciwciał Oxford Molecular AbM. Kontaktowe HVR opierają się na analizie dostępnych struktur krystalicznych kompleksów. Reszty każdego z tych HVR są opisane poniżej. Pętla Kabat AbM Chothia Kontakt L1 L24-L34 L24-L34 L26-L34 L30-L36 L2 L50-L56 L50-L56 L50-L56 L46-L55 L3 L89-L97 L89-L97 L91-L96 L89-L96 H1 H31-H35B H26-H35B H26-H32 H30-H35B (Numeracja Kabat) H1 H31-H35 H26-H35 H26-H32 H30-H35 (Numeracja Chothia) H2 H50-H65 H50-H58 H53-H56 H47-H58 H3 H95-H102 H95-H102 H95-H102 H93-H101 [0077] HVR mogą zawierać rozszerzone HVR w następujący sposób: lub (L1), lub (L2), i lub (L3) w VL i (H1), lub (korzystny przykład wykonania) (H2), i (H3) w VH. Reszty domeny zmiennej są numerowane według Kabata i wsp., supra, dla każdej z tych definicji rozszerzonych HVR. [0078] Reszty zrębowe lub FR są tymi resztami domeny zmiennej innymi niż te zdefiniowane tu reszty HVR.
28 27- [0079] Wyrażenie numeracja reszt domeny zmiennej według Kabata lub numeracja pozycji aminokwasowych według Kabata i ich warianty, odnosi się do systemu numerowania stosowanego dla domen zmiennych łańcucha ciężkiego lub łańcucha lekkiego z domen zmiennych kompilacji przeciwciał w Kabat i wsp., supra. Przy zastosowaniu tego systemu numerowania, rzeczywista liniowa sekwencja aminokwasowa może zawierać dodatkowe aminokwasy, co odpowiada skróceniu, lub insercji do, FR lub HVR domeny zmiennej. Na przykład, domena zmienna łańcucha ciężkiego może zawierać pojedynczą wstawkę aminokwasową (reszta 52a według Kabata) po reszcie 52 w H2 oraz wstawione reszty (np. reszty 82a, 82b, 82c itd. według Kabata) po reszcie FR łańcucha ciężkiego 82. Numerowanie reszt według Kabata może być określane dla danego przeciwciała poprzez nałożenie regionów homologicznych sekwencji przeciwciała ze standardową, numerowaną sekwencją Kabata. [0080] Zrąb ludzkiego akceptora dla celów niniejszego wynalazku jest zrębem zawierającym sekwencję aminokwasów regionów zrębowych VL lub VH pochodzącą z sekwencji zrębowej ludzkiej immunoglobuliny lub ludzkiej konsensusowej sekwencji zrębowej. Ludzka akceptorowa sekwencja zrębowa pochodząca z sekwencji zrębowej ludzkiej immunoglobuliny lub ludzkiej konsensusowej sekwencji zrębowej może zawierać te same aminokwasy, co wymieniona sekwencja, lub może zawierać zmiany wcześniej istniejących sekwencji aminokwasów. W niektórych rozwiązaniach, ilość wcześniej istniejących sekwencji aminokwasów wynoszą 10 lub mniej, 9 lub mniej, 8 lub mniej, 7 lub mniej, 6 lub mniej, 5 lub mniej, 4 lub mniej, 3 lub mniej czy 2 lub mniej. [0081] Ludzki zrąb konsensusowy jest zrębem, która reprezentuje najczęściej występujące reszt aminokwasowych w wyborze sekwencji zrębowych ludzkiej immunoglobuliny VH lub VL. Na ogół, wybór ludzkich sekwencji immunoglobulin VL lub VH zawiera się w podgrupie sekwencji domen zmiennych. Ogólnie, podgrupa sekwencji jest podgrupą zgodna z opisem w Kabat i wsp., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991). W jednym przykładzie wykonania, dla VL, podgrupa oznacza podgrupę kappa I, jak w Kabat i wsp., supra. W jednym przykładzie wykonania, dla VH, podgrupa oznacza podgrupę III, jak w Kabat i wsp., supra. [0082] Zrąb konsensusowy podgrupy III VH zawiera sekwencję konsensusową otrzymaną z sekwencji aminokwasowych w zmiennej ciężkiej podgrupie III według Kabat i wsp., supra. W jednym przykładzie wykonania, sekwencja aminokwasowa zrębu konsensusowego podgrupy III VH zawiera co najmniej część lub całość każdej z poniższych sekwencji: EVQLVESGGGLVQPGGSLRLSCAAS (H1)(SEQ ID NO:55), WVRQAPGKGLEWVA (SEQ ID NO:56)(H2), RFTISRDDSKNTLYLQMNSLRAEDTAVYYCAR (SEQ ID NO:57)(H3), WGQGTLVTVSS (SEQ ID NO:58)(H4). [0083] Zrąb konsensusowy podgrupy I VL zawiera sekwencję konsensusową otrzymaną z sekwencji aminokwasowych w zmiennej lekkiej kappa podgrupie I Kabat i wsp., supra. W jednym przykładzie wykonania, sekwencja aminokwasowa zrębu podgrupy I VH zawiera co najmniej część lub całość każdej z poniższych sekwencji: DIQMTQSPSSLSASVGDRVTITC (SEQ ID NO:59)(L1), WYQQKPGKAPKLLIY (SEQ ID NO:60)(L2),
29 28- GVPSRFSGSGSGTDFTLTISSLQPEDFATYYC (SEQ ID NO:61)(L3), FGQGTKVEIKR (SEQ ID NO:62)(L4). [0084] Modyfikacja aminokwasowa w określonym położeniu, na przykład regionie Fc odnosi się do substytucji lub delecji danej reszty lub insercji co najmniej jednej reszty aminokwasowej w sąsiedztwie danej reszty. Insercja w sąsiedztwie określonej reszty oznacza wprowadzenie jej w jednej do dwóch reszt. Insercja może być N-końcowa lub C- końcowa w stosunku do danej reszty. Korzystną modyfikacją aminokwasową jest tu substytucja. [0085] Przeciwciało o dojrzałym powinowactwie jest przeciwciałem z jedną lub większą liczbą zmian w ich HVR, które prowadzą do poprawy powinowactwa przeciwciała do antygenu, w porównaniu do przeciwciała macierzystego, które nie posiada tej zmiany (tych zmian). W jednym przykładzie wykonania, przeciwciało o dojrzałym powinowactwie ma nanomolarne lub nawet pikomolarne powinowactwo do antygenu docelowego. Przeciwciała o dojrzałym powinowactwie są wytwarzane za pomocą procedur znanych ze stanu techniki. Na przykład, Marks i wsp., Biotechnology 10: (1992) opisuje dojrzewanie powinowactwa przez tasowanie domen VH i VL. Losowa mutageneza reszt HVR i/lub zrębowych została opisana w, na przykład: Barbas i wsp. Proc Nat. A cad. Sci. USA 91: (1994); Schier i wsp. Gene 169: (1995); Yelton i wsp. J. Immunol. 155: (1995); Jackson i wsp., J. Immunol. 154(7): (1995); i Hawkins et al, J. Mol. Biol. 226: (1992). [0086] Przeciwciało, które specyficznie wiąże się z lub jest specyficzne dla określonego polipeptydu lub epitopu na określonym polipeptydzie jest takim, które wiąże się do tego określonego polipeptydu lub epitopu na określonym polipeptydzie bez zasadniczego wiązania się do jakiegokolwiek innego polipeptydu lub epitopu polipeptydu. Na przykład, przeciwciała specyficzne dla M1 według niniejszego wynalazku są specyficzne dla pozakomórkowego odcinka M1 IgE występującego na IgE związanej na komórkach B, lecz niewystępującego na wydzielanej IgE. [0087] Przeciwciałem blokującym lub antagonistycznym jest przeciwciało, które hamuje lub redukuje aktywność biologiczną wiązanego antygenu. W niektórych przykładach wykonania, przeciwciała blokujące lub antagonistyczne zasadniczo lub całkowicie hamują aktywność biologiczną antygenu. Apoptotyczne przeciwciała anty-ige według wynalazku blokują aktywność IgE w mediowaniu odpowiedzi immunologicznej, w taki sposób aby zmniejszyć zarówno wolną IgE w surowicy, jak i całkowitą IgE w surowicy. [0088] Przeciwciała, które indukują apoptozę albo są apoptotycznye są tymi, które indukują programowaną śmierć komórki, co oznaczono standardowymi testami na apoptozę takimi, jak wiązanie aneksyny V, fragmentacja DNA, kurczenie się komórki, rozszerzanie retikulum endoplazmatycznego, fragmentacja komórek i/lub tworzenie pęcherzyków błonowych (nazywanych ciałkami apoptycznymi). Na przykład, aktywność apoptotyczną przeciwciała anty-ige niniejszego wynalazku można przedstawić jako barwienie komórek z IgE związaną na powierzchni aneksyną V.
30 29- [0089] Określenie całkowite IgE w surowicy odnosi się do całkowitej ilości IgE w próbce, w tym zarówno wolnej lub niezwiązanej, oraz IgE, która jest skompleksowana z partnerem wiązania (np. przeciwciało anty-ige, komórka B mająca IgE). Wolna IgE w surowicy odnosi się do IgE, która nie jest związana z partnerem wiążącym. [0090] Określenie IgE specyficzna alergenowo odnosi się do IgE, która jest specyficzna dla danego antygenu, uzyskanej z początkowej ekspozycji na alergen w procesie znanym jako uczulanie alergiczne, i która wiąże się z powierzchnią komórek tucznych i bazofili i która może powodować aktywację komórek tucznych i bazofili po późniejszej ekspozycji na ten sam alergen. Kilka czynników mitogennych w wirusach (np. wirus cytomegalii - CMV), bakteriach (np. Staphylococcus), robakach (np. Ascaris, Schistosoma) i czynników adiuwantowych w zanieczyszczeniu powietrza (np. dym papierosowy i wydech z diesla) stymulują produkcję cząsteczek IgE bez inicjowania specyficznego dla alergenu IgE uczulenia. Tak więc, ponieważ poziomy IgE mogą zwiększyć się w sposób, który nie musi predysponować gospodarza do stania się bardziej podatnym na zaburzenia, w których pośredniczy IgE, poziomy IgE specyficznej alergenowo są czasami stosowane w ocenach klinicznych. [0091] Określenie specyficznie zubaża komórki B produkujące IgE oznacza zdolność do specyficznego zmniejszenia populacji lub skuteczności komórek B, które wydzielają w szczególności IgE (tj. komórek plazmatycznych), jednak, która nie wpływa znacząco na skuteczność lub populację komórek B, które wydzielają inne immunoglobuliny takie, jak lgg1, IgG2, IgG3, IgG4, IgA i IgM. [0092] Określenie faza stała opisuje niewodną macierz, do której przeciwciało według niniejszego wynalazku może przylegać. Przykłady faz stałych objętych zakresem niniejszego wynalazku obejmują te utworzone częściowo lub w całości ze szkła (np. szkła o kontrolowanej porowatości), polisacharydów (np. agarozy), poliakrylamidów, polistyrenu, alkoholu poliwinylowego i silikonów w niektórych przykładach wykonania, w zależności od kontekstu, faza stała może obejmować dołek płytki testowej; w innych jest to kolumna do oczyszczania (np. kolumna do chromatografii powinowactwa) Określenie to obejmuje również nieciągłą fazę stałą odrębnych cząstek takich, jak tych opisanych w patencie US nr 4,275,149. [0093] Funkcje efektorowe przeciwciała odnoszą się do tych aktywności biologicznych, które można przypisać regionowi Fc (sekwencja natywna regionu Fc lub wariant sekwencji aminokwasowej regionu Fc) przeciwciała i różnią się w zależności od izotypu przeciwciała. Przykłady funkcji efektorowych przeciwciał obejmują: wiązanie C1q i cytotoksyczność zależną od dopełniacza; wiązanie receptora Fc; cytotoksyczność komórkowa zależna od przeciwciał (ADCC); fagocytozę; regulację w dół receptorów na powierzchni komórek (np. receptorów komórek B); i aktywację komórek B. [0094] Cytotoksyczność komórkowa zależna od przeciwciał lub ADCC odnosi się do postaci cytotoksyczności, w której wydzielane Ig związane z receptorami Fc (FcR) obecne na niektórych komórkach cytotoksycznych (np. komórki naturalnych zabójców (NK), neutrofile
31 30- i makrofagi) umożliwiają tym cytotoksycznym komórkom efektorowym specyficzne wiązanie się do komórki docelowej niosącej antygen, a następnie zabijają komórkę docelową za pomocą cytotoksyn. Przeciwciała uzbrajają komórki cytotoksyczne i są niezbędne do uśmiercania komórki docelowej w tym mechanizmie. Komórki pierwotne dla mediowania ADCC, komórki NK, eksprymują tylko FcyRIII, natomiast monocyty eksprymują FcyRI, FcyRII i FcyRIII. Ekspresja Fc na komórkach hematopoetycznych jest podsumowana w Tabeli 3 na stronie 464 w publikacji Ravetch i Kinet, Annu. Rev. Immunol. 9: (1991). W celu oszacowania aktywności ADCC cząsteczki będącej przedmiotem zainteresowania, można przeprowadzić test in vitro ADCC taki, jak opisano na przykład w Patencie US Nr 5,500,362 lub 5,821,337. Użyteczne komórki efektorowe do takich testów obejmują komórki jednojądrzaste krwi obwodowej (PBMC) i komórki naturalnych zabójców (NK). Alternatywnie, lub dodatkowo, aktywność ADCC cząsteczki będącej przedmiotem zainteresowania można oceniać in vivo, np. w modelu zwierzęcym takim, jak ujawniony w Clynes i wsp., PNAS USA 95: (1998). [0095] O ile nie wskazano inaczej, numeracja reszt łańcucha ciężkiego immunoglobuliny jest zgodna z indeksem EU, jak w Kabat i wsp., supra. Indeks EU, jak w Kabat odnosi się do numerowania reszt ludzkiego przeciwciała EU IgGl. [0096] Stosowane tu określenie region Fc jest używane do określenia C-końcowego regionu łańcucha ciężkiego immunoglobuliny, w tym regionów sekwencji natywnej Fc i regionów wariantu Fc. Mimo, że granice regionu Fc łańcucha ciężkiego immunoglobuliny mogą się różnić, ludzki ciężki łańcuch regionu Fc IgG jest zazwyczaj definiowany jako rozciągający się od reszty aminokwasowej w pozycji Cys226 lub od Pro230 do jego końca karboksylowego. C-końcowa lizyna (reszta 447 według systemu numeracji EU) regionu Fc może zostać usunięta, na przykład podczas wytwarzania lub oczyszczania przeciwciała lub poprzez inżynierię w drodze rekombinacji kwasu nukleinowego kodującego łańcuch ciężki przeciwciała. Zgodnie z tym, kompozycja nienaruszonych przeciwciał może zawierać populację przeciwciał ze wszystkimi resztami K447 usuniętymi, i populacje przeciwciał, które nie mają usuniętych reszt K447 i populacje przeciwciał mających mieszaninę przeciwciał z lub bez reszty K447. Odpowiednie regiony Fc o natywnej sekwencji do zastosowania przeciwciał według wynalazku obejmują ludzkie IgG1, IgG2, IgG3 i IgG4. [0097] Receptor Fc lub FcR opisuje receptor, który wiąże się do regionu Fc przeciwciała. Korzystnym FcR jest natywna sekwencja ludzkiego FcR. Ponadto, korzystnym FcR jest taki, który wiąże przeciwciało IgG (receptor gamma) i obejmuje receptory podklas FcγRI, FcγRII i FcγRIII, w tym warianty alleliczne i alternatywnie składane postacie tych receptorów, receptory FcγRII obejmują FcγRIIA ( receptor aktywujący ) i FcγRIIB ( receptor hamujący ), które mają podobne sekwencje aminokwasowe, różniące się przede wszystkim w swoich domenach cytoplazmatycznych. Receptor aktywujący FcγRIIA zawiera motyw opartej na tyrozynie aktywacji immunoreceptora (ITAM) w swojej domenie cytoplazmatycznej. Hamujący receptor FcγRIIB zawiera immunoreceptorowy motyw hamowania oparty na tyrozynie (ITIM) w swej domenie cytoplazmatycznej. (patrz M. Daëron, Annu. Rev. Immunol. 15: (1997). FcR omówiono w Ravetch i Kinet, Annu. Rev. Immunol. 9:
32 (1991); Capel i wsp., Immunomethods 4: (1994); i de Haas i wsp., J. Lab. Clin. Med. 126: (1995). Inne FcR, w tym te, które zostaną zidentyfikowane w przyszłości, są tu objęte określeniem FcR. [0098] Określenie receptor Fc lub FcR obejmuje również noworodkowy receptor, FcRn, który jest odpowiedzialny za przenoszenie matczynych IgG do płodu. Guyer i wsp., J. Immunol. 117: 587 (1976) i Kim i wsp., J. Immunol. 24: 249 (1994). Metody pomiaru wiązania do FcRn są znane (patrz np. Ghetie i Ward, Immunol. Today 18: (12): (1997); Ghetie i wsp., Nature Biotechnology 15 (7): (1997); Hinton i wsp., J. Biol. Chem. 279 (8): (2004); WO 2004/92219 (Hinton i wsp.). [0099] Wiązanie do FcRn in vivo oraz czas półtrwania w osoczu ludzkiego FcRn o wysokim powinowactwie wiązania polipeptydów można badać, na przykład, w transgenicznych myszach lub w transfekowanych liniach ludzkich komórek wykazujących ekspresję ludzkiego FcRn, albo u naczelnych, do których polipeptydy posiadające wariant regionu Fc są podawane. WO 2004/42072 (Presta) opisuje warianty przeciwciał, które miały zwiększone lub obniżone wiązanie do FcR. Patrz też np. Shields i wsp., J. Biol. Chem. 9(2): (2001). [0100] Ludzkie komórki efektorowe to leukocyty, które eksprymują jedno lub więcej FcR i które pełnią funkcje efektorowe. Korzystnie, komórki wykazują ekspresję co najmniej FcyRIII i wykonują funkcję efektorową ADCC. Przykłady ludzkich leukocytów, które pośredniczą w ADCC obejmują komórki jednojądrzaste krwi obwodowej (PBMC), komórki naturalnych zabójców (NK), monocyty, komórki T cytotoksyczne i neutrofile, PBMC i korzystnie komórki MNK. Komórki efektorowe można izolować z ich naturalnego źródła, np. krwi. [0101] Cytotoksyczność zależna od dopełniacza lub CDC odnosi się do lizy komórki docelowej w obecności dopełniacza. Aktywacja klasycznego szlaku dopełniacza jest inicjowana przez wiązanie pierwszego składnika układu dopełniacza (C1q) do przeciwciał (odpowiedniej podklasy), które są związane do pokrewnego antygenu. W celu ocenienia aktywacji dopełniacza można przeprowadzić oznaczenie CDC, np. jak opisano w Gazzano- Santoro i wsp., J. Immunol. Methods 202: 163 (1996). [0102] Warianty polipeptydowe o zmienionych sekwencjach aminokwasowych regionu Fc i zwiększonej lub zmniejszonej zdolności wiązania C1q są opisane w patencie US nr 6,194,551B1 i WO99/ Zawartość tych publikacji patentowych jest specyficznie włączona do niniejszego opisu przez odniesienie. Zobacz także, Idusogie i wsp. J. Immunol. 164: (2000). [0103] Miejsce N-glikozylacji w IgG jest przy Asn297 w domenie CH2. Niniejszy wynalazek dostarcza także kompozycje wiążącego CD20, humanizowanego przeciwciała mającego region Fc, przy czym % (a korzystnie 90-99%) przeciwciała w kompozycji zawiera dojrzałą strukturę węglowodanową rdzenia, której brak fukozy, dołączonej do regionu Fc glikoproteiny. Takie kompozycje przedstawiono poniżej wykazują zaskakującą poprawę wiązania FcγRIIIA(F158), które nie jest tak skuteczne jak FcγRIIIA (V158) w interakcji z
33 32- ludzkim IgG. Tak więc, przewiduje się, że ujawnione tu kompozycje są lepsze niż uprzednio opisane kompozycje przeciwciała anty-cd20, w szczególności do leczenia pacjentów będących ludźmi, którzy eksprymują FcγRIIIA (F158). FcγRIIIA (F158) jest bardziej powszechny niż FcγRIIIA (V158) w prawidłowych, zdrowych Afroamerykanów i rasy kaukaskiej. Patrz Lehrnbecher i wsp. Blood 94:4220 (1999). Niniejsze zgłoszenie dodatkowo wykazuje synergistyczny wzrost wiązania FcyRIII i/lub funkcji ADCC wynikającej z połączenia ujawnionych tu wariacji glikozylacji z modyfikacją (modyfikacjami) sekwencji aminokwasowej w regionie Fc glikoproteiny. [0104] Powinowactwo wiązania ogólnie odnosi się do siły sumy niekowalencyjnych oddziaływań pomiędzy pojedynczymi miejscami wiązania cząsteczki (np. przeciwciała) i partnera wiązania (np. antygenu). Jeśli nie wskazano tego inaczej, stosowany tu termin powinowactwo wiązania dotyczy wewnętrznego powinowactwa wiązania, które odzwierciedla oddziaływanie 1:1 pomiędzy składnikami wiązanej pary (np. przeciwciała i antygenu).powinowactwo cząsteczki X do jej partnera Y może ogólnie być wyrażone poprzez stałą dysocjacji (Kd). Powinowactwo może być mierzone za pomocą powszechnych sposobów znanych w danej dziedzinie, włączając te opisane w niniejszym dokumencie. Przeciwciała o niskim powinowactwie na ogół wiążą antygen wolno i wykazują tendencję do łatwej dysocjacji, podczas gdy przeciwciała o wysokim powinowactwie z reguły wiążą antygen szybciej i pozostają związane przez dłuższy czas. W dziedzinie znanych jest wiele sposobów mierzenia powinowactwa wiązania, z których każdy może być stosowany do celów niniejszego wynalazku. Konkretne ilustrujące przykłady wykonania i do pomiaru powinowactwa wiązania, są opisane poniżej. [0105] W jednym przykładzie wykonania, Kd lub wartość Kd według tego wynalazku mierzy się w teście radioznakowanego wiązania antygenu (RIA), wykonanym z wersją Fab przeciwciała będącego przedmiotem zainteresowania oraz jego antygenu, jak opisano w następującym badaniu. Powinowactwo wiązania w roztworze Fab do antygenu zmierzono przez Fab w równowadze ze stężeniem minimalnym ( 125 I)-znakowanego antygenu w obecności serii miareczkowań nieznakowanego antygenu, a następnie wychwycenie antygenu związanego z płytką pokrytą przeciwciałami anty-fab (patrz np. Chen i wsp., J. Mol. Biol. 293: (1999)). W celu stworzenia warunków do przeprowadzenia oznaczenia płytki do mikromiareczkowania (DYNEX Technologies, Inc.) powleka się przez noc 5 µg/ml wychwytującym przeciwciałem anty-fab (Cappel Labs) w 50 mm węglanu sodu (ph 9,6) a następnie blokuje się 2% (w/v) albuminy surowicy bydlęcej w PBS przez dwie do pięciu godzin w temperaturze pokojowej (około 23 C). W płytce nie pochłaniającej (Nunc #269620) miesza się 100 pm lub 26 pm [ 125 I]-antygenu z serią rozcieńczeń Fab będącego przedmiotem zainteresowania (np. zgodnie z oznaczeniem przeciwciała anty-vegf, Fab-12, w Presta i wsp., Cancer Res. 57: (1997)). Fab będący przedmiotem zainteresowania poddaje się inkubacji przez noc; jednak, inkubacja może być kontynuowana przez dłuższy okres (np. około 65 godzin) w celu zapewnienia osiągnięcia stanu równowagi. Następnie, mieszaniny są przenoszone na płytkę wychwytującą celem inkubacji w temperaturze pokojowej (np. przez jedną godzinę). Roztwór jest następnie usuwany, a płytka płukana osiem razy 0.1% surfaktantem TWEEN-20 w PBS. Po wyschnięciu płytek, dodaje
34 33- się 150 µl/dołek scyntylanta (MICROSCINT-20 ; Packard), a płytki zlicza się w liczniku gamma TOPCOUNT (Packard) przez dziesięć minut. Stężenia każdego Fab zapewniające nie więcej niż lub równo 20% maksymalnego wiązania są wybierane do użycia w konkurencyjnych testach wiązania. [0106] Zgodnie z innym przykładem wykonania, Kd mierzy się przy użyciu oznaczenia powierzchniowym rezonansem plazmonowym za pomocą aparatu BIACORE lub BIACORE (BIAcore, Inc., Piscataway, NJ) w 25 C z płytkami CM5 z immobilizowanym antygenem przy 10 jednostkach odpowiedzi (RU). W skrócie, płytki bioczujnikowe z karboksymetylowanego dekstranu (CM5, BIAcore Inc.) aktywuje się chlorowodorkiem N-etylo-N -(3-dimetyloaminopropylo)-karbodiimidu (EDC) oraz N- hydroksysukcynoimidem (NHS) zgodnie z instrukcjami dostawcy. Antygen rozcieńcza się 10 mm octanem sodowym, ph 4.8, do 5 µg/ml ( 0.2 µm) przed wstrzyknięciem z szybkością przepływu 5 µl/minutę, do osiągnięcia około 10 jednostek odpowiedzi (RU) sprzężonego białka. Po wstrzyknięciu antygenu, wstrzykuje się 1 M etanoloaminę w celu zablokowania grup nieprzereagowanych. Dla pomiarów kinetycznych, nastrzykuje się serie dwukrotnych rozcieńczeń Fab (0.78 nm to 500 nm) w PBS z 0.05% surfaktanta TWEEN 20 (PBST) w 25 C z szybkością przepływu około 25 µl/min. Stałe asocjacji (k on ) i stałe dysocjacji (k off ) oblicza się za pomocą prostego modelu wiązania Langmuira typu jeden-do-jednego (BIAcore Evaluation Software version 3.2) poprzez jednoczesne dopasowanie sensorgramów asocjacji i dysocjacji. Stałą równowagi dysocjacji (Kd) oblicza się jako stosunek k off /k on. Patrz np. Chen i wsp., J. Mol. Biol. 293: (1999). Jeśli stała szybkości asocjacji oznaczona rezonansem plazmonowym powyżej przekracza 10 6 M -1 s -1, wówczas szybkość asocjacji można określić stosując techniki wygaszania fluorescencji, która mierzy wzrost lub spadek intensywności emisji fluorescencji (wzbudzanie = 295 nm, emisja = 340 nm, 16 nm filtr pasmowo-przepustowy) w temperaturze 25 C z przeciwciałem antyantygen 20 nm (postać Fab) w PBS, ph 7.2, w obecności rosnących stężeń antygenu, jak mierzono w spektrometrze, takim jak spektrofotometru wyposażony w stop flow (Aviv Instruments) lub spektrofotometru 8000-series SLM-AMINCO (ThermoSpectronic) z mieszaną kuwetą. [0107] Szybkość asocjacji, szybkość asocjacyjna lub k on według niniejszego wynalazku może być również określona w sposób opisany powyżej, stosując układ BIACORE lub BIACORE (BIAcore, Inc., Piscataway, NJ). [0108] Sformułowanie zasadniczo zmniejszony lub zasadniczo różny stosowane w niniejszym opisie, oznacza wystarczająco wysoki stopień różnicy pomiędzy dwiema wartościami liczbowymi (ogólnie jedną związaną z cząsteczką, a drugą związaną z cząsteczką odniesienia/porównawczą), by znawca w danej dziedzinie uznał różnicę pomiędzy obiema wartościami za istotną statystycznie w odniesieniu do parametru biologicznego mierzonego wspomnianymi wartościami (np. wartości Kd). Różnica pomiędzy dwiema wymienionymi wartościami wynosi, na przykład, więcej niż około 10%, więcej niż około 20%, więcej niż około 30%, więcej niż około 40% i/lub więcej niż około 50% jako funkcja wartości dla cząsteczki odniesienia/porównawczej.
35 34- [0109] Określenie zasadniczo podobny lub zasadniczo taki sam, jak użyto w niniejszym opisie, oznacza wystarczająco wysoki stopień podobieństwa pomiędzy dwiema wartościami liczbowymi (na przykład jedną związaną z przeciwciałem według wynalazku, a drugą związaną z przeciwciałem odniesienia/porównawczym), by znawca w dziedzinie uznał różnicę pomiędzy obiema wartościami za posiadającą niewielkie bądź żadne znaczenie biologiczne i/lub statystyczne w kontekście cech biologicznych mierzonych tymi wartościami (np. wartościami Kd). Różnica pomiędzy tymi dwiema wartościami wynosi korzystnie mniej niż około 50%, korzystnie mniej niż około 40%, korzystnie mniej niż około 30%, korzystnie mniej niż około 20%, korzystnie mniej niż około 10% wartości dla przeciwciała odniesienia/porównawczego. [0110] Procentową (%) identyczność sekwencji aminokwasów w odniesieniu do sekwencji peptydu lub polipeptydu definiuje się jako odsetek reszt aminokwasowych sekwencji kandydującej identycznych z resztami aminokwasowymi określonej sekwencji peptydowej lub polipeptydowej po dopasowaniu sekwencji i wprowadzeniu odstępów (jeśli są konieczne) w celu osiągnięcia maksymalnej procentowej identyczności sekwencji, bez uznawania jakichkolwiek konserwatywnych substytucji za element identyczności sekwencji. Dopasowanie do celów ustalenia procentowej zgodności sekwencji aminokwasów można osiągnąć na różne sposoby znane w dziedzinie wynalazku, na przykład poprzez zastosowanie publicznie dostępnego oprogramowania komputerowego takiego jak BLAST, BLAST-2, ALIGN lub Megalign (DNASTAR). Znawcy w dziedzinie są w stanie wyznaczyć odpowiednie parametry pomiaru wyrównania sekwencji, w tym ewentualne algorytmy wymagane do maksymalnego dopasowania sekwencji na pełnej długości porównywanych sekwencji. Dla celów niniejszego wynalazku jednak, wartości % identyczności sekwencji aminokwasów generowane są przy użyciu programu komputerowego do porównywania sekwencji ALIGN-2, opracowanego przez Genentech, Inc. Kod źródłowy ALIGN-2 został złożony wraz z dokumentacją użytkownika w US Copyright Office, Washington DC, 20559, gdzie jest zarejestrowany pod nr. rejestracji praw autorskich TXU Program ALIGN-2 jest publicznie dostępny za pośrednictwem Genentech, Inc., South San Francisco, Kalifornia. Program ALIGN-2 powinien zostać skompilowany do pracy pod systemem operacyjnym UNIX, korzystnie pod systemem Digital UNIX V4.0D. Wszystkie parametry porównania sekwencji ustalane są przez program ALIGN-2 i są niezmienne. [0111] W przypadkach, gdy do porównywania sekwencji aminokwasowych wykorzystuje się program ALIGN-2, % identyczność sekwencji aminokwasów danej sekwencji aminokwasowej A z, względem lub w porównaniu z sekwencją aminokwasową B (co można inaczej sformułować jako posiadanie przez daną sekwencję aminokwasową A określonej % identyczności sekwencji aminokwasowej z, względem lub w porównaniu z sekwencją aminokwasową B) oblicza się w następujący sposób: 100 razy ułamek X/Y gdzie X jest liczbą reszt aminokwasowych zaliczonych jako identyczne dopasowania z programu do dopasowania sekwencji, ALIGN-2 w linii danego programu A i B, i gdzie Y jest całkowitą liczbą reszt aminokwasowych w B. Rozumie się przez to, że w przypadkach, gdy
36 35- długość sekwencji aminokwasowej A nie jest równa długości sekwencji aminokwasowej B, % identyczność sekwencji aminokwasowej A względem B nie będzie równa % identyczności sekwencji aminokwasowej B względem A. [0112] Jeżeli nie podano inaczej, wszystkie wartości % identyczności sekwencji kwasu aminokwasów stosowane w niniejszym dokumencie otrzymuje się jak opisano w poprzednim akapicie z zastosowaniem programu komputerowego ALIGN-2. [0113] Wyizolowana cząsteczka kwasu nukleinowego kodującego przeciwciała według niniejszego wynalazku jest cząsteczką kwasu nukleinowego, która jest zidentyfikowana i oddzielona od przynajmniej jednej zanieczyszczającej cząsteczki kwasu nukleinowego, z którą jest zazwyczaj związana w środowisku, w którym został on wytworzony. Korzystnie, wyizolowany kwas nukleinowy jest wolny od asocjacji z wszystkimi elementami związanymi ze środowiskiem produkcyjnym. Wyizolowane cząsteczki kwasu nukleinowego kodujące polipeptydy i przeciwciała według niniejszego wynalazku są w postaci innej niż postać lub ustawienie, w których występują w naturze. Dlatego też, wyizolowane cząsteczki kwasu nukleinowego odróżniają się od kwasów nukleinowych kodujących polipeptydy i przeciwciała według niniejszego wynalazku naturalnie występujących w komórkach. [0114] Określenie sekwencje kontrolne odnosi się do sekwencji DNA koniecznych do ekspresji funkcjonalnie związanej sekwencji kodującej w danym organizmie gospodarza. Sekwencje kontrolne, które są odpowiednie dla prokariotów, na przykład zawierają promotor, ewentualnie sekwencję operatorową i miejsce wiążące rybosomu. Komórki eukariotyczne wykorzystują promotory, sygnały poliadenylacji i wzmacniacze. [0115] Kwas nukleinowy jest funkcjonalnie połączony, gdy jest umieszczony w funkcjonalnej zależności z inną sekwencją kwasu nukleinowego. Na przykład, DNA dla presekwencji lub lidera wydzielania jest funkcjonalnie związany z DNA polipeptydu, jeśli jest wyrażany jako pre-białko, które uczestniczy w wydzielaniu polipeptydu; promotor lub wzmacniacz jest funkcjonalnie połączony z sekwencją kodującą, jeżeli wpływa na transkrypcję sekwencji; lub miejsce wiązania rybosomu jest połączone funkcjonalnie z sekwencją kodującą, jeśli jest położone w sposób ułatwiający translację. Ogólnie funkcjonalnie połączony oznacza, że połączone sekwencje DNA są ciągłe oraz, w przypadku liderowej sekwencji wydzielniczej, ciągłe i w fazie odczytu. Jednakże, wzmacniacze nie muszą być ciągłe. Połączenie uzyskuje się przez ligację w dogodnych miejscach restrykcyjnych. Jeśli takie miejsca nie istnieją, stosuje się syntetyczne adaptery oligonukleotydowe lub łączniki, zgodnie z konwencjonalną praktyką. [0116] Stosowane tu określenie tagowany epitopem odnosi się do chimerycznego polipeptydu zawierającego opisane tu polipeptyd lub przeciwciało skondensowane ze polipeptydem tagowym. Znakowany polipeptyd ma wystarczająco reszt, aby dostarczyć epitop, względem którego może być wykonane przeciwciało, a jednak jest na tyle krótki, że nie zakłóca aktywności polipeptydu, z którym jest on połączony. Polipeptyd tagowy korzystnie jest unikatowy tak, że przeciwciało zasadniczo nie reaguje krzyżowo z innymi epitopami. Odpowiednie znakowane polipeptydy na ogół mają co najmniej sześć reszt
37 36- aminokwasowych i zazwyczaj pomiędzy około 8 i 50 reszt aminokwasowych (korzystnie między około 10 i 20 reszt aminokwasowych). [0117] W stosowanym tu kontekście, określenie immunoadhezyna oznacza cząsteczki podobne do przeciwciał, które łączą specyficzność wiązania heterologicznego białka ( przyczepność ) z funkcjami efektorowymi domen stałych immunoglobuliny. Strukturalnie, immunoadhezyny zawierają fuzję sekwencji aminokwasowej z pożądaną specyficznością wiązania, która jest inna niż rozpoznawanie antygenu i miejsce wiązania przeciwciała (tj. jest heterologiczna ), oraz sekwencji domeny stałej immunoglobuliny. Adhezynowa część cząsteczki immunoadhezyny zazwyczaj jest ciągłą sekwencją aminokwasową zawierającą co najmniej miejsce wiązania receptora lub ligandu. Sekwencja domeny stałej immunoglobuliny w immunoadhezynie może być otrzymana z dowolnej immunoglobuliny takiej, jak podtypy IgG-1, IgG-2, IgG-3 lub IgG-4, IgA (w tym IgA-1 i IgA-2), IgE, IgD lub IgM. Fuzje Ig korzystnie obejmują substytucję domeny polipeptydu lub przeciwciała tu opisanego w miejscu co najmniej jednego regionu zmiennego w obrębie cząsteczki Ig. W szczególnie korzystnym przykładzie wykonania fuzja immunoglobuliny obejmuje region zawiasowy, CH2 i CH3, albo zawias, regiony CH1, CH2 i regiony CH3 cząsteczki IgG1. Dla produkcji fuzji immunoglobulin zobacz także patent nr US 5,428,130 wydany 27 czerwca, Na przykład, użyteczne immunoadhezyny jako drugie leki użyteczne tu do leczenia skojarzonego obejmują polipeptydy, które zawierają te fragmenty wiążące BLyS receptora BLyS bez transbłony Br3, TACI lub BCMA jest zfuzjowane z domeną stałą sekwencję immunoglobuliny. [0118] Białko fuzyjne i polipeptyd fuzyjny odnosi się do polipeptydu mającego dwie kowalencyjnie połączone ze sobą części, przy czym każda z tych części jest polipeptydem mającym różne właściwości. Właściwość może być właściwością biologiczną taką, jak aktywność in vitro lub in vivo. Właściwość może być prostą chemiczną lub fizyczną właściwością taką, jak wiązanie do docelowej cząsteczki, kataliza reakcji, itd. Dwie części mogą być połączone bezpośrednio przez pojedyncze wiązanie peptydowe lub poprzez łącznik peptydowy będzie w ramce odczytu ze sobą. [0119] Stabilna formulacja jest taką, w której białko w niej zawarte w zasadzie zachowuje swoją stabilność fizyczną i chemiczną oraz swoją integralność podczas przechowywania. Różne techniki analityczne do pomiaru stabilności białka są dostępne w tej dziedzinie i są przedstawione w przeglądzie w Peptide and Protein Drug Delivery, , Vincent Lee Ed., Marcel Dekker, Inc., New York, New York, Pubs. (1991) i Jones, A. Adv. Drug Delivery Rev. 10: (1993). Stabilność można mierzyć przy wybranej temperaturze w ciągu wybranego okresu czasu. Dla szybkiego skriningu, preparat można przechowywać w temperaturze 40 C przez 2 tygodnie do 1 miesiąca, kiedy to mierzy się trwałość. W przypadku, gdy formulacja ma być przechowywana w temp. 2-8 C, zwykle formulacja powinna być stabilna w 30 C lub 40 C przez co najmniej 1 miesiąc i/lub w temp. 2-8 C przez co najmniej 2 lata. W przypadku, gdy formulacja ma być przechowywana w 30 C, zazwyczaj preparat powinien być stabilny przez co najmniej 2 lata w 30 C i/lub stabilna w 40 C przez co najmniej 6 miesięcy. Na przykład, stopień agregacji w czasie przechowywania
38 37- można wykorzystać jako wskaźnik stabilności białka. Tak więc, stabilna formulacja może być taką, w której mniej niż około 10%, a korzystnie mniej niż około 5% białka występuje w postaci agregatu w preparacie. W innych przykładach wykonania można określić zwiększenie tworzenia agregatów w czasie przechowywania kompozycji. [0120] Odtworzona formulacja to taka, która została przygotowana poprzez rozpuszczenie liofilizowanej formulacji białka lub przeciwciała w rozcieńczalniku tak, że białko staje się dokładnie zdyspergowane. Rozpuszczona formulacja jest odpowiednia do podawania (np. podawania pozajelitowego) pacjentowi, który jest leczony białkiem będącym przedmiotem zainteresowania, w pewnych wykonaniach wynalazku może to być formulacja odpowiednia do podawania podskórnego. [0121] Formulacja izotoniczna to taka, która ma zasadniczo takie samo ciśnienie osmotyczne jak ludzka krew. Formulacje izotoniczne, na ogół będą miały ciśnienie osmotyczne od około 250 do 350 mosm. Określenie hipotoniczny opisuje preparat o ciśnieniu osmotycznym poniżej poziomu osmotyczności ludzkiej krwi. Odpowiednio, określenie hipertoniczny jest stosowane do opisania kompozycji o ciśnieniu osmotycznym wyższym od ludzkiej krwi. Izotoniczność można mierzyć na przykład stosując osmometr ciśnienia par lub typu zamrażania lodu. Formulacje według wynalazku są hipertoniczne w wyniku dodania soli i/lub buforu. [0122] Stosowane tu nośniki obejmują farmaceutycznie dopuszczalne nośniki, zaróbki lub stabilizatory, które są nietoksyczne dla komórki lub ssaka wystawionych na ich działanie w stosowanych dawkach i stężeniach. Często fizjologicznie dopuszczalnym nośnikiem jest wodny roztwór o buforowanym ph. Przykładami fizjologicznie dopuszczalnych nośników są bufory takie, jak fosforan, cytrynian i inne kwasy organiczne; przeciwutleniacze, w tym kwas askorbinowy; polipeptyd o niskiej masie cząsteczkowej (mniej niż około 10 reszt); białka takie, jak albumina surowicy, żelatyna lub immunoglobuliny; polimery hydrofilowe takie, jak poliwinylopirolidon; aminokwasy takie, jak glicyna, glutamina, asparagina, arginina lub lizyna; monosacharydy, disacharydy i inne węglowodany, w tym glukoza, mannoza lub dekstryny; środki chelatujące takie, jak EDTA; alkohole cukrowe takie, jak mannitol lub sorbitol; przeciwjony tworzące sole takie, jak sód; i/lub niejonowe środki powierzchniowo czynne takie, jak TWEEN, glikol polietylenowy (PEG) i PLURONICS. [0123] Ulotka informacyjna odnosi się do instrukcji zwyczajowo dołączanych do handlowych opakowań leków, które zawierają informacje o wskazaniach zazwyczaj zawartych w opakowaniach handlowych leków, które zawierają informacje o wskazaniach, stosowaniu, dawkowaniu, podawaniu, przeciwwskazaniach, innych lekach, które mają być połączone z zapakowanym produktem i/lub ostrzeżeniach dotyczących stosowania takich leków, itp. [0124] Farmaceutycznie dopuszczalny kwas obejmuje kwasy nieorganiczne i organiczne, które nie są toksyczne w stężeniu oraz sposobie ich formulacji. Na przykład, odpowiednie kwasy nieorganiczne obejmują kwas chlorowodorowy, nadchlorowy, bromowodorowy, jodowodorowy, azotowy, siarkowy, sulfonową, sulfinową, sulfanilowy, fosforowy, węglowy,
39 38- itd. Odpowiednie kwasy organiczne obejmują prostołańcuchowe i rozgałęzione alkilowe, aromatyczne, cykliczne, cykloalifatyczne, aryloalifatyczne, heterocykliczne, nasycone, nienasycone, mono, di- i tri-karboksylowe, w tym na przykład, mrówkowy, octowy, 2- hydroksyoctowy, trifluorooctowy, fenylooctowy, trimetylooctowy, t-butylooctowy, antranilowy, propionowy, 2-hydroksypropionowy, 2-oksopropionowy, propanodiowy, cyklopentanopropionowy, cyklopentanopropionowy, 3-fenylopropionowy, masłowy, butandiowy, benzoesowy, 3-(4-hydroksybenzoilo)benzoesowy, 2-acetoksy- benzoesowy, askorbinowy, cynamonowy, laurylosiarkowy, stearynowy, śluzowy, migdałowy, bursztynowy, embonowy, fumarowy, jabłkowy, maleinowy, hydroksymaleinowy, malonowy, mlekowy, cytrynowy, winowy, glikolowy, glukonowy, glukonowy, pirogronowy, glioksylowy, szczawiowy, mesylowy, bursztynowy, salicylowy, ftalowy, palmowy, palmeiowy, tiocyjanowy, metanosulfonowy, etanosulfonowy, 1,2-etanodisulfonowy, 2- hydroksyetanosulfonowy, benzenosulfonowy, 4-chorobenzenosulfonowy, naftaleno-2- sulfonowy, p-toluenosulfonowy, kamforosulfonowy, 4-metylobicyklo[2.2.2]-okt-2-eno-1- karboksylowy, glukoheptonowy, 4,4 -metylenobis-3-(kwas hydroksy-2-eno-1-karboksylowy), hydroksynaftowy. [0125] Farmaceutycznie dopuszczalne zasady obejmują zasady nieorganiczne i organiczne, które nie są toksyczne w stężeniu oraz sposobie ich formulacji. Na przykład, odpowiednie zasady obejmują te utworzone z metali tworzących nieorganiczne zasady takich, jak lit, sód, potas, magnez, wapń, amon, żelazo, cynk, miedź, mangan, glin, N-metyloglukamina, morfolina, piperydyna i zasady organiczne nietoksyczne, w tym, pierwszorzędowe, drugorzędowe i trzeciorzędowe aminy, podstawione aminy, aminy cykliczne i zasadowe żywice jonowymienne, [np. N(R ) 4 + (gdzie R oznacza niezależnie H lub C 1-4 alkil, np. jon amoniowy, Tris)], na przykład, izopropyloamina, trimetyloamina, dietyloamina, trietyloamina, tripropyloamina, etanoloamina, 2-dietyloaminoetanol, trimetamina, dicykloheksyloamina, lizyna, arginina, histydyna, kofeina, prokaina, hydrabamina, cholina, betaina, etylenodiamina, glukozamina, metyloglukamina, teobromina, puryny, piperazyna, piperydyna, N-etylopiperydyna, żywice poliaminowe i podobne. Szczególnie korzystnymi organicznymi nietoksycznymi zasadami są izopropyloamina, dietyloamina, etanoloamina, trimetamina, dicykloheksyloamina, cholina i kofeina. [0126] Dodatkowe farmaceutycznie dopuszczalne kwasy i zasady użyteczne w niniejszym wynalazku obejmują te, które pochodzą od aminokwasów, na przykład, glicyna, histydyna, fenyloalanina, kwas asparaginowy, kwas glutaminowy, lizyna i asparagina. [0127] Określenie farmaceutycznie dopuszczalne bufory i sole obejmują te pochodzące z soli addycyjnych zarówno z kwasami i zasadami wyżej wymienionych kwasów i zasad. Szczególne środki buforujące i/lub sole obejmują histydynę, bursztynian i octan. [0128] Farmaceutycznie dopuszczalny cukier to cząsteczka, która po połączeniu z białkiem będącym przedmiotem zainteresowania, w znacznym stopniu zapobiega lub redukuje chemiczną i/lub fizyczną niestabilność białka przy przechowywaniu. Gdy formulacja jest przeznaczona do liofilizacji, a następnie rekonstytuowana, farmaceutycznie dopuszczalne cukry mogą być również znane jako lioprotektanty. Przykładowe cukry oraz
40 39- odpowiadające im alkohole cukrowe obejmują: aminokwasy takie, jak glutaminian sodu czy histydyna; metyloamina taka, jak betaina; sól liotropowa taka, jak siarczan magnezu; poliol taki, jak alkohole cukrowe trójwodorotlenowe lub o wyższej masie cząsteczkowej, np. gliceryna, dekstran, erytryt, glicerol, arabitol, ksylitol, sorbitol oraz mannitol; glikol propylenowy; glikol polietylenowy; PLURONICS ; oraz ich kombinacje. Ponadto przykładowe lioprotektanty obejmują glicerynę oraz żelatynę i cukry: melibiozę, melezitozę, rafinozę i stachiozę, mannotriozę. Przykłady cukrów redukujących obejmują glukozę, maltozę, laktozę, maltulozę, izomaltulozę i laktulozę. Przykłady cukrów nieredukujących to nieredukujące glikozydy związków wielowodorotlenowych dobranych spośród alkoholi cukrów oraz innych polialkoholi o prostym łańcuchu. Korzystne alkohole cukrowe to monoglikozydy, zwłaszcza związki otrzymane na drodze redukcji dwucukrów takich, jak laktoza, maltoza, laktuloza oraz maltuloza. Boczne grupy glikozydowe mogą być grupami glikozydowymi lub galaktozydowymi. Dodatkowe przykłady alkoholi cukrowych to glucitol, maltitol, laktitol oraz izomaltuloza. Korzystne farmaceutycznie dopuszczalne cukry to nieredukujące cukry, trehaloza lub sacharoza. Farmaceutycznie dopuszczalne cukry dodaje się do formulacji w ilości zabezpieczającej (np. przed liofilizacją), co oznacza, że białko zasadniczo zachowuje swoją stabilność fizyczną i chemiczną oraz swoją integralność podczas przechowywania (np. po rozpuszczeniu i przechowywaniu). [0129] Rozcieńczalnik będący tu przedmiotem zainteresowania jest takim, który jest dopuszczalny farmaceutycznie (bezpieczny i nietoksyczny po podaniu człowiekowi) i jest przydatny do wytwarzania ciekłych formulacji takich, jak formulacja odtworzona po liofilizacji. Przykładowe rozcieńczalniki obejmują jałową wodę, wodę bakteriostatyczną do iniekcji (BWFI), roztwór o buforowanym ph (np. soli fizjologicznej buforowanej fosforanami), sterylny roztwór soli fizjologicznej, roztwór Ringera lub roztwór dekstrozy. W alternatywnym przykładzie wykonania, rozcieńczalniki mogą obejmować wodne roztwory soli i/lub bufory. [0130] Środek konserwujący to związek, który można dodawać do kompozycji w niniejszym opisie w celu zmniejszenia aktywności bakteryjnej. Dodawanie środka konserwującego może, na przykład, ułatwiać wytwarzanie formulacji wielokrotnego użytku (wielodawkowej). Przykłady potencjalnych środków konserwujących należą chlorek oktadecylodimetylobenzyloammoniowy, chlorek heksametoniowy, chlorek benzalkoniowy (mieszanina chlorków alkilobenzylodimetyloamoniowych, w których grupy alkilowe są grupami długołańcuchowymi), i chlorek benzetoniowy. Inne typy środków konserwujących obejmują alkohole aromatyczne takie, jak fenol, alkohol butylowy i benzylowy, parabeny alkilowe takie, jak paraben metylowy lub propylowy, katechol, rezorcynol, cykloheksanol, 3- pentanol i m-krezol. Najkorzystniejszym środkiem konserwującym jest alkohol benzylowy. [0131] Leczenie dotyczy interwencji klinicznej mającej na celu zmianę naturalnego przebiegu choroby u osobnika lub w komórce leczonego osobnika, i może być prowadzone albo dla profilaktyki, albo w przebiegu patologii klinicznej. Pożądane efekty leczenia obejmują zapobieganie wystąpieniu lub nawrotowi choroby, zapobieganie przerzutom, zmniejszenie szybkości postępu choroby, polepszanie lub złagodzenie stanu choroby oraz
41 40- remisję lub lepsze rokowanie. W niektórych przykładach wykonania, przeciwciała według wynalazku są stosowane w celu opóźnienia rozwoju choroby lub zaburzenia. Osobnik jest z powodzeniem leczony, na przykład, przy użyciu apoptotycznych przeciwciał anty-ige według wynalazku, jeśli ograniczeniu ulega jeden lub większa liczba objawów związanych z zaburzeniem pośredniczy IgE. [0132] Skuteczna ilość dotyczy co najmniej ilości skutecznej, przy dawkowaniu i przez okresy czasu, niezbędnej do osiągnięcia pożądanego lub wskazanego efektu, w tym terapeutycznego lub profilaktycznego. [0133] Terapeutycznie skuteczna ilość oznacza co najmniej minimalne stężenie wymagane do spowodowania mierzalnej poprawy lub zapobiegania danego zaburzenia. Terapeutycznie skuteczna ilość może zmieniać się w niniejszym opisie w odniesieniu do czynników takich, jak stan choroby, wiek, płeć i waga pacjenta oraz zdolność przeciwciała do wywołania pożądanej odpowiedzi u danego osobnika. Terapeutycznie skuteczna ilość to również taka, w której jakiekolwiek toksyczne lub szkodliwe skutki tego przeciwciała są zrównoważone poprzez uzyskiwane terapeutycznie korzystne efekty. Profilaktycznie skuteczna ilość odnosi się do ilości skutecznej, przy dawkach, i przez okresy czasu, niezbędnej do osiągnięcia pożądanego skutku profilaktycznego. Zazwyczaj, choć niekoniecznie, ponieważ dawka profilaktyczna jest stosowana u osobnika przed albo na wcześniejszym etapie choroby, skuteczna profilaktycznie ilość może być mniejsza niż ilość skuteczna terapeutycznie. [0134] Przewlekłe podawanie odnosi się do podawania leku (leków) w sposób ciągły, w przeciwieństwie do trybu ostrego, aby utrzymać początkowy efekt terapeutyczny (aktywność) przez przedłużony okres czasu. Przerywane podawanie jest leczeniem, które nie jest wykonywane konsekutywnie w sposób ciągły, ale raczej jest z natury cykliczne. [0135] Ssak dla celów traktowania odnosi się do jakiegokolwiek zwierzęcia sklasyfikowanego jako ssak, włącznie z człowiekiem, zwierzętami domowymi i hodowlanymi oraz zwierzętami z zoo, sportowymi lub zwierzętami domowymi takimi, jak psy, konie, króliki, bydło, świnie, chomika, myszy, myszoskoczki, fretki, szczury, koty, itd. Korzystnie, ssakiem jest człowiek. [0136] Określenie formulacja farmaceutyczna odnosi się do preparatu, który znajduje się w takiej postaci, aby umożliwić skuteczność działania biologicznego składnika czynnego i nie zawiera żadnych dodatkowych części, które są zbyt toksyczne dla osobnika, któremu formulacja będzie podawana. Takie formulacje są jałowe. [0137] Formulacja jałowa jest aseptyczna lub wolna od wszelkich żywych mikroorganizmów i ich zarodników. [0138] Określenie około w znaczeniu stosowanym w niniejszym dokumencie odnosi się zwykle do zakresu błędu dla danej wartości dobrze znanej znawcy w tej dziedzinie techniki. [0139] Stosowany tu lek przeciwhistaminowy jest środkiem agonizującym działanie fizjologiczne histaminy. Wiązanie histaminy z receptorami, H 1 i H 2 daje charakterystyczne objawy alergiczne i efekty lub swędzenie, zaczerwienienie, obrzęk itd. Wiele leków
42 41- przeciwhistaminowych działają przez blokowanie wiązania histaminy z receptorami H1, H2; Jednakże inni uważają się, że działają one przez hamowanie uwalniania histaminy. Przykładami leków przeciwhistaminowych są chlorfeniramina, difenhydramina, prometazyna, kromoglikan sodu, astemizol, maleinian azatadyny, maleinian brofeniraminy, maleinian karbinoksaminy, chlorowodorek cetyryzyny, fumaran klemastyny, chlorowodorek cyproheptadyny, maleinian deksbromfeniraminy, maleinian dekschlorfeniraminy, dimenhydrynian, chlorowodorek difenhydraminy, bursztynian doksylaminy, chlorowodorek feksofendadyny, chlorowodorek terfenadyny, chlorowodorek hydroksyzyny, loratydyna, chlorowodorek meklizyny, cytrynian tripelannaminy, chlorowodorek tripelenaminy, chlorowodorek triprolidyny. [0140] Stosowany tu lek rozszerzający oskrzela, opisuje środki, które antagonizują lub odwracają skurcz oskrzeli, fizjologiczne zdarzenie, które występuje zazwyczaj na początku fazy reakcji astmatycznych powodując zmniejszoną pojemność płuc i duszności. Przykłady leków rozszerzających oskrzela obejmują epinefrynę, szeroko działające leki alfa i betaadrenergiczne i leki beta-adrenergiczne, albuterol, pirbuterol, metaproterenol, salmeterol i izoetarynę. Rozszerzanie oskrzeli można osiągnąć przez podawanie ksantyn takich, jak teofilina i aminofilina. [0141] Glikokortykoid w niniejszym opisie określa środki oparte na steroidach o działaniu przeciwzapalnym. Glikokortykoidy są powszechnie stosowane w celu osłabienia późnej fazy reakcji astmatycznych. Przykład glikokortykoidów obejmują prednizon, dipropionian beklometazonu, acetonid triamcynolonu, flunizolid, betametazon, budesonid, deksametazon, desameazon tramcinolon, octan fludrokortyzonu, flunizolid, propionian flutikazonu, hydrokortyzon, prednizolon, [w tym metyloprednizolon (np. SOLU-MEDROL sól sodowa bursztynianu metyloprednizolonu)] i triamcynolon. [0142] Stosowany tu niesteroidowy lek przeciwzapalny lub NSAID, opisuje środki mające aktywność przeciwzapalną, które nie są oparte na steroidach. Przykład NSAID obejmują acematacynę, paracetamol, aspirynę, azapropazon, benorylat, bromfenak sodu, inhibitory cyklooksygenazy (COX)-2 takie, jak GR , MK966, celekoksyb (CELEBREX ; 4-(5- (4- metylofenylo)-3-(trifluorometylo)-1h-pirazol-1-ilo) benzenosulfonamid) i waldekoksyb (BEXTRA ), diklofenak, diklofenak retard, diklofenak sodu, diflunizal, etodolak, fenbufen, fenoprofen wapnia, flurbiprofen, ibuprofen, ibuprofen retard, indometacynę, ketoprofen, meklofenamat sodu, kwas mefenamowy, meloksykam (MOBIC ), nabumeton, naproksen, naproksen sodu, oksyfenobutazon, fenylobutazon, piroksykam, sulindak, tenoksykam, kwas tiaprofenowy, tolmetynę, tolmetynę sodu, w tym ich sole i ich pochodne, itd. [0143] Określenie zaburzenia mediowane IgE obejmuje zaburzenia atopowe, które charakteryzują się ogólną dziedziczną skłonnością do odpowiedzi immunologicznej na wiele typowych naturalnie występujących wziewnych i wchłanianych antygenów oraz ciągłą produkcją przeciwciał IgE. Specyficzne zaburzenia atopowe obejmują astmę alergiczną, alergiczny nieżyt nosa (zapalenie spojówek), atopowe zapalenie skóry, alergię pokarmową, anafilaksję, kontaktowe zapalenie skóry, alergiczną gastroenteropatię, alergiczną aspergilozę oskrzelowopłucną i plamicę alergiczną (Henoch-Schönlein). Pacjenci atopowi często mają
43 42- wiele alergii, co oznacza, że mają przeciwciała IgE przeciwko, i objawy pochodzące z, wielu alergenów środowiskowych, w tym alergenów sezonowych, stałych i zawodowych. Przykłady sezonowych alergenów obejmują pyłki (np. trawa, drzewo, żyto, tymotka, ambrozja), natomiast przykłady stałych alergenów obejmują grzyby (np. pleśnie, zarodniki pleśni), pióra, zwierzęce (np. sierść zwierząt domowych lub innych zwierząt) i pozostałości owadów (np. roztocza kurzu). Przykładowe alergeny zawodowe obejmują także antygeny zwierząt (np. myszy) i roślin oraz leki, detergenty, metale i immunoenhancery, np. izocyjaniany. Bodźce niespecyficzne dla antygenu, które dają reakcję mediowaną IgE obejmują infekcję, czynniki drażniące takie, jak dym, opary cząstki spalin z silników Diesla i dwutlenek siarki, ćwiczenia, zimno i stres. Specyficzne reakcje nadwrażliwości u osobników atopowych i nieatopowych o określonym tle genetycznym może być wynikiem narażenia na białka w pokarmach (np. rośliny strączkowe, orzechy ziemne), jad (np. owady, węże), szczepionki, hormony, enzymy, surowice odpornościowe, lateks, antybiotyki, leki zwiotczające mięśnie, witaminy, cytotoksyny, opioidy oraz polisacharydy takie, jak dekstryny, dekstran żelaza i polygeline. [0144] Inne choroby związane z podwyższonymi poziomami IgE, które wydają się być mediowane IgE i są uleczalne za pomocą formulacji według niniejszego wynalazku obejmują: ataksja-teleangiektazja, zespół Churg-Strauss, wyprysk, zapalenie jelit, gastroenteropatię, reakcję przeszczep przeciwko gospodarzowi, zespół hiper-ige (Joba), nadwrażliwość (np. nadwrażliwość anafilaktyczna, drożdżyca, zapalenie naczyń), szpiczaka IgE, chorobę zapalną jelit (np. choroba Crohna, wrzodziejące zapalenie okrężnicy, nieokreślone zapalenie okrężnicy i zapalenie zakaźne), zapalenie błony śluzowej (np. zapalenie błony śluzowej jamy ustnej, zapalenie błony śluzowej przewodu pokarmowego, zapalenie błony śluzowej nosa i zapalenie odbytu), martwicze zapalenie jelit i przełyku, choroby pasożytnicze (np. trypanosomoza), zapalenie naczyń z nadwrażliwości, pokrzywka i zespół Wiskotta-Aldricha. [0145] Ponadto, zaburzenia, które można leczyć za pomocą obniżenia stężenia IgE, niezależnie od tego, czy same są zaburzeniami związanymi z podwyższonym IgE, a tym samym należy uznać za objęte zakresem zaburzenia mediowane IgE obejmują: chorobę Addisona (przewlekła niewydolność kory nadnerczy), łysienie, dziedziczny obrzęk naczynioruchowy, anigioedemę (chorobę Bannistera, obrzęk naczynioruchowy), zesztywniające zapalenie stawów kręgosłupa, niedokrwistość aplastyczną, zapalenie tętnic, amyloidozę, zaburzenia układu odpornościowego takie, jak autoimmunologiczna anemia hemolityczna, autoimmunologiczne zapalenie jajników, autoimmunologiczne zapalenie jąder, autoimmunologiczna niewydolność poliendokrynna, autoimmunologiczna niedokrwistość hemolityczna, autoimmunocytopenia, autoimmunologiczne zapalenie kłębuszków nerkowych, choroba Behceta, zapalenie oskrzeli, chorobę Buergera, pemfigoid pęcherzowy, zespół Caplana (reumatoidalne zapalenie stawów z pylicą płuc), zapalenie serca, celiakię, zespół Chediak-Higashi, przewlekłą obturacyjną chorobę płuc (COPD), zespół Cogan-Reese (zespół śródbłonkowy tęczówkowo-rogówkowy), zespół CREST, opryszczkowe zapalenie skóry (Choroba Duhringa), cukrzycę, eozynofilowe zapalenie powięzi, eozynofilowe zapalenie nerek, zapalenie nadtwardówki, zewnątrzpochodne alergiczne zapalenie pęcherzyków płucnych, rodzinne napadowe zapalenie błon surowiczych, zespół Felty ego, włókniejące zapalenie pęcherzyków płucnych, kłębuszkowe zapalenie nerek, zespół Goodpasture a,
44 43- granulocytopenię, ziarniniak, ziarniniakowatość, ziarniniakowe zapalenie mięśni, chorobę Gravesa, zespół Guillaina-Barrego (zapalenie wielonerwowe), zapalenie tarczycy Hashimoto (wole limfocytarne), hemochromatoza, histocytozę, zespół hipereozynofilowy, zespół jelita drażliwego, młodzieńcze zapalenie stawów, zapalenie rogówki, trąd, toczeń rumieniowaty, chorobę Lyella, chorobę z Lyme, mieszaną chorobę tkanki łącznej, zapalenie pojedynczych nerwów, mnogie zapalenie pojedynczych nerwów, zespół Muckle-Wellsa, skórnośluzówkowy zespół węzłów chłonnych (choroba Kawasaki), retikulohistiocytozę wieloogniskową, stwardnienie rozsiane, miastenia gravis, ziarniniaka grzybiastego, zapalenie tkanki tłuszczowej, pemfigoid, pęcherzycę zwykłą, zapalenie osierdzia, zapalenie wielonerwowe, guzkowe zapalenie tętnic, łuszczycę, łuszczycowe zapalenie stawów, płucne zmiany w zapaleniu stawów, gruczolakowatości płuc, zwłóknienie płuc, nawracające zapalenie wielochrząstkowe, gorączkę reumatyczną, reumatoidalne zapalenie stawów, zapalenie zatok przynosowych (zapalenie zatok), sarkoidozę, zapalenie twardówki, stwardniające zapalenie dróg żółciowych, chorobę posurowiczą, zespół Sezary ego, zespół Sjögrena, zespół Stevensa-Johnsona, mastocytozę układową, odrzucenie przeszczepu, plamicę małopłytkową, hipoplazję grasicy, zapalenie błony naczyniowej oka, bielactwo, ziarniniakowatość Wegenera. [0146] Choroba autoimmunologiczna jest w niniejszym opisie chorobą lub zaburzeniem pochodzącym lub skierowanym przeciwko własnym tkankom lub organom osobnika lub jej ko-segregacją albo manifestacją bądź stanem z niej wynikającym. W wielu z tych chorób autoimmunologicznych i zapalnych mogą istnieć liczne kliniczne i laboratoryjne wskaźniki, w tym, bez ograniczania do wymienionych, hipergammaglobulinemia, wysokie poziomy autoprzeciwciał, depozyty kompleks przeciwciało-antygen w tkankach, korzyści z leczenia kortykosteroidami lub lekami immunosupresyjnymi i limfatyczne agregaty komórek w tkankach dotkniętych. Nie ograniczając się do żadnej teorii dotyczącej zaburzenia autoimmunologicznego mediowanego komórkami B, uważa się, że komórki B wykazują działanie patogenne w ludzkich chorobach autoimmunologicznych poprzez wiele ścieżek mechanistycznych, w tym produkcję autoprzeciwciał, tworzenie kompleksu immunologicznego, aktywację komórek dendrytycznych i T, syntezę cytokin, bezpośrednie uwalnianie chemokin, i zapewniając ognisko dla ektopowej neo-limgogenezy. Każdy z tych szlaków może w różnym stopniu brać udział w patologii chorób autoimmunologicznych. [0147] Choroba autoimmunologiczna może być chorobą specyficzną dla narządu (tj. odpowiedź immunologiczna jest skierowana konkretnie przeciwko układowi narządu takiemu, jak układ hormonalny, układ krwiotwórczy, skóra, układ krążenia, przewód pokarmowy oraz układy wątroby, układ nerek, układ tarczycy, uszy, układ nerwowomięśniowy, ośrodkowy układ nerwowy, itd.) lub chorobą układową, które może dotykać wiele układów narządów (na przykład toczeń rumieniowaty układowy (SLE), reumatoidalne zapalenie stawów (RA), zapalenie wielomięśniowe, itd.). Korzystnie takie choroby obejmują autoimmunologiczne choroby reumatologiczne (takie, jak na przykład RA, zespół Sjögrena, twardzina skóry, toczeń taki, jak SLE i toczniowe zapalenie nerek, zapalenie skórnomięśniowe, krioglobulinemia, zespół przeciwciał skierowanych przeciwko fosfolipidom i łuszczycowe zapalenie stawów), autoimmunologiczne zapalenie wątroby i zaburzenia
45 44- żołądkowo-jelitowe (jak, na przykład, choroby zapalne jelit (np. wrzodziejące zapalenie okrężnicy i choroba Crohna), autoimmunologiczne zapalenie żołądka i niedokrwistość złośliwą, autoimmunologiczne zapalenie wątroby, pierwotną żółciową marskość wątroby, pierwotne stwardniające zapalenie dróg żółciowych i choroba trzewna), zapalenie naczyń (takie jak, na przykład, ANCA-ujemne zapalenie naczyń i zapalenie naczyń związane z ANCA, tym zapalenie naczyń Churga-Straussa, ziarniniak Wegenera, i mikroskopowe zapalenie naczyń), autoimmunologiczne choroby neurologiczne (jak, na przykład, stwardnienie rozsiane, zespół mioklonii i opsoklonii, miastenia gravis, zapalenie rdzenia i nerwów wzrokowych, choroba Parkinsona, choroba Alzheimera i autoimmunologiczne polineuropatie), zaburzenia nerek (jak, na przykład, zapalenie kłębuszków nerkowych, zespół Goodpasture a i choroba Bergera), autoimmunologiczne zaburzenia dermatologiczne (jak, na przykład, łuszczyca, pokrzywka, pokrzywka, pęcherzyca zwykła, pemfigoid pęcherzowy i skórny toczeń rumieniowaty), zaburzenia hematologiczne (jak, na przykład, małopłytkowość samoistna, zakrzepowa plamica małopłytkowa, plamica poprzetoczeniowa, oraz autoimmunologiczna niedokrwistość hemolityczna), miażdżyca tętnic, zapalenie błony naczyniowej oka, autoimmunologiczne choroby słuchu (jak, na przykład, choroba ucha wewnętrznego i utrata słuchu), choroba Behceta, zespół Raynauda, przeszczep narządu, i autoimmunologiczne zaburzenia endokrynologiczne (jak, na przykład, choroby autoimmunologiczne związane z cukrzycą takie, jak cukrzyca insulinozależna (IDDM), choroba Addisona i autoimmunologiczna choroba tarczycy, (np. choroba Gravesa i zapalenie tarczycy)). Korzystne takie choroby obejmują, na przykład RA, wrzodziejące zapalenie okrężnicy, zapalenie naczyń związane z ANCA, toczeń, stwardnienie rozsiane, zespół Sjögrena, chorobę Gravesa, IDDM, niedokrwistość złośliwą, zapalenie tarczycy i kłębuszkowe zapalenie nerek. [0148] Stosowane tu określenie środek cytotoksyczny odnosi się do substancji, która hamuje lub zapobiega funkcji komórek i/lub powoduje zniszczenie komórek. Określenie to obejmuje izotopy radioaktywne (np. At 211, I 131, I 125, Y 90, Re 186, Re 188, Sm 153, Bi 212, P 32 oraz izotopy radioaktywne Lu), toksyny takie, jak toksyny o małej cząsteczce lub aktywne enzymatycznie toksyny pochodzenia bakteryjnego, grzybowego, roślinnego lub zwierzęcego, bądź ich fragmenty. [0149] Środek chemioterapeutyczny jest związkiem chemicznym przydatnym w leczeniu nowotworu. Przykłady środków chemioterapeutycznych obejmują czynniki alkilujące takie, jak tiotepa i cyklofosfamid (CYTOXAN ); sulfoniany alkilu takie, jak busulfan, improsulfan i piposulfan; azyrydyny takie, jak benzodopa, karbokwon, meturedopa i uredopa; etylenoiminy i metylamelaminy, w tym altretamina, trietylenomelamina, trietylenofosforamid, trietylenotiofosforamid i trimetylolomelamina; acetogeniny (zwłaszcza bullatacyna i bullatacynon); delta-9- tetrahydrokannabinol (dronabinol, MARINOL ); betalapachon; lapachol; kolchicyny; kwas betulinowy; kamptotecynę (w tym syntetyczny analog topotekanu (HYCAMTIN ), CPT-11 (irinotekan, CAMPTOSAR ), acetylokamptotecynę, skopolektynę i 9-aminokamptotecyna); briostatynę; pemetreksed; kallistatynę; CC-1065 (w tym ich syntetyczne analogi adozelesyna, karzelesyna i bizelesyna); podofilotoksynę; kwas podofilinowy; tenipozyd; kryptoficyny (w szczególności kryptoficyna 1 i kryptoficyna 8);
46 45- dolastatynę; duokarmycynę (w tym syntetyczne analogi, KW-2189 i CB1-TM1); eleuterobinę; pankratystatynę; TLK-286; CDP323, doustny inhibitor alfa-4 integryny; sarkodiktyinę; spongistatynę; iperyty azotowe takie, jak chlorambucyl, chlomafazyna, cholofosfamid, estramustyna, ifosfamid, mechloretamina, chlorowodorek tlenku mechloretaminy, melfalan, novembichin, fenesteryna, prednimustyna, trofosfamid, iperyt uracylowy; nitrozomoczniki takie, jak karmustyna, chlorozotocyna, fotemustyna, lomustyna, nimustyna i ranimnustyna; antybiotyki takie, jak antybiotyki enediynowe (np. kalicheamycyna, zwłaszcza kalicheamycyna gamma II i kalicheamicyna omegai1 (patrz np. Nicolaou i wsp., Angew. Chem Intl. Ed. Engl., 33: (1994)); dynemicynę, w tym dynemicyna A; esperamicynę; jak również chromofor neokarzynostatynowy i pokrewne chromofory enediynowych antybiotyków chromobiałkowych), aklacynomyzyny, aktynomycyna, autramycyna, azaseryna, bleomycyny, kaktynomycyna, karabicyna, karminomycyna, karzinofilina, chromomycyny, daktynomycyna, daunorubicyna, detorubicyna, 6-diazo-5-okso-L-norleucyna, doksorubicyna (w tym ADRIAMYCIN, morfolino-doksorubicyna, cyjanomorfolino-doksorubicyna, 2-pirolino-doksorubicyna, doksorubicyna HCI liposomalna do wstrzykiwań (DOXIL ) i deoksydoksorubicyna), ubicyna, ezorubicyna, idarubicyna, marcellomycyna, mitomycyny takie, jak mitomycyna C, kwas mykofenolowy, nogalamycyna, oliwomycyny, peplomycyna, potfiromycyna, puromycyna, kwelamycyna, rodorubicyna, streptonigryna, streptozocyna, tubercydyna, ubenimeks, zinostatyna, zorubicyna; anty-metabolity takie, jak metotreksat, gemcytabina (GEMZAR ), tegafur (UFTORAL ), kapecytabina (XELODA ), epotilon i 5-fluorouracyl (5-FU); analogi kwasu foliowego takie, jak denopteryna, metotreksat, pteropteryna, trimetreksat; analogi purynowe takie, jak fludarabina, 6-merkaptopuryna, tiamipryna, tioguanina; analogi pirymidyny takie, jak ancytabina, azacytydyna, 6-azaurydyna, karmofur, cytarabina, dideoksyurydyna, doksyflurydyna, enocytabina, floksurydyna i imatinib (pochodna 2-fenyloaminopirymidyna), jak również inne inhibitory c-kit; leki działające na korę nadnerczy takie, jak aminoglutetymid, mitotan, trilostan; środki uzupełniające działanie kwasu foliowego taki, jak kwas frolinowy; aceglaton; glikozyd aldofosfamidu; kwas aminolewulinowy; eniluracyl; amsakrynę; bestrabucyl; bisantren; edatraksat; defofaminę; demekolcynę; diazykwon; elfornitynę; octan eliptynium; etoglucyd; azotan galu; hydroksymocznik; lentinan; lonidaininę; majtanzynoidy takie, jak majtanzyna i ansamitocyny; mitoguazon; mitoksantron; mopidanmol; nitraerynę; pentostatynę; fenamet; pirarubicynę; losoksantron; 2-etylohydrazyd; prokarbazynę; kompleks polisacharydowy PSK (JHS Natural Products, Eugene, OR); razoksan; rizoksynę; sizofiran; spirogermanium; kwas tenuazonowy; triazykwon; 2,2,2 -trichlorotrietyloamina; trichoteceny (zwłaszcza toksyna T-2, werakuryna A, rorydyna A i anguidyna); uretan; windezynę (ELDISINE, FILDESIN ); dakarbazynę; mannomustyna; mitobronitol; mitolaktol; pipobroman; gacytozynę; arabinozyd ( Ara-C ); tiotepę; taksoidy, np. paklitaksel (TAXOL ), paklitaksel w postaci nanocząsteczkowej formulacji z albuminą (ABRAXANE ), i doksetaksel (TAXOTERE ); chloranbucil; 6-tioguaninę; merkaptopurynę; metotreksat; analogi platyny takie, jak cisplatyna i karboplatyna; winblastyna (VELBAN ); platyna; etopozyd (VP-16); ifosfamid; mitoksantron; winkrystynę (ONCOVIN ); oksaliplatyna; leukowowinę;
47 46- winorelbinę (NAVELBINE ); nowantron; edatreksat; daunomycynę; aminopterynę; ibandronian; inhibitor topoizomerazy RFS 2000; difluorometyloornitynę (DMFO); retinoidy takie, jak kwas retinowy; farmaceutycznie dopuszczalne sole, kwasy lub pochodne dowolnego z powyższych; jak również kombinacje dwóch lub więcej z wymienionych wyżej CHOP, skrót skojarzonego leczenia cyklofosfamidem, doksorubicyną, winkrystyną i prednizolonem, lub FOLFOX, skrót dla schematu leczenia oksaliplatyną (ELOXATIN ) w połączeniu z 5-FU i leukowowiną. [0150] Również zawarte w tej definicji są środki anty-hormonalne, których działanie polega na regulacji, zmniejszaniu, blokowaniu lub hamowaniu działania hormonów promujących wzrost raka i są często podawane w postaci leczenia układowego lub całego ciała. Same mogą one być hormonami. Przykłady obejmują antyestrogeny oraz selektywne modulatory receptorów estrogenowych (SERM), w tym na przykład tamoksyfen (w tym tamoksyfen NOLVADEX ), raloksyfen (EVISTA ), droloksifen, 4- hydroksytamoksyfen, trioksyfen, keoksyfen, LY117018, onapriston i toremifen (FARESTON ); antyprogesterony; regulatory w dół receptorów estrogenowych (ERD); Antagoniści receptora estrogenowego np. fulwestrant (FASLODEX ); środki o działaniu hamującym lub zamykającym jajniki, na przykład agoniści hormonu uwalniającego hormon (LHRH) tacy, jak octan leuprolidu (LUPRON i ELIGARD ), octan gosereliny, octan busereliny i tripterelina; antyandrogeny takie, jak flutamid, nilutamid oraz bikalutamid; oraz inhibitory aromatazy, które hamują enzym aromatazę, który reguluje wytwarzanie estrogenu w gruczołach nadnercza, jak, na przykład, 4(5)-imidazol, aminoglutetymid, octan megestrolu (MEGASE ), eksemestan (AROMASIN ), formestan, fadrozol, worozol (RIVISOR ), letrozol (FEMARA ) i anastrozol (ARIMIDEX ). Ponadto taka definicja chemioterapeutyków obejmuje bisfosfoniany takie, jak klodronian (na przykład BONEFOS lub OSTAC ), etydronian (DIDROCAL ), NE-58095, kwas zoledronowy/zoledronian (ZOMETA ), alendronian (FOSAMAX ), pamidronian (AREDIA ), tiludronian (SKELID ), lub rizedronian (ACTONEL ); jak również troksacytabina (1,3-dioksolanowy nukleozydowy analog cytozyny); oligonukleotydy antysensowne, zwłaszcza te, które hamują ekspresję genów w ścieżkach sygnalizacyjnych zaangażowanych w nieprawidłową proliferację komórek, jak, na przykład, PKC-alfa, Raf, H-Ras, i naskórkowy receptor czynnika wzrostu (EGF-R); szczepionki takie, jak szczepionka THERATOPE oraz szczepionki terapii genowej, na przykład, szczepionka ALLOVECTIN, szczepionka LEUVECTIN, szczepionka VAXID ; inhibitor topoizomerazy 1 (np. LURTOTECAN ); anty-estrogen taki, jak fulwestrant; inhibitor Kit taki, jak imatinib lub EXEL-0862 (inhibitor kinazy tyrozynowej); Inhibitor EGFR taki, jak erlotynib lub cetuksymab; inhibitor anty-vegf taki, jak bewacizumab; arinotekan; rmrh (np. ABARELIX ); lapatynib i ditosylan lapatynibu (małocząsteczkowy podwójny inhibitor kinazy tyrozynowej ErbB-2 i EGFR również znany jako GW572016); 17AAG (pochodna geldanamycyny, która jest trucizną 90 białka szoku cieplnego (HSP)), oraz ich farmaceutycznie dopuszczalne sole, kwasy lub pochodne dowolnego z powyższych. [0151] Środek hamujący wzrost odnosi się do związku lub kompozycji, która hamuje wzrost komórki, który to wzrost zależy od aktywacji receptora albo in vitro, albo in vivo. Tak
48 47- więc, środek hamujący wzrost, obejmuje taki, który znacząco zmniejsza procent komórek zależnych od receptora w fazie S. Przykłady środków hamujących wzrost obejmują środki, które blokują progresję cyklu komórkowego (w miejscu innym niż faza S) takie, jak środki, które indukują zatrzymanie G1 i zatrzymanie fazy M. Klasyczne blokery fazy M obejmują barwinka i alkaloidy barwinka (winkrystyna i winblastyna), taksany i inhibitory topoizomerazy II takie, jak doksorubicyna, epirubicyna, daunorubicyna, etopozyd i bleomycyna. Te środki, które zatrzymują G1, także rozlewają się na zahamowanie fazy S, na przykład, środki alkilujące DNA takie, jak tamoksyfen, prednizon, dakarbazyna, mechloretamina, cisplatyna, metotreksat, 5-fluorouracyl i ara-c. Więcej informacji można znaleźć w The Molecule Basis of Cancer, Mendelsohn and Israel, eds., Rozdział 1, pod tytułem Cell cycle regulation, oncogenes, and antineoplastic drugs Murakami i wsp. (WB Saunders: Philadelphia, 1995), zwłaszcza str. 13. Taksany (paklitaksel i docetaksel) są lekami przeciwnowotworowymi pochodzącymi z drzewa cisowego. Docetaksel (TAXOTERE, Rhone-Poulenc Rorer), pochodzący z cisu europejskiego, jest półsyntetycznym analogiem paklitakselu (TAXOL, Bristol-Myers Squibb). [0152] Określenie cytokina jest ogólnym terminem dla białek uwalnianych przez jedną populację komórek, które działają na inną komórkę jako mediatory międzykomórkowe. Przykładami takich cytokin są limfokiny, monokiny; interleukiny (IL) takie, jak IL-1, IL-1α, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-11, IL-12, IL-13, IL-15, w tym PROLEUKIN ril-2; czynnik martwicy guza taki, jak TNF-α lub TNF-β; inne czynniki polipeptydowe w tym LIF i ligand kit (KL). W stosowanym tu kontekście, określenie cytokina obejmuje białka ze źródeł naturalnych lub z hodowli komórek rekombinowanych oraz biologicznie czynne równoważniki natywnych sekwencji cytokin, w tym syntetycznie wytworzone małocząsteczkowe indywidua i ich farmaceutycznie dopuszczalne pochodne i sole. [0153] Określenie hormon oznacza hormony polipeptydowe, które są na ogół wydzielane przez organy gruczołowe z kanałami. Wśród hormonów są, na przykład, hormon wzrostu taki, jak ludzki hormon wzrostu, N-metionylo ludzki hormon wzrostu, bydlęcy hormon wzrostu; parathormon; tyroksyna; insulina; proinsulina; relaksyna; estradiol; hormonalna terapia zastępczej; androgeny takie, jak kalusteron, propionian dromostanolonu, epitiostanol, mepitiostan, testolakton; lub prorelaksyna; hormony glikoproteinowe takie, jak hormon folikulotropowy (FSH), hormon tyreotropowy (TSH) oraz hormon luteinizujący (LH); prolaktyna, laktogen łożyskowy, mysi peptyd związany z gonadotropiną, hormon uwalniający gonadotropinę; inhibina; aktywina; hormon antymullerianowski; i trombopoetyna. W stosowanym tu kontekście, określenie hormon obejmuje białka ze źródeł naturalnych lub z hodowli komórek zrekombinowanych i biologicznie aktywne ekwiwalenty natywnej sekwencji hormonu, w tym syntetycznie wytworzone małocząsteczkowe indywidua i ich farmaceutycznie dopuszczalne pochodne i sole. [0154] Określenie czynnik wzrostu odnosi się do białek, które wspomagają wzrost, i obejmują, na przykład, wątrobowy czynnik wzrostu; czynnik wzrostu fibroblastów; czynnik wzrostu śródbłonka naczyniowego; czynniki wzrostu nerwów taki, jak NGF-β; płytkowy
49 48- czynnik wzrostu; transformujące czynniki wzrostu (TGF) takie, jak TGF-α i TGF-β; czynnik wzrostu typu insuliny I i II; erytropoetyna (EPO); czynniki indukcyjne kości; interferony takie, jak interferon -α, -β i -γ; i czynniki stymulujące kolonie (CSF) takie, jak makrofagowy- CSF (M-CSF); granulocyt-makrofag-csf (GM-CSF); i granulocyt-csf (G-CSF). W stosowanym tu kontekście, określenie czynnik wzrostu obejmuje białka ze źródeł naturalnych lub z hodowli komórek zrekombinowanych i biologicznie aktywnych ekwiwalentów natywnej sekwencji czynnika wzrostu, w tym syntetycznie wytworzone małocząsteczkowe indywidua oraz ich farmaceutycznie dopuszczalne pochodne i ich sole. [0155] Określenie integryna odnosi się do białka receptora, które umożliwia komórce zarówno wiązanie się z i reagowanie na macierz zewnątrzkomórkową i bierze udział w wielu funkcjach komórkowych takich, jak gojenie się ran, różnicowanie komórek, osadzanie się komórek nowotworowych i apoptoza. Stanowią one część dużej rodziny receptorów adhezji komórek, które są zaangażowane w oddziaływanie komórka-macierz zewnątrzkomórkową i komórka-komórka. Integryny funkcyjne składają się z dwóch transbłonowych podjednostek glikoproteinowych, zwanych alfa i beta, które nie są związane kowalencyjnie. Wszystkie podjednostki alfa mają pewną wspólną wzajemną homologię, jak i podjednostki beta. Receptory zawsze zawiera jeden łańcuch alfa i jeden łańcuch beta. Przykłady obejmują Alfa6beta1, Alfa3beta1, Alfa7beta1, LFA-1 itd. W stosowanym tu kontekście, określenie integryna obejmuje białka ze źródeł naturalnych lub z hodowli komórek zrekombinowanych i biologicznie aktywnych ekwiwalentów natywnej sekwencji integryny, w tym syntetycznie wytworzone małocząsteczkowe indywidua i ich farmaceutycznie dopuszczalne pochodne i ich sole. [0156] Antagonistę TNF definiuje się tutaj jako cząsteczkę, która zmniejsza, blokuje, hamuje, znosi lub zakłóca aktywność TNFα in vitro, in situ, i/lub korzystnie in vivo. Odpowiedni antagonista TNF może również zmniejszać blokować, znosić, zakłócać, zapobiegać i/lub hamować syntezę RNA, DNA lub białka TNF, uwalnianie TNFα, sygnalizację receptora TNFα, rozszczepienie błony TNFα, aktywność TNFα, produkcję i/lub syntezę TNFα. Tacy antagoniści TNF obejmują, bez ograniczania do wymienionych, przeciwciała anty-tnfα, jego fragmenty wiążące antygen, określone ich mutanty lub domeny, które wiążą się specyficznie do TNFα, które, po związaniu TNFα, zniszczą lub zubażają komórki wyrażające TNFα u ssaka i/lub zaburza jedną lub więcej funkcji tych komórek, rozpuszczalny receptor TNF (np. p55, p70 lub p85) lub jego fragment, jego peptyd fuzyjny, małocząsteczkowego antagonistę TNF, np. białko wiążące TNF I lub II (TBP-I lub TBP-II), nerelimonmab, CDP-571, infliksymab, enteracept (ENBREL ), adalimulab (HUMIRA ), CDP-571, CDP-870, afelimomab, lenercept i podobne), jego fragmenty wiążące antygen, oraz cząsteczki receptora, które specyficznie wiążą TNFα; związki, które zapobiegają i/lub hamują syntezę TNFα, uwalnianie TNFα lub jego działanie na komórki docelowe, takie, jak talidomid, tenidap, inhibitory fosfodiesterazy (np. pentoksyfilina i rolipram), agoniści receptora adenozynowego A2b i wzmacniacze receptora adenozynowego A2b; związki, które zapobiegają i/lub hamują przekazywanie sygnałów przez receptor TNFα takie, jak inhibitory kinazy białkowej aktywowane mitogenem (MAP); związki, które blokują i/ lub hamują rozszczepienie błony TNFα takie, jak inhibitory metaloproteinaz; związki, które
50 49- blokują i/lub hamują aktywność TNFα takie, jak inhibitory enzymu konwertującego angiotensynę (ACE) (np. kaptopryl); oraz związki, które blokują i/lub hamują produkcję i/lub syntezę TNFα takie, jak inhibitory kinazy MAP. Korzystny antagonista obejmuje przeciwciało. [0157] Czynnik martwicy nowotworu alfa, TNF-alfa lub TNFα odnosi się do cząsteczki ludzkiego TNFα zawierającej sekwencję aminokwasową Pennica i wsp., Nature, 312:721 (1984) lub Aggarwal i wsp., JBC, 260:2345 (1985). Tutaj, inhibitor TNFα jest środkiem, który hamuje w pewnym stopniu funkcję biologiczną TNFα, zazwyczaj poprzez wiązanie się z TNFα i neutralizowanie jego aktywności. Przykłady inhibitorów TNFα obejmują tutaj etanercept (ENBREL ), infliksymab (REMICADE ), i adalimumab (HUMIRA ). [0158] Przykłady antagoniści lub przeciwciała integryny obejmują tutaj przeciwciało LFA- 1 takie, jak efalizumab (RAPTIVA ) dostępny w handlu z firmy Genentech, lub przeciwciało integryny alfa-4 takie, jak natalizumab (ANTEGREN ) dostępne od Biogen lub diazacykliczbe pochodne fenyloalaniny (WO 2003/89410), pochodne fenyloalaniny (WO 2003/70709, WO 2002/28830, WO 2002/16329 i WO 2003/53926), pochodne kwasu fenylopropionowego (WO 2003/10135), pochodne enaminowe (WO 2001/79173), pochodne kwasu propionowego (WO 2000/37444), pochodne kwasów alkanowych (WO 2000/32575), podstawione pochodne fenylowe (Pat US Nry 6,677,339 i 6,348,463), pochodne amin aromatycznych ((Pat US Nr 6,369,229), polipeptydy domen ADAM dezintegryny (US2002/ ), przeciwciała do alfavbeta3 integryny (EP ), aza-mostkowane bicykliczne pochodne aminokwasów (WO 2002/02556), itd. [0159] Określenie środek immunosupresyjny odnosi się do substancji, która działa supresyjnie lub maskuje układ odpornościowy leczonego tutaj pacjenta. Obejmuje to substancje, które hamują wytwarzanie cytokin, regulują w dół lub tłumią ekspresję własnego antygenu lub maskują antygeny MHC. Przykłady takich środków obejmują 2-amino-6-arylo- 5-podstawione pirymidyny (patrz US 4,665,077); niesteroidowe leki przeciwzapalne (NSAID); gancyklowir, takrolimus, glikokortykoidy takie, jak kortyzol lub aldosteron, środki przeciwzapalne takie, jak inhibitor cyklooksygenazy, inhibitory 5-lipoksygenazy lub antagoniści receptora leukotrienowego; antagoniści puryny takie, jak azatiopryna lub mykofenolan mofetylu (MMF); trokad (Ro32-355); antagonistę receptora sigma peryferyjnego takiego, jak ISR-31747; środki alkilujące takie, jak cyklofosfamid; bromokryptyna; danazol; dapson; aldehyd glutarowy (maskujący antygeny MHC, co opisano w US 4,120,649); przeciwciała anty-idiotypowe dla antygenów MHC i fragmentów MHC; cyklosporynę A; steroidy takie, jak kortykosteroidy lub glikokortykosteroidy lub analogi glukokortykoidów, np. prednizon, metyloprednizolon, w tym SOLU-MEDROL sól sodowa bursztynianu metyloprednizolonu, rimeksolon i deksametazon; inhibitory reduktazy dihydrofolianowej takie, jak metotreksat (doustnie lub podskórnie); środki przeciwmalaryczne takie, jak chlorochina i hydroksychlorochina; sulfasalazyna; leflunomid; inhibitory uwalniania cytokin takich, jak przeciwciała monoklonalne SB i SB i antagoniści MHC II tacy, jak ZD2315; antagonistę receptora PG1 taki, jak ZD4953; bloker adhezji VLA4 taki, jak ZD7349; przeciwciała anty-cytokiny i anty-receptor cytokiny, w tym
51 50- przeciwciała anty-interferon-alfa, -beta lub -gamma, przeciwciała anty-tnf-α (infliksymab (REMICADE ) lub adalimumab), immunoadhezyna anty-tnf-α (etanercept), przeciwciała anty-tnf-beta, blokery interleukiny-1 (IL-1) takie, jak rekombinowany inhibitor hull-1 Ra i IL-1B, przeciwciała anty-interleukina-2 (IL-2) i przeciwciała receptora anty-il-2; toksyna fuzyjna IL-2; przeciwciało anty-l3t4; leflunomid; heterologiczna globulina antylimfocytowej; OPC-14597; NISV (modyfikator odpowiedzi immunologicznej); niezbędne kwasy tłuszczowe takie, jak kwas gammalinolenowy lub kwas eikozapentaenowy; blokery CD-4, przeciwciała pan-t, korzystnie przeciwciała anty-cd3 lub anty-cd4/cd4a; modyfikatory ko-stymulujące (np. fuzja CTLA4-Fc, znana również jako ABATACEPT ; przeciwciała i antagoniści receptora anty-interleukina-6 (IL-6); przeciwciała anty-lfa-1, w tym przeciwciała anty-cd11a i anty-cd18; rozpuszczalny peptyd zawierający domenę wiążącą LFA-3 (WO 1990/08187); streptokinaza; IL-10; antagoniści anty-il-4, antagoniści anty-il-13 i przeciwciała antagonistyczne bispecyficzne anty-il-4/il-13, transformujący czynnik wzrostu-beta (TGF-beta); streptodornaza; RNA lub DNA z gospodarza; FK506; RS ; enlimomab; CDP-855; inhibitor PNP; CH-3298; GW353430; 4162W94, chlorambucil; dezoksyspergualina; rapamycyna; receptor komórek T (US 5,114,721); fragmenty receptora komórek T (Offner i wsp., Science, 251: (1991); WO 1990/11294; Janeway, Nature, 341: (1989); i WO 1991/01133); antagoniści BAFF tacy, jak przeciwciała BAFF i przeciwciała BR3; antagoniści ztnf4 (Mackay i Mackay, Trends Immunol., 23:113-5 (2002)); środki biologiczne, które zakłócają sygnały pomocnicze komórek T, takie, jak receptor anty-cd40 lub ligand anty-cd40 (CD 154), w tym przeciwciał blokujących ligand CD40-CD40 (np. Durie i wsp., Science, 261: (1993); Mohan i wsp., J. Immunol., 154: (1995)) i CTLA4-Ig (Finck i wsp., Science, 265: (1994)); oraz przeciwciała receptora komórek T (EP 340,109) takie, jak T10B9. Pewne korzystne tu środki immunosupresyjne obejmują cyklofosfamid, chlorambucyl, azatiopryna, leflunomid, MMF, lub metotreksat (MTX). [0160] Leki przeciwreumatyczne modyfikujące przebieg choroby lub DAMARD obejmują, np. chlorochinę, hydroxychlorochinę, miocryzynę, auranofinę, sulfasalazynę, metotreksat, leflunomid, etanercept, infliksymab (oraz doustny i podskórny MTX), azatioprynę, D-penicilaminę, sole złota (doustne), sole złota (domięśniowe), minocyklinę, cyklosporynę, np. cyklosporynę A i miejscową cyklosporynę, gronkowcowe białko A (Goodyear i Silverman, J. Exp. Med., 197: (2003)), w tym ich sole i pochodne, itd. [0161] Komórka B jest limfocytem, który dojrzewa w szpiku kostnym, obejmuje naiwne komórki B, komórki pamięci B lub komórki efektorowe B (komórki plazmatyczne). Tutaj, komórka B może być prawidłowa lub niezłośliwa. [0162] Marker powierzchniowy komórki B lub antygen powierzchniowy komórki B jest tu antygenem ulegającym ekspresji na powierzchni komórki B, którą można celować za pomocą antagonisty wiążącego się z nim. Przykładowe markery powierzchniowe komórki B obejmują markery powierzchniowe CD10, CD19, CD20, CD21, CD22, CD23, CD24, CD37, CD40, CD53, CD72, CD73, CD74, CDw75, CDw76, CD77, CDw78, CD79a, CD79b, CD80, CD81, CD82, CD83, CDw84, CD85 i CD86 (dla opisów, patrz The Leukocyte Antigen Facts
52 51- Book, 2 wydanie. 1997, ed. Barclay i wsp. Academic Press, Harcourt Brace & Co., New York). Inne markery powierzchniowe komórek B obejmują RP105, FcRH2, B-komórkowy CR2, CCR6, P2X5, HLA-DOB, CXCR5, FCER2, BR3, Btig, NAG14, SLGC16270, FcRH1, IRTA2, ATWD578, FcRH3, IRTA1, FcRH6, BCMA i Korzystny marker powierzchni komórek B jest korzystnie eksprymowany na komórkach B w stosunku do innych tkanek nie-b-komórkowych ssaka i może ulegać ekspresji na zarówno prekursorowych i dojrzałych limfocytach B. Najbardziej korzystnymi markerami są CD20 i CD22. [0163] Antygen CD20 lub CD20 jest około 35-kDa, nieglikozylowaną fosfoproteiną występującą na powierzchni ponad niż 90% komórek B z krwi obwodowej lub narządów limfoidalnych. CD20 występuje zarówno na prawidłowych komórkach B oraz złośliwych komórkach B, lecz nie na komórkach macierzystych. Inne nazwy dla CD20 w literaturze obejmują antygen ograniczony do limfocytów B i Bp35. Antygen CD20 jest opisany na przykład w Clark i wsp., Proc. Natl. Acad. Sci. (USA) 82:1766 (1985). [0164] Antygen CD22, lub CD22 znany również jako BL-CAM lub Lyb8, jest integralną glikoproteiną błony typu 1 o masie cząsteczkowej około 130 (zredukowana) do 140kD (niezredukowana). Jest on eksprymowany zarówno w cytoplazmie, jak i błonie komórkowej limfocytów B. Antygen CD22 pojawia się na początku różnicowania limfocytów komórek B, w przybliżeniu na takim samym etapie jak antygen CD19. W przeciwieństwie do innych markerów komórek B, ekspresja błonowa CD22 jest ograniczona do późnych stadiów różnicowania zawartych pomiędzy dojrzałymi komórkami B (CD22+) i komórek plazmatycznych (CD22-). Antygen CD22 jest opisany przykładowo w Wilson i wsp., J. Exp. Med. 173:137 (1991) i Wilson i wsp., J. Immunol. 150:5013 (1993). [0165] Przeciwciało, które wiąże się z markerem powierzchniowym komórki B to cząsteczka, która po związaniu się z markerem powierzchniowym komórki B, niszczy lub zubaża komórki B u ssaka i/lub zaburza jedną lub więcej funkcji komórek B, np. przez zmniejszenie lub zapobieganie odpowiedzi humoralnej wywoływanej przez komórki B. Przeciwciało jest korzystnie w stanie zubożyć komórki B (tj. zmniejszyć poziom krążących komórek B) u ssaków nim leczonych. Takie zubożenie można uzyskać poprzez różne mechanizmy, jak cytotoksyczność komórkowa zależna od przeciwciał (ADCC) i/lub cytotoksyczność zależna od dopełniacza (CDC), hamowanie proliferacji komórek B i/lub indukowanie śmierci komórek B (np. poprzez apoptozę). [0166] Przykłady przeciwciał CD20 obejmują: C2B8, które obecnie nazywa się rytuksymab ( RITUXAN ) (Patent US Nr 5,736,137); znakowane itrem-[90] mysie przeciwciało 2B8 oznaczone Y2B8 lub tiuksetan ibrytumomabu (ZEVALIN ) dostępne w handlu z firmy IDEC Pharmaceuticals, Inc. (Patent US Nr 5,736,137; 2B8 zdeponowane w ATCC pod nr dostępu. HB11388 w dniu 22 czerwca 1993); mysia IgG2a B1, nazywana także tositumomab ewentualnie znakowany 131 I do wytworzenia przeciwciała 131I-B1 lub jod I131 tositumomab (BEXXAR ) dostępny w handlu z firmy Corixa (patrz także Patent US Nr 5,595,721); mysie przeciwciało monoklonalne 1F5 (Press i wsp. Blood 69(2): (1987) i jego warianty w tym 1F5 typu framework patched lub
53 52- humanizowane (WO 2003/002607, Leung, S.; ATCC depozyt HB-96450); mysie przeciwciało 2H7 i chimeryczne 2H7 (Patent US Nr 5,677,180); humanizowane 2H7 (WO 2004/ (Lowman i wsp.) i jak przedstawiono niżej); HUMAX-CD20 w pełni ludzkie przeciwciała o dużym powinowactwie ukierunkowane na cząsteczki CD20 w błonie komórkowej komórek B (Genmab, Denmark; patrz, na przykład, Glennie i van de Winkel, Drug Discovery Today 8: (2003) i Cragg i wsp., Blood 101: (2003)); ludzkie monoklonalne przeciwciała zawarte w WO04/ (Teeling i wsp.); przeciwciała AME-133 (Applied Molecular Evolution); przeciwciało A20 lub jego warianty takie, jak chimeryczne lub humanizowane przeciwciało A20 (odpowiednio ca20, ha20) (US 2003/ , Immunomedics); i monoklonalne przeciwciała L27, G28-2, 93-1B3, B-C1 lub NU-B2 dostępne z International Leukocyte Typing Workshop (Valentine i wsp., In: Leukocyte Typing III (McMichael, Ed., p. 440, Oxford University Press (1987)). Korzystnymi przeciwciałami CD20 tu są chimeryczne, humanizowane lub ludzkie przeciwciała CD20, korzystniej rytuksymab, humanizowane 2H7, chimeryczne lub humanizowane przeciwciało A20 (Immunomedics) i ludzkie przeciwciało CD20 HUMAX- CD20 (Genmab). [0167] Określenia rytuksymab lub RITUXAN odnoszą się tu do monoklonalnego przeciwciała chimerycznego mysiego/ludzkiego uzyskanego metodą inżynierii genetycznej, skierowanego przeciw antygenowi CD20 i oznaczonego C2B8 w Patencie US Nr 5,736,137, w tym ich fragmenty, które zachowują zdolność do wiązania CD20. [0168] Wyłącznie dla celów niniejszego wynalazku i jeśli nie wskazano inaczej, humanizowane 2H7 dotyczy humanizowanego przeciwciała CD20, lub jego fragmentu wiążącego antygen, przy czym przeciwciało jest skuteczne w celu zubażania komórek B naczelnych in vivo. Przeciwciała obejmują te przedstawione na US 2006/ i zamieszczonych tam figurach, i obejmujące wersję 114, sekwencje, które przedstawiono w US 2006/ Patrz także US 2006/ i US 2006/ W podsumowaniu różnych korzystnych przykładów wykonania według wynalazku, region V wariantów na podstawie 2H7 wersji 16, jak ujawniono w US 2006/ będzie miał sekwencje aminokwasowe v16 z wyjątkiem pozycji podstawień aminokwasowych, które są wskazane w poniższej tabeli. O ile nie wskazano inaczej, warianty 2H7 mają ten sam łańcuch L jak w v16. Wersja 2H7 Zmiany łańcucha ciężkiego (VH) Zmiany łańcucha lekkiego (VL) 16 - Zmiany Fc S298A, E333A, K334A 73 N100A M32L 75 N100A M32L S298A, E333A, K334A 96 D56A, N100A S92A 114 D56A, N100A M32L, S92A S298A, E333A, K334A
54 D56A, N100A M32L, S92A S298A, E333A, K334A, E356D, M358L 116 D56A, N100A M32L, S92A S298A, K334A, K322A 138 D56A, N100A M32L, S92A S298A, E333A, K334A, K326A 477 D56A, N100A M32L, S92A S298A, E333A, K334A, K326A, N434W K334L [0169] Jedno korzystne humanizowane 2H7 jest nienaruszonym przeciwciałem lub fragmentem przeciwciała o sekwencji wersji 16. Inne korzystne humanizowane 2H7 ma sekwencje wersji 114. [0170] Antagoniści BAFF stanowią dowolne cząsteczki, które blokują aktywność BAFF lub Br3. Zawierają one immunoadhezyny zawierające część BR3, TACI lub BCMA, która wiąże BAFF, lub ich warianty, które wiążą BAFF. W innych aspektach, antagonista BAFF jest przeciwciałem BAFF. Przeciwciało BAFF jest przeciwciałem, które wiąże BAFF, a korzystnie wiąże BAFF w regionie ludzkiego BAFF zawierającego reszty ludzkiego BAFF. W innym aspekcie, antagonista BAFF jest przeciwciałem BR3. Antagonista BR3 jest przeciwciałem, które wiąże BR3 i korzystnie wiąże BR3 w regionie ludzkiego BR3 zawierającego reszty ludzkiej Br3. Sekwencje ludzkiego BAFF i ludzkiej Br3 można znaleźć, na przykład, w US 2006/ Inne przykłady polipeptydów wiążących BAFF lub przeciwciał BAFF można znaleźć w, np. WO 2002/092620, WO 2003/014294, Gordon i wsp., Biochemistry 42(20): (2003), Kelley i wsp., J. Biol.Chem. 279: (2004), WO 1998/18921, WO 2001/12812, WO 2000/68378 i WO 2000/ [0171] Przeciwciało anty-ige obejmuje dowolne przeciwciało, które wiąże się specyficznie z IgE w taki sposób, aby nie powodować sieciowania, gdy IgE jest związane z receptorem o wysokim powinowactwie na komórkach tucznych i bazofili. Do przykładowych przeciwciał zalicza się przeciwciała według wynalazku, jak również rhumabe25 (E25, XOLAIR ), E26, E27, oraz CGP-5101 (Hu-901) i przeciwciało HA. Sekwencje aminokwasowe domen zmiennych łańcucha ciężkiego i lekkiego humanizowanych przeciwciał anty-ige E25, E26 i E27 ujawniono, na przykład w U.S.P. 6,172,213 i WO99/ Przeciwciało CGP-5101 (Hu-901) jest opisane w Corne i wsp., (1997) J. Clin. Invest. 99(5): , WO 92/17207 i Nr Dep. ATCC BRL-10706, BRL-11130, BRL-11131, BRL i BRL Przeciwciało HA jest opisane w USSN 60/444,229, WO2004/ i WO2004/ II. Sposoby Realizacji Wynalazku A. Wytwarzanie metodą rekombinacji [0172] Wynalazek zapewnia także wyizolowany kwas nukleinowy kodujący apoptotyczne przeciwciała anty-ige, wektory i komórki gospodarza zawierające taki kwas nukleinowy i techniki rekombinacji dla wytwarzania przeciwciała. [0173] W celu rekombinacyjnego wytwarzania przeciwciała, kodujący je kwas nukleinowy izoluje się i wstawia do zdolnego do replikacji wektora do dalszego klonowania (amplifikacji DNA) lub do ekspresji. DNA kodujący przeciwciało monoklonalne łatwo izoluje się i
55 54- sekwencjonuje stosując konwencjonalne procedury (np. używając oligonukleotydów okazuje się, że są zdolne do specyficznego wiązania z genami kodującymi ciężkie i lekkie łańcuchy przeciwciała). Dostępnych jest wiele wektorów. Składniki wektora generalnie obejmują, bez ograniczania do wymienionych, jeden lub więcej z następujących, sekwencję sygnałową, miejsce startu replikacji, jeden lub więcej genów markerowych, element wzmacniacza, promotor i sekwencja terminacji transkrypcji. (1) Składnik sekwencja sygnałowa [0174] Przeciwciała anty-ige według niniejszego wynalazku można wytwarzać rekombinacyjnie nie tylko bezpośrednio, ale także jako polipeptyd fuzyjny z polipeptydem heterologicznym, który jest korzystnie sekwencją sygnałową lub innym polipeptydem posiadającym miejsce specyficznego odszczepiania na końcu N dojrzałego białka lub polipeptydu. Wybrana heterologiczna sekwencja sygnałowa korzystnie jest taką, która jest rozpoznawana i poddawana obróbce (tj. cięta przez peptydazę sygnałową) przez komórkę gospodarza. Dla prokariotycznych komórek gospodarza, które nie rozpoznają i nie przetwarzają natywnej sekwencji sygnałowej ssaków, sekwencja sygnałowa jest zastępowana przez prokariotyczną sekwencję sygnałową wybraną, na przykład, z grupy liderów fosfatazy alkalicznej, penicylinazy, Ipp lub liderów stabilnej termicznie enterotoksyny II. Dla wydzielania z drożdży, natywną sekwencję sygnałową można podstawić np. liderem drożdżowej inwertazy, liderem czynnika (w tym liderem czynnika Saccharomyces i Kluyveromyces), lub liderem kwaśnej fosfatazy, liderem glukoamylazy C. albicans, lub sygnał opisany w WO 90/ W przypadku ekspresji w komórkach ssaczych, dostępne są ssacze sekwencje sygnałowe, jak również wirusowe lidery wydzielnicze, na przykład sygnał gd wirusa opryszczki. [0175] DNA dla takiego regionu prekursora liguje się w ramce odczytu z DNA kodującym przeciwciało wiążące IgE. (2) Miejsce inicjacji replikacji [0176] Zarówno ekspresyjne, jak i klonujące wektory zawierają sekwencję kwasu nukleinowego, która umożliwia wektorowi replikację w jednej lub większej liczbie wybranych komórek gospodarza. Generalnie, w wektorach klonujących sekwencja ta jest taką, która umożliwia wektorowi replikację niezależnie od chromosomalnego DNA gospodarza, i obejmuje miejsce inicjacji replikacji lub sekwencje autonomicznie replikujące. Takie sekwencje są dobrze znane dla szeregu bakterii, drożdży i wirusów. Miejsce inicjacji replikacji z plazmidu pbr322 jest odpowiednie dla większości bakterii Gram-ujemnych bakterii, miejsce inicjacji plazmid 2μ jest odpowiednie dla drożdży, a różne wirusowe miejsca inicjacji replikacji (SV40, polioma, adenowirus, VSV lub BPV) są użyteczne dla wektorów do klonowania w komórkach ssaczych. Ogólnie, komponent miejsca inicjacji replikacji nie jest potrzebny w ssaczych wektorach ekspresyjnych (SV40 zazwyczaj może być używany tylko dlatego, że zawiera wczesny promotor). (3) Składnik gen selekcyjny
56 55- [0177] Wektory ekspresyjne i klonujące mogą zawierać gen selekcyjny, określany również jako marker podlegający selekcji. Typowe geny selekcyjne kodują białka, które (a) nadają oporność na antybiotyki lub inne toksyny, np. ampicylinę, neomycynę, metotreksat lub tetracyklinę, (b) uzupełniają niedobory auksotroficzne lub (c) dostarczają krytycznych substancji odżywczych nie dostępnych w pożywkach złożonych, np. gen kodujący racemazę D-alaniny dla Bacilli. [0178] Jeden z przykładów schematu selekcji wykorzystuje lek do zahamowania wzrostu komórki gospodarza. Te komórki, które zostały z powodzeniem transformowane genem heterologicznym, wytwarzają biało nadające oporność na lek, a zatem przeżywają reżim selekcji. Przykłady takich selekcji dominujących wykorzystują leki neomycynę, kwas mykofenolowy i hygromycynę. [0179] Innym przykładem odpowiednich markerów podlegających selekcji dla komórek ssaczych są te, które umożliwiają identyfikację komórek kompetentnych do wiązania IgE kwasu nukleinowego takie, jak geny DHFR, kinazy tymidynowej, metalotioneiny I i II, korzystnie geny metalotioneiny naczelnych, deaminazy adenozynowej, dekarboksylazy ornityny itd. [0180] Na przykład, komórki poddane transformacji genem selekcyjnym DHFR są najpierw identyfikowane poprzez hodowanie wszystkich transformantów w podłożu hodowlanym, które zawiera metotreksat (Mtx), będący antagonistą kompetycyjnym DHFR. Odpowiednią komórką gospodarza, gdy stosuje się DHFR typu dzikiego, jest linią komórkową jajnika chomika chińskiego (CHO) pozbawioną aktywności DHFR (np. ATCC CRL-9096). [0181] Alternatywnie, komórki gospodarza (szczególnie gospodarze typu dzikiego, którzy zawierają endogenną DHFR) transformowane lub kotransformowane sekwencjami DNA kodującymi przeciwciało wiążące IgE, białko DHFR typu dzikiego lub inne markery podlegające selekcji takie, jak 3 -fosfotransferaza aminoglikozydowa (APH), mogą być selekcjonowane poprzez wzrost komórek w podłożu zawierającym czynnik selekcyjny dla markera podlegającego selekcji takiego, jak antybiotyk aminoglikozydowy, np. kanamycyna, neomycyna lub G418. Patrz Patent US Nr 4,965,199. [0182] Odpowiednim genem selekcyjnym do stosowania w drożdżach jest gen trp1 obecny na drożdżowym plazmidzie YRp7 (Stinchcomb i wsp., Nature, 282:39 (1979)). Gen trp1 zapewnia marker podlegający selekcji dla zmutowanego szczepu drożdży, pozbawionego zdolności wzrostu w obecności tryptofanu, na przykład ATCC nr lub PEP4-1. Jones, Genetics, 85:12 (1977). Obecność uszkodzenia trp1 w genomie komórki gospodarza drożdżowego zapewnia zatem skuteczne środowisko genetyczne do wykrywania transformacji poprzez wzrost w nieobecności tryptofanu. Podobnie szczepy drożdży z niedoborem Leu2 (ATCC 20,622 lub 38,626) uzupełniane są przez znane plazmidy niosące gen Leu2. [0183] Ponadto, wektory pochodzące od kolistego plazmidu 1,6 µm PKD1 mogą być stosowane do transformacji drożdży Kluyveromyces. Alternatywnie, układ ekspresyjny do wytwarzania na dużą skalę zrekombinowanej podpuszczki cielęcej opisano dla K. lactis. Van
57 56- den Berg, Bio/Technology, 8:135 (1990). Ujawniono także stabilne wielokopiowe wektory ekspresyjne do wydzielania dojrzałej, zrekombinowanej albuminy surowicy ludzkiej przez przemysłowe szczepy Kluyveromyces. Fleer i wsp., Bio/Technology, 9: (1991). (4) Składnik Promotor [0184] Wektory ekspresyjne i klonujące zazwyczaj zawierają promotor, który jest rozpoznawany przez organizm gospodarza i jest funkcjonalnie związany z kwasem nukleinowym kodującym przeciwciało wiążące IgE. Do promotorów odpowiednich do użycia z prokariotycznymi gospodarzami należą promotor phoa, - układy promotora laktamazy i laktozy, promotor fosfatazy alkalicznej, układ promotora tryptofanu (trp), oraz promotory hybrydowe takie, jak promotor tac. Jednakże odpowiednie są inne znane promotory bakteryjne. Promotory do stosowania w układach bakteryjnych będą zawierać również sekwencję Shine-Dalgarno (sekwencję SD) połączoną funkcjonalnie z DNA kodującym przeciwciało wiążące IgE. [0185] Znane są sekwencje promotorowe dla organizmów eukariotycznych. Właściwie wszystkie geny eukariotyczne posiadają region bogaty w AT znajdujący się około 25 do 30 zasad przed miejscem, w którym rozpoczynana jest transkrypcja. Inną sekwencją, występującą od 70 do 80 zasad przed miejscem początku transkrypcji wielu genów, jest region CNCAAT, gdzie N może być dowolnym nukleotydem. Na końcu 3 większości genów eukariotycznych znajduje się sekwencja AATAAA, która może być sygnałem dla addycji ogona poli-a do końca 3 sekwencji kodującej. Wszystkie z tych sekwencji są odpowiednie do wstawiania do eukariotycznych wektorów ekspresyjnych. [0186] Przykłady odpowiednich sekwencji promotorowych do stosowania w gospodarzach drożdżowych obejmują promotory dla kinazy 3-fosfoglicerynianowej lub innych enzymów glikolitycznych, np. enolazy, dehydrogenazy gliceraldehydo-3-fosforanowej, heksokinazy, dekarboksylazy pirogronianowej, fosfofruktokinazy, izomerazy glukozo-6-fosforanu, mutazy 3-fosfoglicerynianowej, kinazy pirogronianowej, izomerazy triozofosforanowej, izomerazy fosfoglukozowej i glukokinazy. [0187] Inne promotory drożdżowe, które są promotorami indukowalnymi, posiadającymi dodatkową zaletę kontrolowania transkrypcji przez warunki wzrostu, są regiony promotorowe dla dehydrogenazy alkoholowej 2, izocytochromu C, fosfatazy kwaśnej, enzymów degradacyjne związanych z metabolizmem azotu, metalotioneiny, dehydrogenazy gliceraldehydo-3-fosforanowej, oraz enzymów odpowiedzialnych za wykorzystanie maltozy i galaktozy. Odpowiednie wektory i promotory do użycia przy ekspresji w drożdżach są ponadto opisane w EP 73,657. Drożdżowe enhancery są również korzystnie stosowane z promotorami drożdżowymi. [0188] Transkrypcja przeciwciała wiążącego IgE z wektorów w ssaczych komórkach gospodarza jest kontrolowana, na przykład, przez promotory otrzymane z genomów wirusów takich, jak wirus polyoma, wirus ospy ptasiej, adenowirus (jak Adenowirus 2), wirus brodawczaka bydlęcego, wirus mięsaka ptasiego, wirus cytomegalii, retrowirus, wirus zapalenia wątroby typu B, a najkorzystniej małpi wirus 40 (SV40), z heterologicznych
58 57- promotorów ssaczych, np. promotora aktyny lub promotora immunoglobuliny, z promotorów szoku cieplnego, pod warunkiem, że takie promotory są zgodne z układami komórek gospodarza. [0189] Wczesne i późne promotory wirusa SV40 otrzymuje się dogodnie w postaci fragmentu restrykcyjnego SV40, który zawiera również wirusowe miejsce rozpoczęcia replikacji SV40. Natychmiastowy wczesny promotor ludzkiego cytomegalowirusa otrzymuje się dogodnie w postaci fragmentu restrykcyjnego Hindlll E. Układ do ekspresji DNA w gospodarzach ssaczych z zastosowaniem wirusa brodawczaka bydlęcego jako wektora jest ujawniony w Patencie US Nr 4,419,446. Modyfikację tego układu opisano w Patencie US Nr 4,601,978. Patrz także Reyes i wsp., Nature 297: (1982) odnośnie ekspresji cdna ludzkiego interferonu w komórkach mysich pod kontrolą promotora kinazy tymidynowej z wirusa Herpes simplex. Alternatywnie, długie powtórzenia końcowe wirusa mięsaka Rousa można wykorzystać jako promotor. (5) Składnik elementu wzmacniacza [0190] Transkrypcja DNA kodującego przeciwciało wiążące IgE według tego wynalazku przez wyższe organizmy eukariotyczne często zwiększa wstawienie sekwencji wzmacniacza do wektora. Obecnie znanych jest wiele sekwencji wzmacniających z genów ssaków (globiny, elastazy, albuminy, -fetoproteiny i insuliny). Zazwyczaj jednak stosować się będzie sekwencję wzmacniającą z wirusa komórek eukariotycznych. Przykłady obejmują sekwencję wzmacniającą SV40 na późnej stronie miejsca początku replikacji (bp ), wzmacniacz wczesnego promotora cytomegalowirusa, wzmacniacz polioma na późnej stronie miejsca początku replikacji oraz sekwencje wzmacniające adenowirusów. Zobacz także Yaniv, Nature 297:17-18 (1982) odnośnie elementów wzmacniających dla aktywacji promotorów eukariotycznych. Wzmacniacz może być wstawiony do wektora w pozycji 5 albo 3 od sekwencji wiążącej kodującej przeciwciało IgE, ale korzystnie położona jest ona po stronie 5 promotora. (6) Składnik terminacji transkrypcji [0191] Wektory ekspresyjne stosowane w eukariotycznych komórkach gospodarza (komórkach drożdżowych, grzybowych, owadzich, roślinnych, zwierzęcych, ludzkich lub posiadających jądra komórkach z innych organizmów wielokomórkowych) będą również zawierały sekwencje konieczne do terminacji transkrypcji i do stabilizacji mrna. Takie sekwencje są powszechnie dostępne z 5, a czasami 3, nie ulegających translacji regionów eukariotycznych lub wirusowych DNA lub cdna. Regiony te zawierają odcinki nukleotydowe ulegające transkrypcji jako fragment poliadenylowany w nie ulegającej translacji części mrna kodującej przeciwciało wiążące IgE. Jednym z użytecznych składników terminacji transkrypcji jest region poliadenylacji bydlęcego hormonu wzrostu. Patrz WO94/11026 i ujawniony w nim wektor ekspresyjny. (7) Selekcja i transformacja komórek gospodarza [0192] Odpowiednie komórki gospodarza do klonowania lub ekspresji DNA w wektorach są komórkami prokariotycznymi, drożdży, lub wyższymi komórkami eukariotycznymi
59 58- opisanymi powyżej. Odpowiednie do tego celu organizmy prokariotyczne obejmują bakterie właściwe takie, jak organizmy Gram-ujemne lub Gram-dodatnie, na przykład Enterobacteriaceae takie, jak Escherichia, np. E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, np. Salmonella typhimurium, Serratia, np. Serratia marcescans i Shigella, oraz Bacilli takie, jak B. subtilis i B. licheniformis (np. B. licheniformis 41P ujawnione w DD 266,710 opublikowane 12 kwietnia 1989), Pseudomonas takie, jak P. aeruginosa i Streptomyces. Jednym z korzystnych gospodarzy klonujących E. coli jest E. coli 294 (ATCC 31,446), chociaż odpowiednie są inne szczepy takie, jak E. coli B, E. coli X1776 (ATCC 31,537), i E. coli W3110 (ATCC 27,325). Te przykłady są raczej ilustracyjne niż ograniczające. [0193] Pełnej długości przeciwciała, fragmenty przeciwciała i białka fuzyjne przeciwciał mogą być wytwarzane w bakteriach, zwłaszcza gdy glikozylacja i funkcje efektorowe Fc nie są potrzebne tak, jak wtedy, gdy przeciwciało terapeutyczne jest skoniugowane ze środkiem cytotoksycznym (np. toksyną) i immunokoniugat sam wykazuje skuteczność w niszczeniu komórek guza. Przeciwciała pełnej długości mają dłuższy okres półtrwania w obiegu. Produkcja w E. coli jest szybsza i bardziej opłacalna. Do ekspresji fragmentów przeciwciał i polipeptydów w bakteriach, zobacz, np. U.S. 5,648,237 (Carter i wsp.), U.S. 5,789,199 (Joly i wsp.) i U.S. 5,840,523 (Simmons i wsp.), który opisuje rejon inicjacji translacji (TIR) i sekwencje sygnałowe do optymalizacji ekspresji i wydzielania. Po ekspresji, przeciwciało jest izolowane z pasty komórek E. coli we frakcji rozpuszczalnej i można je oczyszczać poprzez, np. kolumnę z białkiem A lub G w zależności od izotypu. Końcowe oczyszczanie można przeprowadzić podobnie do procesu oczyszczania przeciwciała wyeksprymowanego na przykład w komórkach CHO. [0194] Obok prokariotów, eukariotyczne mikroby takie, jak grzyby nitkowate lub drożdże są odpowiednimi gospodarzami do klonowania lub ekspresji dla wektorów kodujących przeciwciała eksprymujące IgE. Saccharomyces cerevisiae, lub zwykłe drożdże piekarnicze są najczęściej używanym spośród niższych eukariotycznych mikroorganizmów-gospodarzy. Jednakże, wiele innych rodzajów, gatunki i szczepy jest powszechnie dostępnych i użytecznych w niniejszym opisie, takie, jak Schizosaccharomyces pombe; gospodarze Kluyveromyces takie, jak K. lactis, K. fragilis (ATCC 12,424), K. bulgaricus (ATCC 16,045), K. wickeramii (ATCC 24,178), K. waltii (ATCC 56,500), K. drosophilarum (ATCC 36,906), K. thermotolerans, i K. marxianus; yarrowia (EP 402,226); Pichia pastoris (EP 183,070); Candida; Trichoderma reesia (EP 244,234); Neurospora crassa; Schwanniomyces takich, jak Schwanniomyces occidentalis; i grzyby nitkowate takie, jak, na przykład, Neurospora, Penicillium, Tolypocladium, i gospodarze Aspergillus tacy, jak A. nidulans i A. niger. [0195] Odpowiednie komórki gospodarza do ekspresji glikozylowanego przeciwciała wiążącego IgE pochodzą z organizmów wielokomórkowych. Przykłady komórek bezkręgowców obejmują komórki roślin i owadów. Zidentyfikowano liczne szczepy i warianty bakulowirusowe i odpowiadające im zalecane owadzie komórki gospodarza z takich gospodarzy, jak Spodoptera frugiperda (gąsienica), Aedes aegypti (komar), Aedes albopictus (komar), Drosophila melanogaster (muszka owocowa), i Bombyx mori. Szereg szczepów
60 59- wirusowych do transfekcji jest publicznie dostępnych, np. wariant L-1 z NPV Autographa californica i szczep Bm-5 NPV Bombyx mori, i takie wirusy mogą być użyte jako wirus w niniejszym opisie, zgodnie z niniejszym wynalazkiem, w szczególności do transfekcji komórek Spodoptera frugiperda. [0196] Hodowle komórek roślinnych z bawełny, kukurydzy, ziemniaka, soi, petunii, pomidora i tytoniu również mogą być stosowane jako gospodarze. [0197] Jednakże, największe zainteresowanie budziły komórki kręgowców, i namnażanie komórek kręgowców w hodowli (hodowli tkankowej) stało się procedurą rutynową. Przykładami użytecznych ssaczych linii komórek gospodarza są linia nerki małpiej CV1 transformowana przez SV40 (COS-7, ATCC CRL 1651); linia ludzkich komórek embrionalnych nerki (komórki 293 lub 293 subklonowane do wzrostu w hodowli zawiesinowej, Graham i wsp., J. Gen Virol. 36:59 (1977)) ; komórki nerki noworodka chomika (BHK, ATCC CCL 10); komórki jajnika chomika chińskiego /-DHFR (CHO, Urlaub i wsp., Proc. Natl. Acad. Sci. USA 77:4216 (1980)) ; mysie komórki Sertoliego (TM4, Mather, Biol. Reprod. 23: (1980)); małpie komórki nerkowe (CV1 ATCC CCL 70); komórki nerki afrykańskich małp zielonych (VERO-76, ATCC CRL-1587); komórki raka szyjki macicy ludzkiej (HELA, ATCC CCL 2); komórki nerki psa (MDCK, ATCC CCL 34); komórki wątroby szczura buffalo (BRL 3A, ATCC CRL 1442); ludzkie komórki płuc (W138, ATCC CCL 75); ludzkie komórki wątroby (Hep G2, HB 8065); mysie komórki guza sutka (MMT , ATCC CCL51); komórki TRI (Mather i wsp., Annals N.Y. Acad. Sci. 383:44-68 (1982)); komórki MRC 5; komórki FS4; i ludzka linia komórek hepatoma (Hep G2). [0198] Komórki gospodarza transformowane są wyżej opisanymi wektorami ekspresyjnymi lub klonowania do produkcji przeciwciał wiążących IgE i hoduje się je w konwencjonalnych pożywkach odżywczych odpowiednio zmodyfikowanych dla indukcji promotorów, selekcji transformantów lub amplifikacji genów kodujących pożądane sekwencje. (8) Hodowanie komórek gospodarza [0199] Komórki gospodarza stosowane do produkcji przeciwciała wiążącego IgE według tego wynalazku można hodować w szeregu pożywek. Dostępne na rynku pożywki takie, jak Ham s F10 (Sigma), Minimal Essential Medium ((MEM), (Sigma), RPMI-1640 (Sigma) i Dulbecco s Modified Eagle s Medium ((DMEM), Sigma) są odpowiednie do hodowania komórek gospodarza. Dodatkowo którakolwiek z pożywek opisanych w Ham i wsp., Meth. Enz. 58:44 (1979), Barnes i wsp., Anal. Biochem.102:255 (1980), US Pat. Nry 4,767,704; 4,657,866; 4,927,762; 4,560,655; lub 5,122,469; WO 90/03430; WO 87/00195; lub Patent US Re. 30,985 mogą być stosowane jako pożywka do hodowli komórek gospodarza. Każda z tych pożywek może być wzbogacona w miarę potrzeb hormonami i/lub innymi czynnikami wzrostu (jak insulina, transferyna lub naskórkowy czynnik wzrostu), sole (jak chlorek sodowy, wapnia, magnez i fosforan), bufory (jak HEPES), nukleotydy (jak adenozyna i tymidyna), antybiotyki (jak lek GENTAMYCIN ), pierwiastki śladowe (zdefiniowane jako związki nieorganiczne zazwyczaj obecne w stężeniach końcowych w zakresie
61 60- mikromolowym) i glukoza bądź równoważne źródło energii. Jakiekolwiek inne niezbędne suplementy mogą także być włączone w odpowiednich stężeniach, które powinny być znane znawcom w tej dziedzinie. Warunki hodowli takie, jak temperatura, ph i tym podobne, są tymi stosowanymi wcześniej w odniesieniu do komórek gospodarza wybranej do ekspresji i będą oczywiste dla przeciętnego specjalisty w dziedzinie. (9) Oczyszczanie przeciwciała [0200] Podczas korzystania z technik rekombinacji, przeciwciało może być wytwarzane wewnątrzkomórkowo, w przestrzeni peryplazmatycznej, lub bezpośrednio wydzielane do pożywki. Jeśli przeciwciało jest wytwarzane wewnątrzkomórkowo, na pierwszym etapie usuwa się cząstkowe pozostałości, albo komórki gospodarza albo zlizowane fragmenty, na przykład przez wirowanie lub ultrafiltrację. Carter i wsp., Bio/Technology 10: (1992) opisują procedurę izolowania przeciwciał, które są wydzielane do przestrzeni periplazmatycznej E. coli. Krótko, masę komórkową rozmraża się w obecności octanu sodowego (ph 3.5), EDTA i fluorku fenylometylosulfonylu (PMSF) w ciągu około 30 min. Pozostałości komórkowe można usunąć przez wirowanie. Gdy przeciwciało wydzielane jest do pożywki, supernatanty z takich układów ekspresyjnych generalnie najpierw zatęża się stosując komercjalnie dostępny sączek do zatężania białek, na przykład, jednostkę do ultrafiltracji Amicon lub Millipore Pellicon. Inhibitor proteazy taki, jak PMSF może być zawarty w każdym z powyższych etapów w celu zahamowania proteolizy, a antybiotyki mogą być włączone w celu zapobiegania wzrostowi przypadkowych zanieczyszczeń. [0201] Kompozycję przeciwciała przygotowaną z komórek można oczyszczać stosując, na przykład, chromatografię na hydroksyapatycie, elektroforezę żelową, dializę i chromatografię powinowactwa, przy czym chromatografia powinowactwa jest korzystną techniką oczyszczania. Przydatność białka A jako liganda powinowactwa zależy od gatunku i izotypu domeny Fc immunoglobuliny, która jest obecna w przeciwciele. Białko A można stosować do oczyszczania przeciwciał, które oparte są na ludzkich ciężkich łańcuchach 1, 2 lub 4 (Lindmark i wsp., J. Immunol. Meth. 62:1-13 (1983)). Białko G jest zalecane dla wszystkich izotypów mysich oraz ludzkiego 3 (Guss i wsp., EMBO J. 5: (1986)). Matrycą, do której przyłączony jest ligand powinowactwa, najczęściej jest agaroza, ale dostępne są inne matryce. Mechanicznie stabilne matryce takie, jak szkło o kontrolowanej porowatości lub poli (styren-diwinylo)benzen, pozwalają na wyższe szybkości przepływu i krótsze czasy obróbki niż jest to możliwe do uzyskania w przypadku agarozy. Gdy przeciwciało zawiera a domenę C H 3, żywica Bakerbond ABX (J. T. Baker, Phillipsburg, NJ) jest przydatna do oczyszczania. Dopuszczalne są inne techniki oczyszczania białek takie, jak frakcjonowanie na kolumnie jonowymiennej, wytrącanie etanolem, HPLC w układzie faz odwróconych, chromatografia na krzemionce, chromatografia na heparynie SEPHAROSE chromatografia na żywicy anionowymiennej lub kationowymiennej (jak kolumna z kwasem poliaspartylowym), ogniskowanie chromatograficzne SDS-PAGE i wytrącanie siarczanem amonu w zależności od przeciwciała, które ma być odzyskane. [0202] Po każdym wstępnym etapie (etapach) oczyszczania, mieszaninę zawierającą przeciwciało będące przedmiotem zainteresowania i zanieczyszczenia można poddać
62 61- chromatografii oddziaływań w niskim ph z użyciem buforu do hydrofobowej do elucji o ph pomiędzy około 2,5-4,5, korzystnie prowadzonej przy niskim stężeniu soli (np. od około 0 do 0.25 M soli). B. Wytwarzanie przeciwciała 1) Poliklonalne przeciwciała [0203] Przeciwciała poliklonalne zazwyczaj się u zwierząt przez wielokrotne iniekcje podskórne (sc) lub dootrzewnowe (ip) odpowiedniego antygenu i adiuwanta. Użyteczne może być skoniugowanie odpowiedniego antygenu z białkiem, które jest immunogenne u gatunku przeznaczonego do immunizacji, np. hemocyjaniną skałoczepa (KLH), albuminą surowicy, tyroglobuliną bydlęcą lub inhibitorem trypsyny sojowej, z zastosowaniem środka bifunkcyjnego lub derywatyzującego, np. ester maleimidobenzoilosulfosukcynimidowy (koniugacja poprzez reszty cysteiny), N-hydroksysukcynimid (poprzez reszty lizyny), glutaraldehyd, bezwodnik bursztynowy, SOCl 2, lub R 1 N=C=NR, gdzie R i R 1 niezależnie oznaczają niższe grupy alkilowe. Przykłady adiuwantów, które mogą być stosowane, obejmują kompletny adiuwant Freunda i adiuwant MPL-TDM (monofosforylo-lipid A, syntetyczny dikorynomykolan trehalozy). Protokół immunizacji może być wybrany przez specjalistę w dziedzinie bez nadmiernego eksperymentowania. [0204] Zwierzęta immunizuje się antygenem, immunogennymi koniugatami lub pochodnymi przez połączenie np. 100 µg lub 5 µg albo białka, albo koniugatu (odpowiednio dla królików lub myszy) z 3 objętościami kompletnego adiuwanta Freunda i wstrzykiwanie roztworu śródskórnie w kilku miejscach. Miesiąc później zwierzętom podaje się dawkę przypominającą od 1/5 do 1/10 pierwotnej ilości peptydu lub koniugatu w kompletnym adiuwancie Freunda przez podskórne iniekcje w kilku miejscach. Siedem do czternastu dni później, zwierzętom pobiera się krew i w surowicy oznacza się miano przeciwciał. Zwierzętom podaje się dawki przypominające do plateau miana. Koniugaty można sporządzać w hodowli zrekombinowanych komórek jako białka fuzyjne. Ponadto, czynniki agregujące takie, jak ałun, są odpowiednie do wzmocnienia odpowiedzi immunologicznej. 2) Monoklonalne przeciwciała [0205] Przeciwciała monoklonalne uzyskuje się z populacji zasadniczo homogennych przeciwciał, tj. poszczególne przeciwciała tworzące populację są identyczne, z wyjątkiem możliwych naturalnie występujących mutacji i/lub modyfikacji posttranslacyjnych (np. izomeryzacje, amidowania), które mogą być obecne w niewielkich ilościach. Tak więc, modyfikator monoklonalne wskazuje na charakter przeciwciała, jako nie będącego mieszaniną różnych przeciwciał. [0206] Na przykład, przeciwciała monoklonalne można otrzymywać stosując metodę hybrydom, po raz pierwszy opisaną przez Kohler i wsp., Nature, 256:495 (1975), lub mogą być wytworzone metodami rekombinacji DNA (Patent US Nr 4,816,567). [0207] W metodzie hybrydom mysz lub inne odpowiednie zwierzę gospodarza takie, jak chomik, immunizuje się w opisany powyżej sposób w celu pobudzenia limfocytów, które
63 62- wytwarzają lub są zdolne do produkcji przeciwciał, które będą specyficznie wiązać się z białkiem zastosowanym do immunizacji. Alternatywnie, limfocyty można immunizować in vitro. Limfocyty są następnie poddawane fuzji z komórkami szpiczaka, stosując odpowiedni czynnik indukujący fuzję taki, jak glikol polietylenowy, tworząc komórki hybrydoma (Goding, Monoclonal Antibodies: Principles and Practice, strony (Academic Press, 1986). [0208] Czynnik immunizujący będzie typowo zawierać białko antygenowe lub jego wariant fuzyjny. Ogólnie albo stosuje się limfocyty krwi obwodowej ( PBL ), jeżeli pożądane są komórki pochodzenia ludzkiego, albo stosowane są komórki śledziony lub węzłów komórki chłonnych, gdy pożądane są źródła ssaków innych niż człowiek. Limfocyty są następnie poddawane fuzji z unieśmiertelnioną linią komórkową, stosując odpowiedni czynnik indukujący fuzję taki, jak glikol polietylenowy, tworząc komórki hybrydoma. Goding, Monoclonal Antibodies: Principles and Practice, Academic Press (1986), strony [0209] Można też stosować unieśmiertelnione linie komórkowe zazwyczaj transformowanych komórek ssaczych, szczególnie komórki szpiczaka pochodzące od gryzoni, bydła i pochodzenia ludzkiego. Zazwyczaj, wykorzystywane są szczurze lub mysie linie komórkowe szpiczaka. W ten sposób przygotowane komórki hybrydom wysiewa się i hoduje w odpowiedniej pożywce hodowlanej, która korzystnie zawiera jedną lub więcej substancji hamujących wzrost lub przeżycie nie poddanych fuzji, macierzystych komórek szpiczaka. Na przykład, jeśli macierzyste komórki szpiczaka pozbawione są enzymu fosforybozylotransferazy guaninohipoksantynowej (HGPRT lub HPRT), pożywka hodowlana dla hybrydom będzie typowo zawierać hipoksantynę, aminopterynę i tymidynę (podłoże HAT), które to substancje zapobiegają wzrostowi HGPRT niedoborem komórek. [0210] Korzystne unieśmiertelnione komórki szpiczaka są tymi, które wydajnie ulegają fuzji, podtrzymują stabilny, wysoki poziom produkcji przeciwciała przez wyselekcjonowane komórki wytwarzające przeciwciała i są wrażliwe na pożywkę taką, jak pożywka HAT. Spośród nich, korzystne są mysie linie szpiczaka takie, jak te pochodzące z guzów mysich MOPC-21 i MPC-11, dostępne z Salk Institute Cell Distribution Center, San Diego, California USA, i komórki SP-2 (oraz ich pochodne, np.,x63-ag8-653) dostępne z American Type Culture Collection, Manassas, Virginia USA. Linie komórkowe szpiczaka ludzkiego i mysio-ludzkiego heteroszpiczaka opisano uprzednio dla celów wytwarzania ludzkich przeciwciał monoklonalnych (Kozbor, J. Immunol., 133:3001 (1984); Brodeur i wsp., Monoclonal Antibody Production Techniques and Applications, strony (Marcel Decker, Inc., New York, 1987)). [0211] W pożywce hodowlanej, w której hodowano komórki hybrydom, oznacza się wytwarzanie przeciwciał monoklonalnych skierowanych przeciw antygenowi. Korzystnie, specyficzność wiązania przeciwciał monoklonalnych wytwarzanych przez komórki hybrydoma, jest określona przez immunoprecypitację lub przez oznaczenie wiązania in vitro, jak testem radioimmunologicznym (RIA) lub enzymatycznym testem immunosorpcyjnym (ELISA).
64 63- [0212] Pożywkę hodowlaną, w której hoduje się komórki hybrydoma, można testować na obecność przeciwciał monoklonalnych skierowanych przeciwko pożądanemu antygenowi. Korzystnie, powinowactwo wiązania i specyficzność przeciwciała monoklonalnego można określić przez immunoprecypitację lub przez oznaczenie wiązania in vitro, jak testem radioimmunologicznym (RIA) lub enzymatycznym testem immunosorpcyjnym (ELISA). Takie techniki i testy są znane w tej dziedzinie. Na przykład, powinowactwo wiązania można określić na podstawie analizy Scatcharda z Munson i wsp., Anal. Biochem., 107:220 (1980). [0213] Po zidentyfikowaniu komórek hybrydom, które wytwarzają przeciwciała o pożądanej specyficzności, powinowactwie i/lub aktywności, klony można subklonować zgodnie z procedurą kolejnych rozcieńczeń i hodować standardowymi metodami (Goding, supra). Pożywki hodowlane odpowiednie do tego celu obejmują, na przykład, pożywkę D-MEM lub RPMI Ponadto, komórki hybrydom można hodować in vivo jako nowotwory u ssaka. [0214] Przeciwciała monoklonalne wydzielane przez subklony odpowiednio wyodrębnia się z pożywki hodowlanej, płynu wysiękowego lub surowicy zgodnie z konwencjonalnymi procedurami oczyszczania immunoglobulin, jak, na przykład, białko A-sefaroza, chromatografia na hydroksyapatycie, elektroforeza żelowa, dializa lub chromatografia powinowactwa. [0215] Przeciwciała monoklonalne można również wytwarzać metodami rekombinacji DNA takimi, jak opisanymi w Patent US Nr 4,816,567 i jak opisano powyżej, DNA kodujące przeciwciała monoklonalne łatwo izoluje się i sekwencjonuje stosując konwencjonalne procedury (np. przez zastosowanie sond oligonukleotydowych, które są zdolne do specyficznego wiązania z genami kodującymi ciężkie i lekkie łańcuchy mysich przeciwciał). Komórki hybrydoma służą jako korzystne źródło takiego DNA. Po wyizolowaniu, DNA można umieszczać w wektorach ekspresyjnych, które następnie transfekuje się do komórek gospodarza, takich, jak komórki E. coli, małpie komórki COS, komórki jajnika chomika chińskiego (CHO) lub komórki szpiczaka, które inaczej nie wytwarzają białka immunoglobuliny, w celu syntezy przeciwciał monoklonalnych w rekombinowanych komórkach gospodarza, takiego. Artykuły przeglądowe dotyczące rekombinacyjnej ekspresji w bakteriach DNA kodującego przeciwciała obejmują Skerra i wsp., Curr. Opinion in Immunol., 5: (1993) i Plückthun, Immunol. Revs. 130: (1992). [0216] W dalszym przykładzie wykonania, przeciwciała można wyizolować z fagowych bibliotek przeciwciał, utworzonych z zastosowaniem technik opisanych w McCafferty i wsp., Nature, 348: (1990). Clackson i wsp., Nature, 352: (1991) i Marks i wsp., J. Mol. Biol., 222: (1991) opisują odpowiednio izolowanie przeciwciał mysich i ludzkich, z zastosowaniem bibliotek fagowych. W późniejszych publikacjach opisano wytwarzanie ludzkich przeciwciał o wysokim powinowactwie (zakres nm) przez tasowanie łańcuchów (Marks i wsp., Bio/Technology, 10: (1992)), jak również kombinatoryczne zakażenie i rekombinacja in vivo jako strategię konstruowania bardzo dużych bibliotek fagowych (Waterhouse i wsp., Nucl. Acids Res., 21: (1993)). A zatem, techniki te stanowią dobre alternatywy tradycyjnych technik hybrydom dla przeciwciał monoklonalnych do izolowania przeciwciał monoklonalnych.
65 64- [0217] DNA można również modyfikować, na przykład przez podstawienie sekwencji kodującej ludzkich łańcuchów ciężkich i lekkich łańcuchów domen stałych w miejsce homologicznych sekwencji mysich (Patent US Nr 4,816,567; Morrison, i wsp., Proc. Natl Acad. Sci. USA, 81:6851 (1984)), lub poprzez kowalencyjne połączenie z sekwencją kodującą immunoglobulinę całej lub części sekwencji kodującej dla polipeptydu nie będącego immunoglobuliną. Zazwyczaj takie polipeptydy niebędące immunoglobulinami podstawia się za stałe domeny przeciwciała, bądź podstawia się je za domeny zmienne jednego miejsca wiążącego antygen przeciwciała, do wytworzenia chimerycznego przeciwciała dwuwartościowego, zawierającego jedno miejsce łączące antygen o specyficzności dla antygenu i drugie miejsce wiążące antygen wykazujące specyficzność wobec innego antygenu. [0218] Monoklonalne przeciwciała opisane w niniejszym dokumencie mogą być jednowartościowe, których wytwarzanie jest dobrze znane w tej dziedzinie. Na przykład, jeden sposób obejmuje rekombinacyjną ekspresję łańcucha lekkiego immunoglobuliny i zmodyfikowanego łańcucha ciężkiego. Łańcuch ciężki jest obcinany na ogół w dowolnym punkcie w regionie Fc w celu zapobieżenia sieciowaniu łańcucha ciężkiego. Alternatywnie, odpowiednie reszty cysteinowe mogą być podstawione inną resztą aminokwasową i są usuwane w taki sposób, aby uniknąć usieciowania. Metody in vitro są również odpowiednie do wytwarzania przeciwciał jednowartościowych. Trawienie przeciwciał w celu wytworzenia ich fragmentów, szczególnie fragmentów Fab, można wykonać przy użyciu rutynowych technik znanych w dziedzinie. [0219] Chimeryczne lub hybrydowe przeciwciała można również wytwarzać in vitro, przy zastosowaniu znanych sposobów chemii syntezy białek, w tym tych, które wykorzystują środki sieciujące. Na przykład immunotoksyny można konstruować przy pomocy reakcji wymiany dwusiarczków lub poprzez utworzenie wiązania tioeterowego. Przykłady odczynników odpowiednich do tego celu obejmują iminotiolan i 4-merkaptoiminomaślan metylu. 3) Humanizowane przeciwciała. [0220] Przeciwciała według wynalazku mogą zawierać ponadto przeciwciała humanizowane lub ludzkie. Humanizowane postacie przeciwciał innych niż ludzkie (np. mysich) to chimeryczne immunoglobuliny, łańcuchy immunoglobulin lub ich fragmenty (jak Fv, Fab, Fab, F(ab ) 2 lub inne wiążące antygen subsekwencje przeciwciał), które zawierają minimalną sekwencję pochodzącą z nie-ludzkiej immunoglobuliny. Humanizowane przeciwciała zawierają ludzkie immunoglobuliny (przeciwciało biorcy), w których reszty z regionu determinującego komplementarność (CDR) biorcy są zastąpione przez reszty z CDR gatunku innego niż człowiek (przeciwciało dawcy) takiego, jak mysz, szczur lub królik, posiadające pożądaną specyficzność, powinowactwo i zdolność. W niektórych przypadkach, reszty zrębowe Fv ludzkiej immunoglobuliny są zastępowane odpowiednimi resztami pochodzenia innego niż od człowieka. Przeciwciała humanizowane mogą także obejmować reszty, które nie występują ani w przeciwciele biorcy ani w importowanych CDR lub sekwencjach zrębowych. Na ogół, humanizowane przeciwciało będzie zawierać zasadniczo wszystkie z co
66 65- najmniej jednej, a typowo dwóch, domen zmiennych, w których wszystkie lub zasadniczo wszystkie regiony CDR odpowiadają tym z nie-ludzkiej immunoglobuliny oraz wszystkie lub zasadniczo wszystkie regiony FR są tymi o sekwencji immunoglobuliny ludzkiej konsensusowej. Humanizowane przeciwciało optymalnie będzie również zawierać co najmniej część regionu stałego immunoglobuliny (Fc), typowo z ludzkiej immunoglobuliny. Jones i wsp., Nature 321: (1986); Riechmann i wsp., Nature 332: (1988) i Presta, Curr. Opin. Struct. Biol. 2: (1992). [0221] Sposoby humanizowania nie-ludzkich przeciwciał są dobrze znane w tej dziedzinie. Ogólnie, humanizowane przeciwciało ma jedną lub więcej reszt aminokwasowych wprowadzonych do niego ze źródła innego niż człowiek. Te nie-ludzkie reszty aminokwasowe są często określane jako reszty importowane, które zazwyczaj uzyskuje się z importowej domeny zmiennej. Humanizację można zasadniczo prowadzić zgodnie z metodą opisaną przez Winter i współpracowników, Jones i wsp., Nature 321: (1986); Riechmann i wsp., Nature 332: (1988); Verhoeyen i wsp., Science 239: (1988), lub przez wymienienie CDR gryzonia lub sekwencji CDR na odpowiadające sekwencje przeciwciała ludzkiego. Zgodnie z tym, takie humanizowane przeciwciała są przeciwciałami chimerycznymi (Patent US Nr 4,816,567), w których zasadniczo mniej niż nienaruszona ludzka domena zmienna została zastąpiona odpowiadającą jej sekwencją pochodzącą od gatunku innego niż człowiek. W praktyce, humanizowane przeciwciała są typowo ludzkimi przeciwciałami, w których niektóre reszty CDR i ewentualnie niektóre reszty FR podstawiono resztami z analogicznych miejsc w przeciwciałach gryzoni. [0222] Wybór ludzkich domen zmiennych, zarówno lekkich, jak i ciężkich, do stosowania w przygotowywaniu przeciwciał humanizowanych, jest bardzo ważny dla obniżania antygenowości. Zgodnie z tak zwaną metodą najlepszego dopasowania, sekwencję domeny zmiennej przeciwciała gryzonia stosuje się do przeszukiwania całej biblioteki znanych ludzkich sekwencji domen zmiennych. Ludzką sekwencję, która jest najbardziej podobna do tej gryzonia, akceptuje się następnie jako ludzki zrąb (FR) przeciwciała humanizowanego. Sims i wsp., J. Immunol., 151:2296 (1993); Chothia i wsp., J. Mol. Biol., 196:901 (1987). W innych sposobach wykorzystuje się szczególny zrąb pochodzący z sekwencji konsensusowej wszystkich ludzkich przeciwciał z określonej podgrupy łańcuchów lekkich lub ciężkich. Wspomniany zrąb można stosować dla kilku różnych przeciwciał humanizowanych. Carter i wsp., Proc. Natl. Acad. Sci. USA, 89:4285 (1992); Presta i wsp., J. Immunol., 151:2623 (1993). [0223] Ważne jest ponadto, aby przeciwciała były humanizowane z zachowaniem wysokiego powinowactwa do antygenu oraz innych korzystnych właściwości biologicznych. Dla osiągnięcia tego celu, według korzystnego sposobu, przeciwciała humanizowane przygotowuje się dokonując analizy sekwencji macierzystych i różnych teoretycznych produktów humanizowanych z zastosowaniem trójwymiarowych modeli macierzystej i humanizowanej sekwencji. Trójwymiarowe modele immunoglobulin są powszechnie dostępne i są znane specjalistom w tej dziedzinie. Dostępne są programy komputerowe, które ilustrują i prezentują prawdopodobne trójwymiarowe struktury konformacyjne wybranych
67 66- kandydatów sekwencji immunoglobulin. Analiza tych struktur umożliwia analizę prawdopodobnej roli reszt w działaniu potencjalnej sekwencji immunoglobuliny, tj. analizę reszt, które wpływają na zdolność potencjalnej immunoglobuliny do wiązania jej antygenu. W ten sposób można wybrać reszty FR i połączyć z sekwencjami biorcy i importowanymi tak, że osiąga się pożądane cechy przeciwciała, jak zwiększone powinowactwo do docelowych antygenu (antygenów). Ogólnie, reszty CDR bezpośrednio i w największym stopniu uczestniczą we wpływaniu na wiązanie antygenu. [0224] Rozważa się różne postacie przeciwciała humanizowanego. Na przykład, humanizowane przeciwciało może być fragmentem przeciwciała takim, jak Fab, który jest opcjonalnie sprzężony z jednym lub większą liczbą środków cytotoksycznych, w celu wygenerowania immunokoniugatu. Alternatywnie, humanizowane przeciwciało może być nienaruszonym przeciwciałem takim, jak nienaruszone przeciwciało lgg1. 4) Ludzkie przeciwciała [0225] Jako alternatywę do humanizacji, można generować przeciwciała ludzkie. Na przykład, obecnie możliwe jest wytwarzanie zwierząt transgenicznych (np. myszy), które są zdolne, po immunizacji, do wytwarzania pełnego repertuaru przeciwciał ludzkich, przy braku endogennej produkcji immunoglobulin. Na przykład, opisano, że homozygotyczna delecja genu łańcucha ciężkiego przeciwciała genu regionu łączącego (J H ) u chimerycznych i germinalnych mutantów myszy skutkuje całkowitym zahamowaniem produkcji endogennych przeciwciał. Transfer ludzkiego germinalnego układu genów immunoglobulin u takich myszy o zmutowanej linii płciowej będzie powodować wytwarzanie przeciwciał ludzkich po stymulacji antygenem. Patrz np. Jakobovits i wsp., Proc. Natl. Acad. Sci. USA, 90:2551 (1993); Jakobovits i wsp., Nature, 362: (1993); Bruggermann i wsp., Year w Immuno., 7:33 (1993); Patenty US Nr 5,591,669 i WO 97/ [0226] Alternatywnie, można stosować technologię prezentacji fagowej do wytwarzania ludzkich przeciwciał i fragmentów przeciwciał in vitro z repertuaru genów domeny zmiennej (V) immunoglobuliny od nieimmunizowanych dawców. McCafferty i wsp., Nature 348: (1990); Hoogenboom i Winter, J. Mol. Biol. 227: 381 (1991). Zgodnie z tą techniką, geny domen V przeciwciał klonuje się w jednej ramce odczytu z genem większego lub mniejszego białka płaszcza bakteriofaga nitkowatego takiego, jak M13 lub fd, i prezentuje jako funkcjonalne fragmenty przeciwciała na powierzchni cząstki faga. Ponieważ włókienkowata cząstka zawiera jednoniciową kopię DNA genomu faga, wynikiem selekcji opartej na funkcjonalnych właściwościach przeciwciała będzie również selekcja genu kodującego przeciwciało wykazujące te właściwości. Tak więc, fag naśladuje pewne właściwości komórki B. Prezentację na fagu można prowadzić w wielu formatach, których przegląd można znaleźć w np., Johnson, Kevin S. i Chiswell, David J., Curr. Opin Struct. Biol. 3: (1993). Kilka źródeł segmentów genu V można stosować do prezentacji fagowej. Clackson i wsp., Nature 352: (1991) wyizolowali różne przeciwciała skierowane przeciw oksazolonowi z małej losowej kombinatorycznej biblioteki genów V pochodzących ze śledzion immunizowanych myszy. Można skonstruować repertuar genów V z nieimmunizowanych ludzkich dawców i przeciwciała skierowane wobec szeregu rozmaitych
68 67- antygenów (włącznie z autoantygenami) można wyizolować zasadniczo zgodnie z technikami opisanymi Marks i wsp., J. Mol. Biol. 222: (1991) lub Griffith i wsp., EMBO J. 12: (1993). Patrz także Patent Us Nry 5,565,332 i 5,573,905. [0227] Techniki Cole i wsp., i Boerner i wsp., dostępne są również do wytwarzania ludzkich przeciwciał monoklonalnych (Cole i wsp., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985) i Boerner i wsp., J. Immunol. 147(1): (1991). Podobnie, ludzkie przeciwciała mogą być wykonane przez wprowadzenie loci ludzkiej immunoglobuliny do zwierząt transgenicznych, np. myszy, w których endogenne geny immunoglobulin zostały częściowo lub całkowicie inaktywowane. Po prowokacji, obserwuje się produkcję ludzkich przeciwciał, która bardzo przypomina tą obserwowaną u ludzi we wszystkich aspektach, w tym rearanżacją genów, montowaniem i repertuarem przeciwciał. To podejście opisano, na przykład, w Patenty US Nr 5,545,807; 5,545,806, 5,569,825, 5,625,126, 5,633,425, 5,661,016 i w następujących publikacjach naukowych: Marks i wsp., Bio/Technology 10: (1992); Lonberg i wsp., Nature 368: (1994); Morrison, Nature 368: (1994), Fishwild i wsp., Nature Biotechnology 14: (1996), Neuberger, Nature Biotechnology 14: 826 (1996) i Lonberg i Huszar, Intern. Rev. Immunol. 13: (1995). [0228] Wreszcie, ludzkie przeciwciała mogą również być wytwarzane przez aktywowane in vitro komórki B (patrz Patent US Nos 5,567,610 i 5,229,275). 5) Fragmenty Przeciwciał [0229] W pewnych okolicznościach korzystniejsze jest na zastosowanie fragmentów przeciwciał niż całych przeciwciał. Mniejsze rozmiary fragmentów pozwalają na szybki klirens i mogą prowadzić do lepszego dostępu do guzów litych. [0230] Różne techniki zostały opracowane do produkcji fragmentów przeciwciał. Tradycyjnie, fragmenty te otrzymywano poprzez trawienie proteolityczne nienaruszonych przeciwciał (patrz np. Morimoto i wsp., J Biochem Biophys. Method. 24: (1992); i Brennan i wsp., Science 229:81 (1985)). Jednakże, obecnie fragmenty te mogą być bezpośrednio wytwarzane przez zrekombinowane komórki gospodarza. Fragmenty przeciwciał Fab, Fv i scfv mogą ulegać ekspresji i wydzielaniu przez E. coli, umożliwiając w ten sposób łatwą produkcję dużych ilości tych fragmentów. Fragmenty przeciwciał można izolować z fagowych bibliotek przeciwciał omówionych powyżej. Alternatywnie, fragmenty Fab -SH mogą być bezpośrednio odzyskiwane z E. coli i chemicznie sprzęgane z utworzeniem fragmentów F(ab ) 2 (Carter i wsp., Bio/Technology 10: (1992)). Zgodnie z innym podejściem, fragmenty F(ab ) 2 można izolować bezpośrednio z hodowli rekombinowanych komórek gospodarza. Fab i F(ab ) 2 ze wzrostem okresem półtrwania in vivo jest opisany w patencie US Nr 5,869,046. W innych przykładach wykonania, przeciwciałem z wyboru jest jednołańcuchowy fragment Fv (scfv). Patrz WO 93/16185; Patent US Nr 5,571,894 i Patent US Nr 5,587,458. Fragment przeciwciała może być również przeciwciałem liniowym, na przykład, jak opisanym w Patencie US 5,641,870. Takie liniowe fragmenty przeciwciał mogą być monospecyficzne lub bispecyficzne.
69 68-6) Zależna od Przeciwciał Mediowana Enzymem Terapia Prolekiem (ang. Antibody Dependent Enzyme-Mediated Prodrug Therapy, ADEPT) [0231] Przeciwciała według niniejszego wynalazku można również stosować w ADEPT, przez koniugowanie przeciwciała do enzymu aktywującego prolek, który przekształca prolek (na przykład peptydowy środek chemioterapeutyczny, patrz WO 81/01145) do aktywnego leku przeciwnowotworowego. Patrz na przykład WO 88/07378 i Patent US Nr 4,975,278. [0232] Enzymatyczna składowa immunokoniugatu użytecznego w ADEPT obejmuje jakikolwiek enzym zdolny do działania na prolek w taki sposób, aby przekształcić go do jego bardziej aktywnej, cytotoksycznej formy. [0233] Enzymy, które są użyteczne w sposobie według tego wynalazku obejmują, bez ograniczania do wymienionych, glikozydazę, oksydazę glukozy, lizozym ludzki, ludzkie glukuronidazy, fosfatazę alkaliczną, użyteczną do przekształcania proleków zawierających fosforan w wolne leki; arylosulfatazę użyteczną do przekształcania proleków zawierających siarczany do wolnych leków; deaminazę cytozynową użyteczną do przekształcania nietoksycznej 5-fluorocytozyny do przeciwnowotworowego leku 5-fluorouracylu; proteazę taką, jak proteaza Serratia, termolizyna, subtylizyna, karboksypeptydazy (np. karboksypeptydaza G2 i karboksypeptydaza A) i katepsyny. [0234] (takie jak katepsyny B i L), które są przydatne do przekształcania proleków zawierających peptyd w wolne leki; D-alanylokarboksypeptydazy, użyteczne do przekształcania proleków zawierających podstawniki D-aminokwasowe; enzymy rozszczepiające węglowodany takie, jak β-galaktozydaza i neuraminidaza, użyteczne do przekształcania proleków glikozylowanych w wolne leki; β-laktamazy przydatne do przekształcania leków derywatyzowanych β-laktamami w wolne leki; i amidazy penicylinowe takie, jak amidaza penicyliny V lub amidaza penicyliny G, przydatne do przekształcania leków derywatyzowanych przy atomach azotu grup aminowych z grupami fenoksyacetylowymi lub fenyloacetylowymi, odpowiednio, w wolne leki. Alternatywnie, można stosować przeciwciała o aktywności enzymatycznej, znane również w nauce jako abzymy, do konwersji proleków według wynalazku w wolne aktywne leki (patrz np. Massey, Nature 328: (1987)). Koniugaty przeciwciało-abzym można wytwarzać w sposób opisany w niniejszym opisie w celu dostarczania abzymu do populacji komórek nowotworowych. [0235] Powyższe enzymy mogą być kowalencyjnie związane z opisanymi tu polipeptydem lub przeciwciałami przy użyciu technik dobrze znanych w dziedzinie, jak stosowanie heterobifunkcjonalnych czynników sieciujących opisanych powyżej. Alternatywnie, białka fuzyjne zawierające co najmniej region wiążący antygen przeciwciała według wynalazku, połączony z co najmniej funkcjonalnie aktywną częścią enzymu według wynalazku, można konstruować z zastosowaniem technik rekombinacji DNA, dobrze znanych w tej dziedzinie techniki (patrz np. Neuberger i wsp., Nature 312: (1984)). 7) Bispecyficzne i polispecyficzne przeciwciała
70 69- [0236] Bispecyficzne przeciwciała (BsAb) są przeciwciałami, które posiadają specyficzności wiązania wobec co najmniej dwóch różnych epitopów, w tym na tym samym lub innym białku. Alternatywnie, ramię może zostać uzbrojone do wiązania docelowego antygenu, a drugie ramię może być połączone z ramieniem, które wiąże się z pobudzającą cząsteczką na leukocycie taką, jak cząsteczka receptora komórek T (np. CD3) lub receptorami Fc dla IgG (FcγR) takimi, jak FcγR1 (CD64), FcγRII (CD32) i FcγRIII (CD 16) tak, aby skoncentrować i zlokalizować mechanizmy obrony komórkowej na komórkach z ekspresją antygenu docelowego. Przeciwciała takie można uzyskać z pełnej długości przeciwciał lub fragmentów przeciwciał (np. przeciwciała bispecyficzne F(ab ) 2 ). [0237] Przeciwciała bispecyficzne można również stosować do dostarczania czynników cytotoksycznych do komórek, w których zachodzi ekspresja antygenu docelowego. Takie przeciwciała posiadają jedno ramię, które wiąże się z pożądanym antygenem i drugie ramię, które wiąże czynnik cytotoksyczny (np. saporynę, anty-interferon-a, alkaloid barwinka, łańcuch A rycyny, metotreksat lub hapten z izotopem promieniotwórczym). Przykłady znanych bispecyficznych przeciwciał obejmują anty-erbb2/anty-fcgriii (WO 96/16673), anty-erbb2/anty-fcgri (U.S.P. 5,837,234), anty-erbb2/anty-cd3 (U.S.P. 5,821,337). [0238] Sposoby wytwarzania przeciwciał bispecyficznych są znane w stanie techniki. Tradycyjne wytwarzanie bispecyficznych przeciwciał o pełnej długości opiera się na koekspresji dwóch par łańcuch ciężki/łańcuch lekki immunoglobuliny, gdzie oba łańcuchy posiadają różne specyficzności. Millstein i wsp., Nature, 305: (1983). Ze względu na losowy dobór immunoglobulinowych łańcuchów ciężkich i lekkich, te hybrydomy (kwadromy) wytwarzają potencjalną mieszaninę 10 różnych cząsteczek przeciwciał, z których tylko jedna posiada prawidłową strukturę bispecyficzną. Oczyszczanie właściwej cząsteczki, którego zazwyczaj dokonuje się dzięki etapom chromatografii powinowactwa, jest raczej niewygodne, a wydajności produktu są niskie. Podobne procedury są ujawnione w WO 93/08829 oraz w Traunecker i wsp., EMBO J., 10: (1991). [0239] Według innego podejścia, domeny zmienne przeciwciał o pożądanych specyficznościach (miejscach wiązania przeciwciało-antygen) poddaje się fuzji z sekwencjami domen stałych immunoglobulin. Fuzję korzystnie prowadzi się z łańcucha ciężkiego immunoglobuliny, domeny stałej, zawierającej co najmniej część zawiasu, regiony CH2 i CH3. Korzystne jest, aby pierwszy ciężki łańcuch regionu stałego (CH1) zawierający miejsce konieczne do wiązania łańcucha lekkiego, był obecny w co najmniej jednej z fuzji. DNA kodujące fuzje łańcucha ciężkiego immunoglobuliny i, jeśli to pożądane, łańcucha lekkiego immunoglobuliny, wstawia się do oddzielnych wektorów ekspresyjnych i kotransfekuje do odpowiedniego organizmu gospodarza. Zapewnia to dużą elastyczność pod względem doprowadzania wzajemnych proporcji trzech łańcuchów polipeptydowych w rozwiązaniach, w których nierówne stosunki trzech łańcuchów polipeptydowych stosowanych w konstruowaniu zapewniają optymalną wydajność. Jest jednak możliwe wstawienie sekwencji kodujących dwa lub wszystkie trzy łańcuchy polipeptydowe do jednego wektora ekspresyjnego, jeśli wynikiem ekspresji co najmniej dwóch łańcuchów polipeptydowych w równych stosunkach jest wysoka wydajność lub jeśli stosunki nie mają większego znaczenia.
71 70- [0240] W korzystnym przykładzie wykonania tego podejścia, przeciwciała bispecyficzne składają się z hybrydowego łańcucha ciężkiego immunoglobuliny o pierwszej specyficzności wiązania w jednym ramieniu i hybrydowej pary łańcuchów ciężkiego i łańcucha lekkiego immunoglobuliny (zapewniając drugą specyficzność wiązania) w drugim ramieniu. Stwierdzono, że ta asymetryczna struktura ułatwia oddzielanie pożądanego związku bispecyficznego od niepożądanych kombinacji łańcuchów immunoglobulin, ponieważ obecność łańcucha lekkiego immunoglobuliny tylko w jednej połowie bispecyficznej cząsteczki zapewnia łatwy sposób oddzielania. To podejście jest ujawnione w WO 94/ Dla dalszych szczegółów tworzenia przeciwciał bispecyficznych patrz, na przykład, Suresh i wsp., Methods in Enzymology 121: 210 (1986). [0241] Według innego podejścia, opisanego w WO 96/27011 lub U.S.P. 5,731,168, można skonstruować interfejs pomiędzy parą cząsteczek przeciwciał zapewniając maksymalizację procentu heterodimerów, które odzyskuje się z hodowli komórek rekombinowanych. Korzystny interfejs zawiera co najmniej część regionu CH3 domeny stałej przeciwciała. W tej metodzie jeden lub więcej małych łańcuchów bocznych aminokwasu z interfesju pierwszej cząsteczki przeciwciała zastępuje się większymi łańcuchami bocznymi (np. tyrozyny lub tryptofanu). Kompensacyjne wgłębienia o identycznej lub podobnej wielkości z dużym łańcuchem (dużymi łańcuchami) bocznymi są utworzone na interfejsie drugiej cząsteczki przeciwciała poprzez zastąpienie dużych łańcuchów bocznych aminokwasów mniejszymi (np. alaniny lub treoniny). Zapewnia to mechanizm zwiększania wydajności heterodimeru w stosunku do niepożądanych produktów końcowych takich, jak homodimery. [0242] Techniki wytwarzania bispecyficznych przeciwciał z fragmentów przeciwciał zostały opisane w literaturze. Na przykład, przeciwciała bispecyficzne można przygotowywać stosując wiązanie chemiczne. Brennan i wsp., Science 229: 81 (1985) opisują procedurę, w której nienaruszone przeciwciała rozszczepia się proteolitycznie do wytworzenia fragmentów F(ab ) 2. Fragmenty te są redukowane w obecności czynnika kompleksującego ditiole, arseninu sodowego, w celu stabilizacji sąsiednich ditioli i zapobiegania tworzeniu międzycząsteczkowych mostków dwusiarczkowych. Wytworzone fragmenty Fab są następnie przekształcane do pochodnych tionitrobenzoesanu (TNB). Jedna z pochodnych Fab -TNB jest następnie ponownie przekształcana do pochodnej Fab -TNB z utworzeniem przeciwciała bispecyficznego. Utworzone przeciwciała bispecyficzne można stosować jako czynniki do selektywnej immobilizacji enzymów. [0243] Fragmenty Fab można bezpośrednio odzyskiwać z E. coli i chemicznie sprzęgać z utworzeniem przeciwciał bispecyficznych. Shalaby i wsp., J. Exp. Med. 175: (1992) opisuje wytwarzanie w pełni humanizowanego bispecyficznego cząsteczki przeciwciała F(ab ) 2. Każdy z fragmentów Fab jest oddzielnie wydzielany z E. coli i poddawany bezpośredniemu sprzęganiu chemicznemu in vitro w celu wytworzenia przeciwciała bispecyficznego. Tak utworzone przeciwciało bispecyficzne było zdolne do wiązania się z komórkami z nadekspresją receptora ErbB2 i normalnymi ludzkimi komórkami T, jak również wyzwalania aktywności litycznej ludzkich limfocytów cytotoksycznych wobec stanowiących cel ludzkich nowotworów sutka.
72 71- [0244] Opisano różnorodne techniki wytwarzania i izolowania fragmentów przeciwciał dwuwartościowe bezpośrednio z hodowli komórek rekombinowanych. Na przykład, dwuwartościowe heterodimery zostały wyprodukowane przy zastosowaniu suwaków leucynowych. Kostelny i wsp., J. Immunol., 148(5): (1992). Peptydy suwaka leucynowego z białek Fos i Jun połączono z częściami Fab dwóch różnych przeciwciał przez fuzję genów. Homodimery przeciwciał redukowano w regionie zawiasowym z utworzeniem monomerów, a następnie ponownie utleniano tworząc heterodimery przeciwciał. Technika diaciał opisana w Hollinger i wsp., Proc. Natl. Acad. Sci. USA, 90: (1993) dostarczyła alternatywnego mechanizmu do wytwarzania fragmentów przeciwciało bispecyficzne/dwuwartościowe. Fragmenty zawierają domenę zmienną łańcucha ciężkiego (V H ) połączoną z domeną zmienną łańcucha lekkiego (V L ) przez łącznik, który jest zbyt krótki, aby umożliwiał parowanie pomiędzy dwiema domenami na tym samym łańcuchu. Zgodnie z tym, domeny V H i V L jednego fragmentu są zmuszone do parowania z komplementarnymi domenami V L i V H innego fragmentu, tworząc w ten sposób dwa miejsca wiązania antygenu. Opisano także inną strategię wytwarzania fragmentów przeciwciał bispecyficznych/dwuwartościowych przez zastosowanie pojedynczego łańcucha Fv (sfv) dimerów opisywano. Patrz Gruber i wsp., J. Immunol., 152:5368 (1994). [0245] Rozważane są przeciwciała o więcej niż dwóch wartościowościach. Na przykład, można przygotować przeciwciała trójspecyficzne. Tutt i wsp., J. Immunol. 147: 60 (1991). [0246] Przykładowe bispecyficzne przeciwciała mogą wiązać się z dwoma różnymi epitopami na danej cząsteczki. Alternatywnie, ramię anty-białko może być połączone z ramieniem, które wiąże się z pobudzającą cząsteczką na leukocycie taką, jak cząsteczka receptora komórek T (np. CD2, CD3, CD28 lub B7), lub receptory dla Fc IgG (FcγR) takie, jak FcγRI (CD64), FcγRII (CD32) i FcγRIII (CD 16) tak, aby skoncentrować mechanizmy obrony komórkowej na komórkach eksprymujących określone białka. Przeciwciała bispecyficzne można również stosować do dostarczania czynników cytotoksycznych do komórek, w których zachodzi ekspresja konkretnego białka. Przeciwciała te mają ramię wiążące białka oraz ramię, które wiąże środek cytotoksyczny lub chelator radionuklidu taki, jak EOTUBE, DPTA, DOTA lub TETA. Kolejnym przedmiotem zainteresowania przeciwciało bispecyficzne wiąże białko i ponadto wiąże czynnik tkankowy (TF). 8) Przeciwciała wielowartościowe [0247] Przeciwciało wielowartościowe może być internalizowane (i/lub katabolizowane) szybciej niż przeciwciało dwuwartościowe przez komórkę wykazującą ekspresję antygenu, do którego wiąże się to przeciwciało. Przeciwciała według niniejszego wynalazku mogą być przeciwciałami wielowartościowymi (innymi niż klasa IgM) z trzema lub większą liczbą miejsc wiązania antygenu (np. przeciwciała czterowartościowe), które mogą być łatwo produkowane poprzez rekombinowaną ekspresję kwasów nukleinowych kodujących łańcuchy polipeptydowe przeciwciała. Wielowartościowe przeciwciała mogą zawierać domenę dimeryzacyjną i trzy lub więcej miejsc wiązania antygenu. Korzystna domena dimeryzacyjna zawiera (lub składa się z) regionu Fc lub regionu zawiasowego. W tym scenariuszu, przeciwciało będzie obejmować region Fc i trzy lub więcej miejsc wiązania antygenu na
73 72- końcu aminowym regionu Fc. Korzystne tu wielowartościowe przeciwciało obejmuje (lub składa się z) trzech do około ośmiu, ale korzystnie czterech, miejsc wiązania antygenu. Przeciwciało wielowartościowe zawiera co najmniej jeden łańcuch polipeptydowy (i korzystnie dwa łańcuchy polipeptydowe), przy czym łańcuch polipeptydowy (łańcuchy polipeptydowe) zawierają dwie lub więcej domen zmiennych. Na przykład, łańcuch polipeptydowy (łańcuchy polipeptydowe) może zawierać VD1-(X1) n -VD2-(X2) n -Fc, w którym VD1 jest pierwszą domeną zmienną, VD2 jest drugą domeną zmienną, Fc jest jednym łańcuchem polipeptydowym regionu Fc, X1 i X2 reprezentują aminokwas lub polipeptyd, a n wynosi 0 lub 1. Na przykład, łańcuch polipeptydowy (łańcuchy polipeptydowe) mogą zawierać: VH-CH1-elastyczny łącznik-vh-ch1-łańcuch regionu Fc; lub H-CH1-VH-CH1- łańuch regionu Fc. Wielowartościowe przeciwciało obejmuje tu korzystnie dodatkowo co najmniej dwa (a korzystnie cztery) polipeptydy domeny zmiennej łańcucha lekkiego. Wielowartościowe przeciwciała w niniejszym dokumencie mogą, na przykład, zawierać od około dwóch do około ośmiu polipeptydów domeny zmiennej łańcucha lekkiego. Te rozważane tutaj polipeptydy domeny zmiennej łańcucha lekkiego obejmują domenę zmienną łańcucha lekkiego oraz, opcjonalnie, dodatkowo zawierają domenę CL. 9) Przeciwciała heterokoniugatowe [0248] Heterokoniugaty przeciwciał są także objęte zakresem niniejszego wynalazku. Heterokoniugaty przeciwciał są złożone z dwóch kowalencyjnie połączonych przeciwciał. Na przykład, jedno przeciwciało w heterokoniugacie może być sprzężone z awidyną, a drugie z biotyną. Takie przeciwciała, na przykład, zaproponowano dla celowania komórkami układu immunologicznego w niepożądane komórki, U.S.P. 4,676,980, i do leczenia zakażeń HIV. WO 91/00360, WO 92/ i EP Uważa się, że przeciwciała można wytwarzać in vitro, przy zastosowaniu znanych sposobów chemii syntezy białek, w tym te, które dotyczą środków sieciujących. Na przykład immunotoksyny można konstruować przy pomocy reakcji wymiany mostków dwusiarczkowych lub poprzez utworzenie wiązania tioeterowego. Przykłady odczynników odpowiednich do tego celu obejmują iminotiolan i 4- merkaptoiminomaślan metylu i te ujawnione, na przykład, w opisie patentowym US Nr 4,676,980. Przeciwciała heterokoniugatowe mogą być wytwarzane za pomocą jakiejkolwiek dogodnej metody sieciowania. Odpowiednie czynniki sieciujące są dobrze znane w dziedzinie i są ujawnione w patencie US Nr 4,676,980, wraz z szeregiem technik sieciowania. 10) Modyfikacja metodami inżynierii genetycznej funkcji efektorowych [0249] Może być pożądane modyfikowanie przeciwciała według wynalazku pod względem funkcji efektorowej, np. w celu zwiększenia cytotoksyczności komórkowej zależnej od antygenu (ADCC) i/lub cytotoksyczności zależnej od dopełniacza (CDC) przeciwciała. Można to uzyskać przez wprowadzenie jednej lub więcej substytucji aminokwasowych w regionie Fc przeciwciała. Alternatywnie lub dodatkowo, reszta (reszty) cysteiny może być wprowadzona do regionu Fc, umożliwiając w ten sposób tworzenie międzyłańcuchowych wiązań dwusiarczkowych w tym regionie. Tak utworzone przeciwciało homodimeryczne może mieć poprawioną zdolność internalizacji i/lub zwiększone zabijanie komórek z udziałem dopełniacza i zależnej od przeciwciał cytotoksyczności komórkowej (ADCC). Patrz
74 73- Caron i wsp., J. Exp Med. 176: (1992) i Shopes, B. J. Immunol. 148: (1992). Homodimeryczne przeciwciała o wzmocnionej aktywności przeciwnowotworowej można również przygotowywać stosując heterobifunkcjonalne czynniki sieciujące, jak opisano w Wolff i wsp., Cancer Research 53: (1993). Alternatywnie, można skonstruować przeciwciało, które posiada podwójne regiony Fc i dlatego może posiadać wzmocnione możliwości zależnej od dopełniacza lizy i ADCC. Patrz Stevenson i wsp., Anti- Cancer Drug Design 3: (1989). [0250] W celu zwiększenia okresu półtrwania w surowicy do przeciwciała można na przykład wprowadzić epitop wiążący receptor ratunkowy w przeciwciele (zwłaszcza fragment przeciwciała) jak opisano w patencie US 5,739,277. W stosowanym tu kontekście, określenie epitop wiążący receptor ratunkowy odnosi się do epitopu regionu Fc cząsteczki IgG (np. IgG 1, IgG 2, IgG 3, lub IgG 4 ), który jest odpowiedzialny za zwiększenie in vivo okresu półtrwania w surowicy cząsteczki IgG. 11) Immunokoniugaty [0251] Wynalazek dotyczy również immunokoniugatów lub koniugatów przeciwciało-lek (ADC), zawierających przeciwciało skoniugowane z czynnikiem cytotoksycznym, takim, jak czynnik chemioterapeutyczny, toksyna (np. enzymatycznie aktywna toksyna pochodzenia bakteryjnego, grzybowego, roślinnego lub zwierzęcego lub jej fragmenty), albo radioaktywny izotop (tj. radiokoniugat). Takie ADC musi pokazać akceptowalny profil bezpieczeństwa. [0252] Zastosowanie ADC do lokalnego dostarczania czynników cytotoksycznych lub cytostatycznych, np. leków do niszczenia lub hamowania komórek nowotworowych w leczeniu nowotworu [Syrigos i Epenetos, Anticancer Research 19: (1999); Niculeascu-Duvaz i Springer, Adv. Drug Del. Rev. 26: (1997); US 4,975,278] teoretycznie pozwala na celowane dostarczanie ugrupowania leku do nowotworów, i wewnątrzkomórkową akumulację, tam, gdzie podawanie układowe tych nieskoniugowanych leków może skutkować niedopuszczalnymi poziomami toksyczności dla prawidłowych komórek, jak też komórek guza, które starano się wyeliminować (Baldwin i wsp., Lancet, (1986); Thorpe, (1985) Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review, in Monoclonal Antibodies 84: Biological And Clinical Applications, A. Pinchera i wsp. (eds), pp ). Poszukuje się w ten sposób maksymalnej skuteczności przy minimalnej toksyczności. Zarówno przeciwciała poliklonalne i przeciwciała monoklonalne opisano jako użyteczne w tych strategiach (Rowland i wsp., Cancer Immunol. Immunother. 21: (1986)). Leki stosowane w tych metodach obejmują daunomycynę, doksorubicynę, metotreksat i windezynę. Toksyny stosowane w koniugatach przeciwciało-toksyna obejmują toksyny bakteryjne, jak toksyna błonicy, toksyny roślinne, jak rycyna, cząsteczki toksyny, jak geldanamycyna (Mandler i wsp. J. Nat. Cancer Inst. 92(19): (2000); Mandler i wsp., Bioorganic & Med. Chem. Letters 10: (2000); Mandler i wsp. Bioconjugate Chem. 13: (2002)), majtansynoidy (EP ; Liu i wsp., Proc. Natl. Acad. Sci. USA 93: (1996)) i kalicheamycynę (Lode i wsp., Cancer Res. 58:2928 (1998); i Hinman i wsp., Cancer Res. 53: (1993)). Toksyny mogą wywierać ich efekty cytotoksyczne i cytostatyczne, poprzez mechanizmy obejmujące wiązanie, wiązanie DNA lub inhibicję
75 74- topoizomerazy tubuliny. Niektóre leki cytotoksyczne wydają się być nieaktywne lub mniej aktywne, kiedy są sprzężone z dużymi przeciwciałami lub białkami receptorów ligandów. [0253] Środki chemioterapeutyczne użyteczne w tworzeniu takich immunokoniugatów opisano powyżej, obejmują BCNU, streptozoicynę, winkrystynę, winblastynę, adriamycynę i 5-fluorouracyl. [0254] Enzymatycznie aktywne toksyny i ich fragmenty, które można stosować, obejmują łańcuch A błonicy, niewiążące aktywne fragmenty toksyny błonicy, łańcuch A egzotoksyny (z Pseudomonas aeruginosa), rycyny A, łańcuch abryny A, łańcuch A modekcyny, alfasarcynę, białka Aleurites fordii, białka diantyny, białka Phytolaca americana (PAPI, PAPU i PAP-S), inhibitor Momordica charantia, kurcynę, krotynę, inhibitor Saponaria officinalis, geloninę, mitożelinę, restryktocynę, fenomycynę, enomycynę i trikoteceny. Różne radionuklidy dostępne są do produkcji przeciwciał sprzężonych z izotopami. Przykłady obejmują 212 Bi, 131 I, 131 In, 90 Y i 186 Re. Koniugaty przeciwciała i środka cytotoksycznego wytwarza się stosując szereg dwufunkcyjnych środków sprzęgających białka takich, jak propionian N-sukcynoimidylo-3-(2-pirydylotiolu) (SPDP), iminotiolan (IT), bifunkcjonalne pochodne imidoestrów (jak chlorowodorek iminoadypinianu dimetylu), aktywne estry (jak suberynian disukcynoimidylu), aldehydy (jak aldehyd glutarowy), związki bis-azydowe (jak bis (p-azydobenzoilo)heksanodiamina), pochodne bis-diazoniowe (jak bis-(pdiazoniobenzoilo)-etylenodiamina), diizocyjaniany (jak 2,6-diizocyjanianotoluen) i bisaktywne związki fluoru (jak 1,5-difluoro-2,4-dinitrobenzen). Na przykład, immunotoksynę rycynową można przygotować w sposób opisany w Vitetta i wsp., Science, 238: 1098 (1987). Znakowany węglem-14 kwas 1-izotiocyjanianobenzylo-3- metylodietylenotriaminopentaoctowy (MX-DTPA) jest przykładowym środkiem chelatującym do koniugacji radionukleotydu z przeciwciałem. Patrz, na przykład, WO 1994/ [0255] Koniugaty przeciwciała i środka cytotoksycznego wytwarza się stosując szereg dwufunkcyjnych środków sprzęgających białka, takich jak propionian N-sukcynoimidylo-3- (2-pirydylotiolu) (SPDP), iminotiolan (IT), bifunkcjonalne pochodne imidoestrów (takie jak chlorowodorek iminoadypinianu dimetylu HCl), aktywne estry (jak suberynian disukcynoimidylu), aldehydy (jak aldehyd glutarowy), związki bis-azydowe (jak bis (pazydobenzoilo)heksanodiamina), pochodne bis-diazoniowe (jak bis-(p-diazoniobenzoilo)- etylenodiamina), diizocyjaniany (jak 2,6-diizocyjanianotoluen) i dwuaktywne związki fluorowe (jak 1,5-difluoro-2,4-dinitrobenzen). Na przykład, immunotoksynę rycynową można przygotować w sposób opisany w Vitetta i wsp., Science, 238: 1098 (1987). Znakowany węglem-14 kwas 1-izotiocyjanianobenzylo-3-metylodietylenotriaminopentaoctowy (MX- DTPA) jest przykładowym środkiem chelatującym do koniugacji radionukleotydu z przeciwciałem. Patrz, na przykład, WO 1994/ Łącznik może być łącznikiem rozszczepialnym, ułatwiającym uwalnianie leku cytotoksycznego w komórce. Na przykład, można stosować łącznik labilny w kwasach, łącznik wrażliwy na peptydazy, łącznik dimetylowy lub łącznik zawierający dwusiarczek (Chari i wsp., Cancer Res. 52: (1992)).
76 75- [0256] Ponadto, toksyny małocząsteczkowe takie, jak kalicheamycyna, majtanzyna (USP 5,208,020), trichoten i CC1065 są również rozważane jako dające koniugat toksyn do zastosowania w formulacji według wynalazku. W jednym przykładzie wykonania, przeciwciało pełnej długości lub jego fragment wiążący antygen można koniugować z jedną lub większą liczbą cząsteczek majtanzynoidu (np. około 1 do około 10 cząsteczek majtanzynoidu na cząsteczkę przeciwciała). Majtanzynoidy są inhibitorami mitotycznymi, które działają poprzez hamowanie polimeryzacji tubulin. Majtanzynoidy, izolowane z naturalnych źródeł lub wytwarzane syntetycznie, włączając majtanzyny, majtanzynal i ich pochodne i analogi są opisane, patrz np. Patent US Nr 5,208,020 i cytowana tam literatura (patrz kol. 2, wiersz 53 do kol. 3, wiersz 10) oraz Patenty US 3,896,111 i 4,151,042. Sposoby wytwarzania koniugatów przeciwciało-majtanzynoid są również opisane Pat. US Nr 5,208,020. W korzystnym przykładzie wykonania, majtanzynoid jest połączony z przeciwciałem poprzez dwusiarczek lub inną grupą łącznikową zawierającą siarkę. Majtanzynę można, na przykład, przekształcić w May-SS-Me, którą można zredukować do May-SH3 i poddać reakcji ze zmodyfikowanym przeciwciałem w celu wygenerowania immunokoniugatu majtanzynoid-przeciwciało. Chari i wsp., Cancer Res. 52: (1992). Przeciwciało może być modyfikowany znanymi sposobami, a przeciwciało zawierające wolne lub chronione grupy tiolowe poddaje się następnie reakcji z majtanzynoidem zawierającym dwusiarczek w celu wytworzenia koniugatu. Cytotoksyczność koniugatu przeciwciałomajtanzynoid można mierzyć in vitro lub in vivo znanymi metodami i określić wartości IC 50. [0257] Kalicheamycyna jest kolejnym immunokoniugatem będącym przedmiotem zainteresowania. Rodzina antybiotyków kalicheamicynowych jest zdolna do wytwarzania dwuniciowego DNA w stężeniach sub-pikomolowych. Strukturalne analogi kalicheamycyny, które można stosować, obejmują, bez ograniczania do wymienionych, γ 1 1, α 2 1, α 3 1, N-aceytloγ 1 1, PSAG i θ 1 1 (Hinman i wsp., Cancer Res. 53: (1993) i Lode i wsp., Cancer Res. 58: (1998)). Inne leki przeciwnowotworowe, które mogą być sprzężone z przeciwciałem, obejmują QFA, który jest antyfolianem. Zarówno kalicheamycyna jak i QFA mają wewnątrzkomórkowe miejsca działania i nie przechodzą łatwo przez błonę plazmatyczną. Dlatego wychwyt komórkowy tych czynników przez internalizację za pośrednictwem przeciwciała znacznie zwiększa ich działanie cytotoksyczne. [0258] Immunokoniugaty utworzone pomiędzy przeciwciałem a związkiem o aktywności nukleolitycznej (np. rybonukleazą lub endonukleazą DNA taką, jak dezoksyrybonukleaza, DNazy) są także rozważane. [0259] W ADC według wynalazku, przeciwciało (Ab) jest skoniugowane z jedną lub większą liczbą wyodrębnionych części cząsteczki leku (D), np. około 1 do około 20 ugrupowań leku na przeciwciało, poprzez łącznik (L). ADC o wzorze I można otrzymywać kilkoma sposobami, wykorzystującymi reakcje chemii organicznej, warunki i odczynniki znane specjalistom w dziedzinie, w tym: (1) reakcja grupy nukleofilowej przeciwciała z reagentem dwuwartościowego łącznika, w celu utworzenia Ab-L, poprzez wiązanie kowalencyjne, z następującą reakcją z ugrupowaniem leku D; oraz (2) reakcja grupy nukleofilowej
77 76- ugrupowania leku z reagentem dwuwartościowego łącznika, w celu utworzenia D-L, poprzez wiązanie kowalencyjne, po czym następuje reakcja z nukleofilową grupą przeciwciała. Ab-(L-D) p Wzór I [0260] Grupy nukleofilowe przeciwciała obejmują, bez ograniczania do wymienionych: (i) N- końcowe grupy aminowe, (ii) grupy aminowe łańcucha bocznego np. lizyny, (iii) grupy tiolowe łańcucha bocznego, np. cysteiny, i (iv) cukrowe grupy hydroksylowe lub aminowe, gdy przeciwciało jest glikozylowane. Grupy aminowe, tiolowe i hydroksylowe są nukleofilowe i zdolne do reagowania w celu utworzenia wiązań kowalencyjnych z grupami elektrofilowymi na resztach łącznikowych i odczynnikach łącznikowych, w tym: (i) aktywne estry takie, jak estry NHS, estry HOBt, halomrówczany i halogenki kwasowe; (ii) halogenki alkilowe i benzylowe takie, jak haloacetamidy; oraz (iii) grupy aldehydowe, ketonowe, karboksylowe i maleimidowe. Niektóre przeciwciała mają ulegające redukcji dwusiarczki, tj. mostki cysteinowe. Przeciwciała można uczynić reaktywnymi przy koniugacji z reagentami łącznika, przez traktowanie środkiem redukującym takim, jak DTT (ditiotreitol). Każdy mostek cysteinowy może więc teoretycznie utworzyć dwa reaktywne nukleofile tiolowe. Dodatkowe grupy nukleofilowe można wprowadzić do przeciwciała poprzez reakcję lizyn z 2-iminotiolanem (odczynnikiem Trauta), co prowadzi do przekształcenia grup aminowych do grup tiolowych. [0261] ADC według wynalazku można również produkować przez modyfikację przeciwciała w celu wprowadzenia ugrupowania eletrofilowego, które mogą reagować z nukleofilowymi podstawnikami na reagencie łącznika lub leku. Cukry przeciwciał glikozylowanych można utleniać, np. odczynnikami utleniającymi nadjodanowymi, w celu utworzenia grup aldehydowych lub ketonowych, które mogą reagować z grupami aminowymi reagentów łącznika lub ugrupowań leku. Powstałe grupy iminowe zasady Schiffa mogą utworzyć stabilne wiązanie, lub mogą zostać zredukowane, np. przez reagenty borowodorkowe, dzięki czemu utworzą stabilne wiązania aminowe. W jednym przykładzie wykonania, reakcja fragmentu węglowodanowego glikozylowanego przeciwciała albo z oksydazą galaktozy albo meta-nadjodanem sodu może dać grupy karbonylowe (aldehydów i ketonów) w białku, które można poddać reakcji z odpowiednimi grupami na leku (Domen i wsp., J. Chromatog., 510: (1990)). W innym przykładzie wykonania, białka zawierające N-końcowe reszty seryny lub treoniny mogą reagować z meta-nadjodanem sodu, co skutkuje produkcją aldehydu w miejscu pierwszego aminokwasu (Geoghegan i Stroh, Bioconjugate Chem. 3: (1992); US 5,362,852). Taki aldehyd może reagować z ugrupowaniem leku lub nukleofilem łącznika. [0262] Podobnie grupy nukleofilowe na ugrupowaniu leku obejmują, bez ograniczania do wymienionych: grupy aminowe, tiolowe, hydroksylowe, hydrazydowe, oksymowe, hydrazynowe, tiosemikarbazonowe, karboksylohydrazynowe oraz arylohydrazydowe zdolne do reakcji z utworzeniem wiązań kowalencyjnych z grupami elektrofilowymi w resztach łącznikowych i odczynnikach łącznikowych, w tym: (i) aktywne estry takie, jak estry NHS, estry HOBt, halomrówczany i halogenki kwasowe; (ii) halogenki alkilowe i benzylowe takie jak haloacetamidy; oraz (iii) grupy aldehydowe, ketonowe, karboksylowe i maleimidowe.
78 77- [0263] Alternatywnie, białko fuzyjne, zawierające przeciwciało i środek cytotoksyczny można wykonać, na przykład, za pomocą technik rekombinacji lub syntezy peptydów. Długość DNA może obejmować odpowiednie regiony kodujące dwie części koniugatu, albo sąsiadujące ze sobą, albo rozdzielone regionem kodującym peptyd łącznikowy, który nie niszczy pożądanych właściwości koniugatu. [0264] W jeszcze innym przykładzie wykonania, przeciwciało może być skoniugowane z receptorem (jak streptawidyna) w celu zastosowania do wstępnego celowania w guz, przy czym pacjentowi jest podawany koniugat przeciwciało-receptor, po czym następuje usunięcie niezwiązanego koniugatu z obiegu, przy zastosowaniu środka czyszczącego, a następnie podawanie ligandu (np. awidyny), który jest skoniugowany ze środkiem cytotoksycznym (np. radionukleotydem). [0265] Tutaj ADC są ewentualnie wytwarzane z zastosowaniem reagentów sieciujących: BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-emcs, sulfo-gmbs, sulfo-kmus, sulfo-mbs, sulfo-siab, sulfo- SMCC i sulfo-smpb, oraz SVSB (sukcynoimidylo-(4-winylosulfonobenzoesan)benzoesan), które są dostępne w handlu (np. Pierce Biotechnology, Inc., Rockford, IL). [0266] Przeciwciało może także być skoniugowane z atomem o wysokiej promieniotwórczości. Różne radionuklidy są dostępne dla produkcji przeciwciał skoniugowanych z izotopami. Przykłady obejmują At 211, Bi 212, I 131, In 131, Y 90, Re 186, Re 188, Sm 153, P 32 i Pb 212 oraz izotopy radioaktywne Lu. Gdy koniugat wykorzystywany jest do diagnozowania, może zawierać atom promieniotwórczy do badań scyntygraficznych, na przykład Tc 99 lub I 123, lub znacznik spinowy do obrazowania jądrowym rezonansem magnetycznym (NMR), (znane także jako obrazowanie rezonansem magnetycznym, mri), taki, jak jod-123, jod-131, ind-111, fluor-19, węgiel-13, azot-15, tlen-17, gadolin, mangan lub żelazo. [0267] Radio- lub inne znaczniki mogą być wprowadzane do koniugatu znanymi sposobami. Na przykład, peptyd może być biosyntetyzowany lub może być syntetyzowany poprzez syntezę chemiczną aminokwasów, z użyciem odpowiednich prekursorów aminokwasów zawierających na przykład fluor-19 w miejsce wodoru. Znaczniki takie, jak Tc 99 lub I 123 Re 186, Re 188 i In 111 mogą być przyłączone poprzez resztę cysteiny w peptydzie. Itr-90 może zostać przyłączony przez resztę lizynową. Metoda IODOGEN może być użyta do włączenia jodu-123, Fraker i wsp., Biohem. Biophys. Res. Commun. 80:49-57 (1978). Inne sposoby koniugowania radionuklidów opisano w Monoclonal Antibodies in Immunoscintigraphy, (Chatal, CRC Press 1989). [0268] Alternatywnie, białko fuzyjne, zawierające przeciwciało oraz środek cytotoksyczny można wykonać za pomocą technik rekombinacji lub syntezy peptydów. Długość DNA może obejmować odpowiednie regiony kodujące dwie części koniugatu, albo sąsiadujące ze sobą i oddzielone regionem kodującym peptyd łącznikowy, który nie niszczy pożądanych właściwości koniugatu.
79 78- [0269] W innym przykładzie wykonania, przeciwciało może być skoniugowane z receptorem (jak streptawidyna) w celu zastosowania do wstępnego celowania w guz, przy czym pacjentowi jest podawany koniugat przeciwciało-receptor, po czym następuje usunięcie niezwiązanego koniugatu z obiegu, przy zastosowaniu środka czyszczącego, a następnie podawanie ligandu (np. awidyny), który jest skoniugowany ze środkiem cytotoksycznym (np. radionukleotydem). 12) Inne modyfikacje sekwencji aminokwasowej [0270] Rozważane są modyfikacja (modyfikacje) sekwencji aminokwasowej opisanych tu przeciwciał. Na przykład, może być pożądana poprawa powinowactwa wiązania i/lub innych właściwości biologicznych przeciwciała. Warianty sekwencji aminokwasowych przeciwciała wytwarza się przez wprowadzenie odpowiednich zmian nukleotydowych do kwasu nukleinowego przeciwciała, lub metodą syntezy peptydu. Takie modyfikacje obejmują, na przykład, delecje i/lub insercje i/lub substytucje reszt w sekwencjach aminokwasowych przeciwciała. Jakakolwiek kombinacja delecji, insercji i substytucji wykonywana jest, aby uzyskać ostateczny konstrukt, pod warunkiem, że końcowy konstrukt posiada pożądane cechy. Zmiany aminokwasów mogą również zmieniać procesy post-translacyjne przeciwciała takie, jak zmiana liczby lub pozycji miejsc glikozylacji. [0271] Użyteczną metodę identyfikowania pewnych reszt lub regionów przeciwciała, które są korzystnymi miejscami dla mutagenezy, nazwano mutagenezą skaningową alaniny, jak opisano w Cunningham and Wells in Science, 244: (1989). Tutaj resztę lub grupę docelowych reszt (np. reszty obdarzone ładunkiem takie jak arg, asp, his, lys i glu) i zastępuje obojętnymi lub ujemnie naładowanymi aminokwasami (najkorzystniej alanina lub polialanina) w celu wpływania na oddziaływanie antygen aminokwasów. Te lokalizacje aminokwasów, wykazujące wrażliwość funkcjonalną na substytucję, są bliżej charakteryzowane poprzez wprowadzenie dalszych lub innych wariantów w, bądź za, miejsca substytucji. Tak więc, podczas gdy miejsce wprowadzania wariacji sekwencji aminokwasowej jest z góry ustalone, charakter mutacji per se nie wymaga ustalenia. Na przykład, w celu zanalizowania działania mutacji w danym miejscu, przeprowadza się skaninig ala lub losową mutagenezę w docelowym kodonie lub w regionie, a ulegające ekspresji warianty przeciwciała przeszukuje się pod kątem pożądanej aktywności. [0272] Insercje sekwencji aminokwasowej obejmują amino- i/lub karboksy-końowe fuzje o długości w zakresie od jednej reszty do polipeptydów zawierających sto lub więcej reszt, jak również insercje wewnątrzsekwencyjne pojedynczych lub wielu reszt aminokwasowych. Przykłady insercji końcowych obejmują przeciwciało z resztą metionylową N-końcową lub przeciwciało poddane fuzji z polipeptydem cytotoksycznym. Inne insercyjne warianty cząsteczki przeciwciała obejmują fuzje do N- lub C-końca przeciwciała do enzymu (np. dla ADEPT) lub polipeptydu, który zwiększa okres półtrwania w surowicy przeciwciała. [0273] Innym rodzajem wariantu jest wariant z substytucją aminokwasu. Warianty te posiadają co najmniej jedną resztę aminokwasową w cząsteczce przeciwciała zastąpioną inną resztą. Miejsca najbardziej interesujące dla substytucyjnej mutagenezy obejmują regiony
80 79- hiperzmienne, ale są także rozważane zmiany FR. Konserwatywne podstawienia przedstawiono w Tabeli A poniżej w rubryce korzystne substytucje. Jeżeli takie substytucje prowadzą do zmian w aktywności biologicznej, to można wprowadzać bardziej znaczące zmiany, nazwane przykładowymi substytucjami w Tabeli A lub jak dodatkowo opisano poniżej w odniesieniu do klas aminokwasów, a produkty poddawane skriningowi. TABELA A Substytucje Aminokwasowe Oryginalna Reszta Przykładowe Substytucje Korzystne Substytucje Ala (A) val; leu; ile val Arg (R) lys; gln; asn lys Asn (N) gln; his; asp, lys; arg gln Asp (D) glu; asn glu Cys (C) ser; ala ser Gln (Q) asn; glu asn Glu (E) asp; gln asp Gly (G) ala ala His (H) asn; gln; lys; arg arg Ile (I) leu; val; met; ala; phe; norleucyna leu Leu (L) norleucyna; ile; val; met; ala; phe ile Lys (K) arg; gln; asn arg Met (M) leu; phe; ile leu Phe (F) leu; val; ile; ala; tyr tyr Pro (P) Ala ala Ser (S) Thr thr Thr (T) Ser ser Trp (W) tyr; phe tyr Tyr (Y) trp; phe; thr; ser phe Val (V) ile; leu; met; phe; ala; norleucyna leu [0274] Istotne modyfikacje właściwości biologicznych przeciwciał osiąga się poprzez wybór substytucji różniących się znacznie wpływem na (a) strukturę szkieletu polipeptydowego w obszarze substytucji, na przykład jako konformacja kartki lub helisy, (b) ładunek lub hydrofobowość cząsteczki w miejscu docelowym, lub (c) zmianę objętości łańcucha
81 80- bocznego. Naturalnie występujące reszty podzielono na grupy w oparciu o wspólne właściwości łańcuchów bocznych: (1) hydrofobowe: norleucyna, met, ala, val, leu, ile; (2) obojętne hydrofilowe: cys, ser, thr; (3) kwasowe: asp, glu; (4) zasadowe: asn, gln, his, lys, arg; (5) reszty, które wpływają na orientację łańcucha: gly, pro; i (6) aromatyczne: trp, tyr, phe. [0275] Podstawienia niekonserwatywne wymagają wymiany przedstawiciela jednej z tych klas na inną klasę. [0276] Każdą resztę cysteiny niebiorącą udziału w utrzymywaniu właściwej konformacji przeciwciała można również podstawić generalnie seryną, w celu poprawienia stabilności oksydacyjnej cząsteczki i zapobieżenia nieprawidłowemu sieciowaniu. Natomiast, wiązanie (wiązania) cysteinowe można dodać do przeciwciała w celu polepszenia jego trwałości (w szczególności gdy przeciwciało jest fragmentem przeciwciała takim, jak fragment Fv). [0277] Szczególnie korzystny rodzaj wariantu substytucyjnego obejmuje substytucję jednej lub więcej reszt regionu hiperzmiennego przeciwciała macierzystego (np. humanizowane lub ludzkie przeciwciało). Generalnie, otrzymany wariant (warianty) wybrane do dalszego opracowywania będą posiadały poprawione właściwości biologiczne w stosunku do przeciwciała macierzystego, z którego go utworzono. Dogodny sposób generowania takich substytucyjnych wariantów obejmuje dojrzewanie powinowactwa z zastosowaniem prezentacji przez fagi. W skrócie, kilka miejsc regionu hiperzmiennego (np. 6-7 miejsc) poddaje się mutacjom w celu utworzenia wszystkich możliwych podstawień aminokwasowych w każdym z miejsc. Warianty przeciwciał wytworzone w ten sposób podane są prezentowane w sposób jednowartościowy z nitkowatymi cząstkami faga jako fuzje z produktem III genu M13 upakowanego w każdej cząstce. Te warianty prezentowane na fagach przeszukuje się następnie pod kątem ich aktywności biologicznej (np. powinowactwa wiązania), jak ujawniono w niniejszej publikacji. W celu zidentyfikowania potencjalnych miejsc regionu hiperzmiennego do modyfikacji można przeprowadzić mutagenezę skaningową alaniny w celu zidentyfikowania reszt regionów hiperzmiennych, uczestniczących w znaczącym stopniu w wiązaniu antygenu. Alternatywnie, lub dodatkowo, korzystne może być analizowanie struktury krystalicznej kompleksu antygen-przeciwciało w celu identyfikacji punktów kontaktu pomiędzy przeciwciałem i IgE. Takie reszty kontaktowe i reszty sąsiadujące są kandydatami do substytucji zgodnie z technikami opracowanymi w niniejszym opisie. Po wygenerowaniu takich wariantów, panel wariantów poddaje się przeszukiwaniu w sposób opisany w niniejszym opisie i do dalszego rozwoju można wybrać przeciwciała o ulepszonych właściwościach w jednym lub większej liczbie odpowiednich testów.
82 81- [0278] Inny typ wariantu aminokwasowego przeciwciała zmienia wyjściowy wzór glikozylacji przeciwciała. Przez zmianę rozumie się usunięcie jednej lub większej liczby ugrupowań węglowodanowych obecnych w przeciwciele i/lub dodanie jednego lub więcej miejsc glikozylacji, które nie są obecne w przeciwciele. [0279] Glikozylacja przeciwciał jest zazwyczaj N-glikozylacją lub O-glikozylacją. N- glikozylacja odnosi się do przyłączenia ugrupowania węglowodanowego do łańcucha bocznego reszty asparaginy. Sekwencje tripeptydów asparagina-x-seryna i asparagina-xtreonina, gdzie X to dowolny aminokwas z wyjątkiem proliny, są sekwencjami rozpoznawanymi do enzymatycznego przyłączenia ugrupowania węglowodanowego do łańcucha bocznego asparaginy. Zatem obecność którejkolwiek z tych sekwencji tripeptydowych w polipeptydzie tworzy potencjalne miejsce glikozylacji. O-glikozylacja odnosi się do przyłączenia jednego z cukrów N-acetylogalaktozaminy, galaktozy lub ksylozy, do kwasu hydroksyaminowego, najczęściej seryny lub treoniny, chociaż może również być wykorzystywana 5-hydroksyprolina lub 5-hydroksylizyna. [0280] Dodanie miejsc glikozylacji do przeciwciała dogodnie przeprowadza się przez zmianę sekwencji aminokwasowej tak, że zawiera ona jedną lub więcej z opisanych powyżej sekwencji tripeptydowych (dla miejsc N-glikozylacji). Zmiany można również dokonać przez dodanie, bądź podstawienie, jednej lub więcej reszt seryny lub treoniny do sekwencji oryginalnego przeciwciała (dla miejsc O-glikozylacji). [0281] Cząsteczki kwasów nukleinowych kodujące warianty sekwencji aminokwasowych przeciwciała anty-ige, wytwarza się różnymi sposobami znanymi w dziedzinie. Metody te obejmują, bez ograniczania do wymienionych, wydzielanie z naturalnego źródła (w przypadku naturalnie występujących wariantów sekwencji aminokwasowych) lub wytwarzanie za pomocą mediowanej oligonukleotydami (lub ukierunkowanej) mutagenezy, mutagenezy PCR oraz mutagenezy kasetowej wcześniej przygotowanej wersji wariantowej lub niewariantowej przeciwciała anty-ige. 13) Inne modyfikacje przeciwciał [0282] Przeciwciała według niniejszego wynalazku mogą być dalej modyfikowane aby zawierały dodatkowe ugrupowania niebiałkowe, które są znane w tej dziedzinie i łatwo dostępne. Korzystnie, ugrupowania odpowiednie do derywatyzacji przeciwciała są polimerami rozpuszczalnymi w wodzie. Nieograniczające przykłady rozpuszczalnych w wodzie polimerów obejmują bez ograniczania do wymienionych, glikol polietylenowy (PEG), kopolimery glikol etylenowy/propylenowy, karboksymetylocelulozę, dekstran, alkohol poliwinylowy, poliwinylopirolidon, poli-1,3-dioksolan, poli-1,3,6-trioksan, kopolimer etylen/bezwodnik kwasu maleinowego, poliaminokwasy (homopolimery albo losowe kopolimery), oraz dekstran lub poli(n-winylopirolidon), glikol polietylenowy, homopolimery glikolu polipropylenowego, tlenek propylenu/etylenu i ko-polimery, polioksyetylowane poliole (np. glicerol), alkohol poliwinylowy i ich mieszaniny. Aldehyd propionowy glikol polietylenowy może przynieść korzyści w produkcji ze względu na trwałość w wodzie. Polimer może mieć dowolną masę cząsteczkową, może być rozgałęziony lub nierozgałęziony.
83 82- Liczba polimerów połączonych z przeciwciałem może być różna i przy więcej niż jednym polimerze, mogą to być takie same lub różne cząsteczki. Ogólnie, liczba i/lub rodzaj polimerów użytych do derywatyzacji można określić w oparciu o przesłanki obejmujące, bez ograniczania do wymienionych, szczególne właściwości lub funkcje przeciwciał, które mają być udoskonalone, czy pochodne przeciwciał mają być użyte w terapii w ramach określonych warunków, itd. Takie techniki i inne odpowiednie preparaty są ujawnione w Remington: The Science and Practice of Pharmacy, 20th Ed., Alfonso Gennaro, Ed., Philadelphia College of Pharmacy and Science (2000). C. Formulacje Farmaceutyczne [0283] Formulacje terapeutyczne są przygotowywane do przechowywania poprzez zmieszanie składnika czynnego o pożądanym stopniu czystości z opcjonalnymi farmaceutycznie dopuszczalnymi nośnikami, zaróbkami lub stabilizatorami (Remington: The Science and Practice of Pharmacy, 20th Ed., Lippincott Williams & Wiklins, Pub., Gennaro Ed., Philadelphia, PA 2000). Dopuszczalne nośniki, zaróbki lub substancje stabilizujące są nietoksyczne dla biorców w stosowanych dawkach i stężeniach, i obejmują bufory, przeciwutleniacze, w tym kwas askorbinowy, metioninę, witaminę E, pirosiarczyn sodu; środki konserwujące, stabilizatory, środki izotonizujące, kompleksy metali (np. kompleksy Zn-białko); środki chelatujące takie, jak EDTA i/lub niejonowe środki powierzchniowo czynne. [0284] Gdy środek terapeutyczny jest fragmentem przeciwciała, korzystny jest najmniejszy fragment hamujący, który specyficznie wiąże się do domeny wiążącej docelowego białka. Na przykład, w oparciu o sekwencje regionu zmiennego przeciwciała, można zaprojektować fragmenty przeciwciał lub nawet peptydowe cząsteczki, które zachowują zdolność do wiązania sekwencji docelowego białka. Takie peptydy można syntetyzować chemicznie i/lub wytwarzać z wykorzystaniem technologii rekombinacji DNA (patrz np. Marasco i wsp., Proc. Natl. Acad. Sci. USA 90: [1993]). [0285] Bufory są wykorzystywane do kontrolowania ph w zakresie, który optymalizuje skuteczność terapeutyczną zwłaszcza, gdy stabilność jest zależna od ph. Bufory są korzystnie obecne w stężeniach wynoszących od około 50 mm do około 250 mm. Odpowiednie środki buforujące do stosowania w niniejszym wynalazku obejmują zarówno kwasy organiczne, jak i nieorganiczne i ich sole. Przykładowo, cytrynian, fosforan, bursztynian, winian, fumaran, glukonian, szczawian, mleczan, octan. Ponadto, bufory mogą zawierać sole histydyny i trimetyloaminy takie, jak Tris. [0286] Środki konserwujące dodaje się w celu zahamowania wzrostu drobnoustrojów i są zazwyczaj obecne w ilości w zakresie od 0,2% - 1,0% (w/v). Odpowiednie środki konserwujące do zastosowania w niniejszym wynalazku obejmują chlorek oktadecylodimetylobenzyloamoniowy, chlorek heksametoniowy; halogenki benzalkoniowe (np. chlorek, bromek, jodek), chlorek benzetoniowy; timerosal, fenol, alkohol butylowy lub benzylowy; parabeny alkilowe takie, jak paraben metylu lub propylu; katechol; rezorcynol; cykloheksanol, 3-pentanol i m-krezol.
84 83- [0287] Środki tonizujące, czasem znane jako stabilizatory są obecne w celu dostosowania lub utrzymywania ciśnienia osmotycznego cieczy w kompozycji. Gdy są stosowane z dużymi naładowanymi cząsteczkami biologicznymi takimi, jak białka i przeciwciała, są one często określane jako środki stabilizujące, ponieważ mogą one oddziaływać z naładowanymi grupami łańcuchów bocznych aminokwasów, w ten sposób zmniejszając potencjalne interakcje między- i wewnątrz-cząsteczkowe. Środki tonizujące mogą być obecne w dowolnej ilości od 0.1% do 25% wagowych, korzystnie 1 do 5%, biorąc pod uwagę względne ilości innych składników. Korzystne środki regulujące toniczność obejmują wielowodorotlenowe alkohole cukrowe, korzystnie trójwodorotlenowe lub wyższe alkohole cukrowe takie, jak gliceryna, erytryt, arabitol, ksylitol, sorbitol i mannitol. [0288] Dodatkowe zaróbki obejmują środki, które mogą służyć jako jeden lub więcej z następujących: (1) wypełniacze, (2), środki zwiększające rozpuszczalność, (3), stabilizatory i (4) i środki zapobiegające denaturacji lub przyleganiu do ścianki pojemnika. Takie substancje pomocnicze obejmują: wielowodorotlenowe alkohole cukrowe (wymienione powyżej); aminokwasy takie, jak alanina, glicyna, glutamina, asparagina, histydyna, arginina, lizyna, ornityna, leucyna, 2-fenyloalanina, kwas glutaminowy, treonina, itd.; cukry organiczne lub alkohole cukrowe takie, jak sacharoza, laktoza, trehaloza, laktitol, stachioza, mannoza, sorboza, ksyloza, ryboza, rybitol, myoinisitose, myoinisitol, galaktozy, gliceryny, galaktyt, cyklitole (np. inozytol), glikol polietylenowy; zawierające siarkę środki redukujące takie, jak mocznik, glutation, kwas tioktowy, tioglikolan sodu, tioglicerol, α-monotioglicerol i tiosiarczan sodu; białka o niskiej masie cząsteczkowej takie, jak albumina surowicy ludzkiej, albumina surowicy bydlęcej, żelatyna lub inne immunoglobuliny; polimery hydrofilowe takie, jak poliwinylopirolidon; monosacharydy (np. ksyloza, mannoza, fruktoza, glukoza; disacharydy (np. laktoza, maltoza, sacharoza); trisacharydy takie, jak rafinoza; i polisacharydy takie, jak dekstryny i dekstran. [0289] Niejonowe środki powierzchniowo czynne lub detergenty (znane także jako środki zwilżające ) są obecne w celu pomocy w rozpuszczeniu środka terapeutycznego, jak i do ochrony białka terapeutycznego przeciwko agregacji wywołanej mieszaniem, co również pozwala formulacji na bycie narażoną na naprężenie ścinające powierzchni bez powodowania denaturacji aktywnego białka lub przeciwciała terapeutycznego. Niejonowe środki powierzchniowo czynne są obecne w zakresie od około 0.05 mg/ml do około 1.0 mg/ml, korzystnie około 0.07 mg/ml do około 0.2 mg/ml. [0290] Odpowiednie niejonowe środki powierzchniowo czynne obejmują polisorbaty (20, 40, 60, 65, 80, itd.), poloksamery (184, 188, itd.), poliole PLURONIC, TRITON, polioksyetylenowane monoetery sorbitolu (TWEEN -20, TWEEN -80, itd.), lauromakrogol 400, stearynian polioksylu 40, polioksyetylenowany uwodorniony olej rycynowy 10, 50 i 60, monostearynian glicerolu, ester kwasu tłuszczowego i sacharozy, metyloceluloza i karboksymetyloceluloza. Anionowe środki powierzchniowo czynne, które mogą być stosowane, obejmują laurylosiarczan sodu, dokuzynian sodu i dioktylosulfonian sodu. Kationowe detergenty obejmują chlorek benzalkonium lub chlorek benzetonium.
85 84- [0291] W celu zapewnienia, aby formulacje były przewidziane do podawania in vivo muszą być jałowe. Formulację można wyjałowić za pomocą filtracji przez jałowe błony filtracyjne. Omówione tu kompozycje terapeutyczne są umieszczane w pojemniku posiadającym jałowy port dostępowy, na przykład, torba z roztworem dożylnym lub fiolka mającą korek do przekłucia igłą do iniekcji podskórnej. [0292] Droga podawania jest zgodna ze znanymi i przyjętymi metodami taka, jak przez jeden lub wiele bolusów lub wlewów w ciągu dłuższego okresu czasu w odpowiedni sposób, na przykład, drogą iniekcji lub wlewu drogą dożylną, podskórną, dootrzewnową, domięśniową, dotętniczą, do zmiany chorobowej lub dostawową, za pomocą podawania miejscowego, inhalacji lub podtrzymywanego uwalniania bądź przedłużonego uwalniania. [0293] Formulacja według wynalazku może również zawierać więcej niż jeden związek czynny, w zależności od konieczności dla konkretnego leczonego wskazania, korzystnie związki o uzupełniających się aktywnościach, które nie wpływają niekorzystnie na siebie. Alternatywnie, lub dodatkowo, kompozycja może zawierać środek cytotoksyczny, cytokinę lub środek hamujący wzrost. Takie cząsteczki są odpowiednio obecne w kombinacji w ilościach, które są skuteczne dla zamierzonego celu. [0294] Składniki aktywne mogą być również uwięzione w mikrokapsułkach, przygotowanych na przykład technikami koascerwacji lub przez polimeryzację międzyfazową, na przykład, odpowiednio hydroksymetyloceluloza lub mikrokapsułki żelatynowe oraz mikrokapsułki poli-(metylometacylanowe), w koloidalnych układach dostarczania leków (na przykład liposomy, mikrokulki albuminowe, mikroemulsje, nanocząstki i nanokapsułki) lub w makroemulsjach. Takie techniki są ujawnione w Remington s Pharmaceutical Sciences wydanie 18, supra. [0295] Stabilność opisanych tu białek i przeciwciał można zwiększyć poprzez zastosowanie nietoksycznych soli wielowartościowych metali rozpuszczalnych w wodzie. Przykłady obejmują Ga 2+, Mg 2+, Zn 2+, Fe 2+, Fe 3+, Cu 2+, Sn 2+, Sn 4+, Al 2+ i Al 3+. Przykładami anionów, które tworzą rozpuszczalne w wodzie sole z powyższymi wielowartościowymi kationami metali obejmują sole utworzone z kwasami nieorganicznymi i/lub kwasami organicznymi. Takie sole rozpuszczalne w wodzie mają rozpuszczalność w wodzie (w 20 C) co najmniej około 20 mg/ml, alternatywnie przynajmniej ok. 100 mg/ml, alternatywnie przynajmniej ok. 200 mg/ml. [0296] Odpowiednie kwasy nieorganiczne, które można stosować w celu wytworzenia rozpuszczalnych w wodzie, wielowartościowych soli metali obejmują kwas solny, octowy, siarkowy, azotowy, fosforowy i tiocyjanowy. Odpowiednie kwasy organiczne, które mogą być stosowane, obejmują alifatyczne kwasy karboksylowe i kwasy aromatyczne. Kwasy alifatyczne, zgodnie z tą definicją można zdefiniować jako nasycone lub nienasycone kwasy C 2-9 karboksylowe (np. alifatyczne kwasy mono-, di- i tri-karboksylowe). Na przykład, przykładowe kwasy monokarboksylowe, zgodnie z tą definicją obejmują nasycone kwasy C 2-9 monokarboksylowe octowy, propionowy, masłowy, walerianowy, kapronowy, enantowy, kaprylowy pelargonowy i capryonic, i nienasycone kwasy C 2-9 monokarboksylowe kwas
86 85- akrylowy, proprioliowy, metakrylowy, krotonowy i izokrotonowy. Przykłady kwasów dikarboksylowych obejmują nasycone kwasy C 2-9 dikarboksylowe kwas malonowy, bursztynowy, glutarowy, adypinowy i pimelinowy, natomiast nienasycone kwasy C 2-9 dikarboksylowe obejmują kwas maleinowy, fumarowy, kwas cytrakonowy i mezakonowy. Przykładowe kwasy trikarboksylowe obejmują nasycone kwasy C 2-9 trikarboksylowe kwas trikarbalilowy i kwas 1,2,3-butanotrikarboksylowy. Dodatkowo, kwasy karboksylowe zgodne z tą definicją, mogą także zawierać jedną lub dwie grupy hydroksylowe, z wytworzeniem hydroksykwasów karboksylowych. Przykładowe hydroksykwasy karboksylowe obejmują kwas glikolowy, mlekowy, glicerynowy, tartronowy, jabłkowy, winowy i kwas cytrynowy. Kwasy aromatyczne, zgodnie z tą definicją obejmują kwas benzoesowy i kwas salicylowy. [0297] Powszechnie stosowane rozpuszczalne w wodzie sole wielowartościowych metali, które można stosować, aby pomóc ustabilizować kapsułkowane polipeptydy według niniejszego wynalazku, obejmują, na przykład: (1) nieorganiczne sole metali z halogenkami (np. chlorek cynku, chlorek wapnia), siarczany, azotany, fosforany i tiocyjaniany; (2) sole metali alifatycznych kwasów karboksylowych (np. octan wapnia, octan cynku, propionian wapnia, cynku, glikolan, mleczan wapnia, mleczan cynku, winian cynku); oraz (3) sole metali aromatycznych kwasów karboksylowych benzoesanów (np. benzoesan cynku) oraz salicylanów. D. Sposoby leczenia: [0298] W celu zapobiegania lub leczenia choroby, odpowiednia dawka substancji czynnej będzie zależała od rodzaju leczonej choroby, jak zdefiniowano powyżej, ciężkości i przebiegu choroby, tego, czy środek podaje się w celach zapobiegawczych, czy leczniczych, wcześniejszego leczenia, historii choroby pacjenta i reakcji na środek, oraz uznania lekarza prowadzącego. Środek korzystnie podaje się pacjentowi jednorazowo lub jako serię leczeń. [0299] Korzystnym sposobem traktowania jest leczenie zaburzeń mediowanych IgE. Zaburzenia mediowane IgE obejmują zaburzenia atopowe, które charakteryzują dziedziczną skłonnością do odpowiedzi immunologicznej na wiele typowych naturalnie występujących inhalowanych i wchłanianych antygenów i ciągłym wytwarzaniem przeciwciał IgE. Specyficzne zaburzenia atopowe obejmują astmę alergiczną, alergiczny nieżyt nosa, atopowe zapalenie skóry i alergiczną gastroenteropatię. Pacjenci atopowi często mają wiele alergii, co oznacza, że mają przeciwciała IgE i objawy powodowane przez wiele alergenów środowiskowych, w tym pyłków roślin, grzybów (np. pleśni), szczątków zwierząt i owadów oraz niektórych pokarmów. [0300] Jednakże zaburzenia powiązane z podwyższonymi poziomami IgE nie są ograniczone do tych z dziedziczną (atopową) etiologią. Inne zaburzenia związane z podwyższonym poziomem IgE, które wydają się być mediowane IgE i które są uleczalne za pomocą preparatów według niniejszego wynalazku obejmują nadwrażliwość (np. nadwrażliwość anafilaktyczną), egzemę, pokrzywkę, alergiczną aspergilozę oskrzelowopłucną, choroby pasożytnicze, zespół hiper-ige, ataksja-teleangiektazja, zespół Wiskotta-Aldricha, alimfoplazję tymiczną, szpiczaka IgE i reakcję przeszczep przeciwko gospodarzowi.
87 86- [0301] Alergiczny nieżyt nosa, znany również jako alergiczne zapalenie spojówek łącznie z alergicznym nieżytem nosa lub katar sienny, jest najczęstszym objawem reakcji atopowej na alergeny wziewne, których nasilenie i czas trwania jest często skorelowany z intensywnością i długością ekspozycji na alergen. Jest to przewlekła choroba, która może wystąpić po raz pierwszy w każdym wieku, ale początek jej zazwyczaj ma miejsce w dzieciństwie lub w okresie dojrzewania. Typowy atak składa się z gęstego wodnistego wycieku z nosa, kichania, napadowej niedrożności nosa oraz swędzenia nosa i podniebienia. Drenaż śluzu po tylnej ścianie gardła powoduje także ból gardła, chrząkanie i kaszel. Mogą wystąpić także objawy alergicznego zapalenia powiek, z intensywnym swędzeniem spojówki i powiek, zaczerwienienie, łzawienie i światłowstręt. Ciężkim atakom często towarzyszy złe samopoczucie układowe, osłabienie, zmęczenie, a czasem, bolesność mięśni po intensywnych okresach kichania. [0302] Astma, znana także jako nawrotowa obturacyjna choroba dróg oddechowych, charakteryzuje się nadreaktywnością drzewa tchawiczo-oskrzelowego na podrażnienia dróg oddechowych i środki zwężające oskrzela, dając ataki sapania, duszności, ucisk w klatce piersiowej i kaszel, które są odwracalne samoistnie lub z zastosowaniem leczenia. Jest to przewlekła choroba dróg oddechowych z udziałem całych dróg oddechowym, ale zmienia się w przebiegu od sporadycznych łagodnych epizodów przejściowych do ciężkiej przewlekłej niedrożność oskrzeli zagrażającej życiu. Astma i atopia mogą współistnieć, ale tylko połowa chorych na astmę jest również atopiczna, a jeszcze mniejszy odsetek pacjentów atopowych ma także astmę. Jednak, atopia i astma nie są całkowicie niezależne, przy tym astma występuje częściej u osób z atopią niż bez atopii, zwłaszcza w okresie dzieciństwa. Astma została ponadto historycznie podzielona na dwie podgrupy, astmę zewnętrzpochodną i astmę wewnętrzpochodną. [0303] Astmę zewnętrzpochodną, znaną także jako alergiczna astma atopowa lub immunologiczna, można opisać tym, że u pacjentów na ogół astma rozwija się we wczesnym okresie życia, zwykle w okresie niemowlęctwa i dzieciństwa. Inne objawy atopii, w tym egzema lub alergiczny nieżyt nosa często współistnieją. Ataki astmy mogą wystąpić podczas pylenia, w obecności zwierząt lub przy wystawieniu na działanie kurzu, pierze z poduszki lub inne alergeny. Testy skórne pokazują pozytywne reakcje w postaci bąbla i zaczerwienia skóry na alergeny wywołujące. Co ciekawe, całkowite stężenia IgE w surowicy są często podwyższone, ale czasami są prawidłowe. [0304] Astma wewnątrzpochodna, znana również jako astma niealergiczna lub idiopatyczna, zazwyczaj najpierw pojawia się w dorosłym życiu, po pozornej infekcji dróg oddechowych. Objawy obejmują przewlekłe lub nawracające niedrożności oskrzeli niezwiązane z sezonowymi pyłkami lub ekspozycją na inne alergeny. Testy skórne są negatywne na typowe alergeny atopowe, stężenie IgE w surowicy jest prawidłowe. Dodatkowe objawy to krew w plwocinie i eozynofilia. Inne systemy klasyfikacji astmy w podgrupach takich, jak wrażliwa na aspirynę, powysiłkowa, zakaźna i psychologiczna jedynie definiują zewnętrzne czynniki wyzwalające, które wpływają na niektórych pacjentów bardziej niż inne.
88 87- [0305] Wreszcie, ważne jest, aby pamiętać, że niektóre klasyfikacje historycznie kojarzyły astma alergiczną tylko z zależnością od IgE, obecnie istnieją silne dane wykazujące statystycznie istotną korelację między IgE i astmą (zarówno alergiczną, jak i niealergiczną). Rozdział 27, The Atopic Diseases, A.I. Terr w Medical Immunology, 9th Ed., Simon i Schuster, Stites et al, Ed. (1997). W rezultacie, określenie zaburzenia mediowane IgE, dla celów niniejszego zgłoszenia patentowego, obejmuje zarówno astmę alergiczną, jak i niealergiczną. [0306] Fizyczne objawy ataku astmy obejmują szybkie oddychanie, słyszalny świszczący oddech oraz korzystanie z dodatkowych mięśni oddychania. Typowo obecne są szybki puls i podwyższone ciśnienie krwi, jak i podwyższone są poziomy eozynofili we krwi obwodowej i wydzieliny z nosa. Funkcje płuc pokazują spadek przepływów i 1-sekundowej natężonej objętości wydechowej (FEV 1 ). Całkowita pojemność płuc i funkcjonalna resztkowa pojemność są zwykle prawidłowe lub nieco zwiększone, ale może być zmniejszona ekstremalnym skurczem oskrzeli. [0307] Patologię astmy można rozróżnić przez reakcję wczesnej fazy i reakcję późnej fazy. Wczesna faza cechuje się skurczem mięśni gładkich, obrzękiem i nadmiernym wydzielaniem, a reakcje późnej fazy charakteryzują się zapaleniem komórkowym. Astma może być indukowana przez różne nieswoiste wyzwalacze, w tym infekcję (np. wirusowe infekcje dróg oddechowych), czynniki fizjologiczne (np. ćwiczenia, hiperwentylacja, głębokie oddychanie, czynniki psychologiczne), czynniki atmosferyczne (np. dwutlenek siarki, amoniak, zimne powietrze, ozon, pary wody destylowanej), środki połykane (np. propranolol, aspiryna, niesteroidowe leki przeciwzapalne), eksperymentalne środki inhalacyjne (np. hipertoniczne roztwory, kwas cytrynowy, histamina, metacholina, prostaglandyna F 2α ) i zawodowe czynniki inhalacyjne (np. izocyjaniany). Różne dodatkowe alergeny zawodowe lub środowiskowe, które powodują astmę alergiczną, mogą obejmować produkty zwierzęce, pyły owadów, stworzenia morskie, produkty roślinne, owoce, nasiona, liście i pyłki, barwniki organiczne i tusze, czynniki mikrobiologiczne, enzymy, środki terapeutyczne, środki do sterylizacji, jak i nieorganiczne i organiczne substancje chemiczne. [0308] Atopowe zapalenie skóry, znane także jako egzema, neurodermit, atopowa egzema lub świerzbiączka Besniera, jest przewlekłą chorobą skóry, specyficzną dla podzbioru pacjentów z rodzinnymi lub immunologicznymi cechami atopii. Zasadniczą cechą jest swędząca skórna reakcja zapalna, co wywołuje charakterystyczne symetrycznie rozłożone wykwity skórne ze skłonnościami do pewnych miejsc. Jest także obecna nadprodukcja IgE przez limfocyty B. Podczas gdy, atopowe zapalenie skóry klasyfikuje się jako postać atopii skórnej, ponieważ jest to związane z alergicznym nieżytem nosa i astmą i wysokimi poziomami IgE, nasilenie zapalenia skóry jednak nie zawsze koreluje z ekspozycją na alergeny podczas testu skórnego i odczulanie (w przeciwieństwie do innych chorób alergicznych) nie jest skutecznym leczeniem. Podczas gdy wysokie IgE w surowicy potwierdza diagnozę astmy alergicznej, prawidłowe poziomy jej nie wykluczają. Początek choroby może wystąpić w każdym wieku, i zmiany zaczyna się ostro od grudki lub płytki rumienia obrzękowego z łuszczeniem. Swędzenie prowadzi do płaczu i strupów, a następnie do przewlekłej lichenifikacji. Na
89 88- poziomie komórkowym, ostre uszkodzenie jest obrzękiem i skóra właściwa jest naciekana komórkami jednojądrzastymi limfocytami CD4. Neutrofile, eozynofile, komórki plazmatyczne i bazofile są rzadkie, ale degranulowane komórki tuczne są obecne. Przewlekłe zmiany cechują się rozrostem naskórkowym, nadmiernym rogowaceniem i parakeratozą, a skóra właściwa jest naciekana przez komórki jednojądrzaste, komórki Langerhansa i komórki tuczne. Może wystąpić również ogniskowe obszary zwłóknienia, w tym zajęcie onerwia małych nerwów. [0309] Alergiczna gastroenteropatia, znana także jako eozynofilowa gastroenteropatia, jest nietypowym objawem atopowym, w którym wiele wrażliwości spożywczych IgE wiąże się z lokalną reakcją śluzówki przewodu pokarmowego. Jest ona rzadka u dorosłych, ale bardziej powszechna, ale przemijająca, u niemowląt. Stan powstaje, gdy spożyte alergeny pokarmowe reagują z lokalnymi przeciwciałami IgE w błonie śluzowej jelita czczego i uwalniają mediatory komórek tucznych, powodując objawy ze strony przewodu pokarmowego tuż po posiłku. Ciąg dalszy ekspozycji daje przewlekłe zapalenie, co skutkuje utratą białka pokarmowego oraz obrzękiem hipoproteinemicznym. Utrata krwi przez zapalnie błony śluzowej jelita może być na tyle istotna, aby spowodować niedokrwistość z niedoboru żelaza. Reakcja alergiczna występuje lokalnie w górnej błony śluzowej przewodu pokarmowego po ekspozycji na alergen, ale zanika wraz z unikaniem alergenów. [0310] Anafilaksja i pokrzywka są wyraźnie mediowane IgE, ale brakuje im uwarunkowań genetycznych, i nie dotykają szczególnie osób atopowych. Anafilaksja jest ostrą, uogólnioną reakcją alergiczną z jednoczesnym udziałem kilku narządów, zazwyczaj układu sercowonaczyniowego, oddechowego, skóry i przewodu pokarmowego. Reakcja jest mediowana immunologicznie i zjawisko to występuje po ekspozycji na alergen, na który pacjent był uprzednio uczulonego. Pokrzywka i obrzęk naczynioruchowy odnosi się do fizycznego obrzęku, rumienia i świądu, wynikającego z receptora stymulowanego histaminą w powierzchownych skórnych naczyniach krwionośnych, i jest cechą charakterystyczną skórną układowej anafilaksji. Układowa anafilaksja jest występowaniem reakcji mediowanej IgE jednocześnie w wielu narządach wynikająca z leku, jadu owada lub pokarmu. Jest spowodowana niespodziewanie przez komórkę tuczną załadowaną IgE indukowaną alergenem, powodując głębokie i zagrażające życiu zmiany w funkcjonowaniu różnych ważnych narządów. Naczyniowa zapaść, ostra niedrożność dróg oddechowych, obrzęk skóry i rozszerzenie naczyń i skurcz mięśni przewodu pokarmowego i układu moczowo-płciowego występują niemal równocześnie, chociaż nie zawsze w tym samym stopniu. [0311] Patologia anafilaksji obejmuje obrzęk naczynioruchowy i nadmiernie rozdęte płuca, z zatykaniem śluzem dróg oddechowych i ogniskową niedodmą. Na poziomie komórkowym, płuca wyglądają podobnie, jak podczas ostrego ataku astmy, z nadmiernym wydzielaniem przez gruczoły podśluzówkowe oskrzeli, obrzękiem śluzówkowym i obrzękiem podśluzówkowym, okołooskrzelowym przekrwieniem naczyń i eozynofilią w ścianach oskrzeli. Może być obecny obrzęk płuc i krwotok. Może być obecny oskrzelowy skurcz mięśni, hiperinflacja, a nawet rozerwanie pęcherzyków. Ważne cechy ludzkiej anafilaksji
90 89- obejmują obrzęk, przekrwienie naczyń i eozynofilię w blaszce właściwej krtani, tchawicy, nagłośni i gardła. [0312] Ekspozycja na alergen może odbyć się poprzez spożycie, wstrzyknięcie, wdychanie lub kontakt ze skórą lub błoną śluzową. Reakcja rozpoczyna się w ciągu sekund lub minut po ekspozycji na alergen. Może wystąpić początkowy strach lub poczucie zbliżającej się katastrofy, a następnie gwałtowne objawy w jednym lub większej liczbie narządów docelowych: układu krążenia, układ oddechowy, skóra lub przewód pokarmowy. [0313] Alergeny odpowiedzialne za anafilaksję odbiegają od powszechnie związanych z atopią. Żywność, leki, jady owadów lub lateks są typowymi źródłami. Alergeny pokarmowe obejmują te w skorupiakach, mięczakach (np. homary, krewetki, kraby), rybach, roślinach strączkowych (np. orzeszki ziemne, groch, fasola, lukrecja), nasionch (np. sezam, nasiona bawełny, kminek, gorczyca, len, słonecznik), orzechach, jagodach, białkach jaj, kaszy gryczanej i mleku. Alergeny lekowe obejmują te w heterologicznych białkach i polipeptydach, polisacharydach i lekach haptenicznych. Owadzie alergeny obejmują owady błonkoskrzydłe, w tym pszczoła miodna, osa o angielskiej nazwie yellow jacket, szerszeń, osa i mrówka ogniowa. [0314] Podczas gdy epinefryna jest typowym leczeniem anafilaksji, lek przeciwhistaminowy lub inne blokery histaminowe są zazwyczaj przepisywane dla mniej ciężkiej pokrzywki lub reakcji obrzęku naczynioruchowego. E. Terapie Skojarzone [0315] Sposób według wynalazku może być łączony ze znanymi metodami leczenia zaburzeń mediowanych IgE, albo jako skojarzone lub dodatkowe etapy leczenia, albo jako dodatkowe składniki formulacji leczniczej. [0316] Na przykład, leki przeciwhistaminowe, zwłaszcza niesedacyjne leki przeciwhistaminowe można podawać wcześniej, przed lub jednocześnie z przeciwciałami anty-ige według wynalazku. Odpowiednie leki przeciwhistaminowe obejmują te z alkiloaminy (np. chlorfeniramina), etanolaminy (np. difenhydramina) oraz fenotiazyny (np. prometazyna). Podczas gdy leki przeciwhistaminowe antagonizują farmakologiczne działanie histaminy poprzez blokowanie jej receptorów na komórkach efektorowych, inne typowe leki przeciwhistaminowe działają poprzez blokowanie uwalniania histaminy z komórek tucznych, które zostały uczulone i uzbrojone w IgE specyficzną alergenowo (np. kromoglikan sodowy). Przykłady leków przeciwhistaminowych obejmują astemizol, maleinian azatadyny, maleinian brofeniraminy, maleinian karbinoksaminy, chlorowodorek cetyryzyny, fumaran klemastyny, chlorowodorek cyproheptadyny, maleinian deksbromfeniraminy, maleinian dekschlorfeniraminy, dimenhydrinat, chlorowodorek difenhydraminy, bursztynian doksylaminy, chlorowodorek feksofendadyny, chlorowodorek terfenadyny, chlorowodorek hydroksyzyny, loratadynę, chlorowodorek meklizyny, cytrynian tripelannaminy, chlorowodorek tripelenaminy, chlorowodorek triprolidyny. [0317] Niektóre objawy zaburzeń mediowanych IgE (np. reakcje fazy wczesnej) można złagodzić sympatykomimetykami lub lekami mającymi działanie rozszerzające oskrzela.
91 90- Epinefryna jest szeroko działającym lekiem beta-adrenergicznym, często podawanym się podskórnie, w dawce ml wodnego roztworu 1:100. Stosuje się również dłużej działającą postać epinefryny (tj. terbutalinę) w 1:200 zawesinie, gdy pożądane jest dłuższe działanie. Odpowiednie dodatkowe leki beta-adrenergiczne obejmują albuterol, pirbuterol metaproterenol, salmeterol, izoetarynę i formeterol do podawania donosowego (np. ręczny nebulizator, urządzenie pracujące w sposób przerywany z oddychaniem w nadciśnieniu lub inhalatory ciśnieniowe z dozownikiem) lub doustnego. [0318] Działanie rozszerzające oskrzela można osiągnąć przez podawanie ksantyn, szczególnie, gdy są one podawane w połączeniu z opisanymi powyżej lekami sympatykomimetycznymi. Przykładowe ksantyny obejmują aminofilinę (iv mg) i teofilinę (doustnie, µg/ml stężenie w surowicy). [0319] Inne objawy różnych zaburzeń mediowanych IgE (np. reakcje fazy późnej) można zmniejszyć leczeniem glukokortykoidami i innymi lekami o działaniu przeciwzapalnym. Prednizon (30-60 mg na dobę) podaje się systemowo dla ciężkich ataków, natomiast dipropionian beklometazonu, acetonid triamcynolonu i flunizolid są podawane w postaci aerozoli jako długoterminowe leczenie podtrzymujące. Dodatkowe kortykosteroidy, które posiadają działanie przeciwzapalne obejmują: betametazon, budezonid, deksametazon, octan fludrokortyzonu, flunizolid, propionian flutikazonu, hydrokortyzon, metyloprednizolon, prednizolon, prednizon, triamcynolon. [0320] Niesteroidowe leki przeciwzapalne, które mogą być również stosowane w połączeniu z metodami leczenia według wynalazku, obejmują acetaminofen, aspirynę, bromfenak sodu, diklofenak sodu, diflunizal, etodolak, fenoprofen wapnia, flurbiprofen, ibuprofen, indometacynę, ketoprofen, meklofenamat sodowy, kwas mefenamowy, nabumeton, naproksen, naproksen sodu, oksyfenobutazon, fenylobutazon, piroksykam, sulindak, tolmetynę sodu. [0321] Ponadto, maksymalne korzyści terapeutyczne mogą być również osiągnięte przy podawaniu leków zmniejszających przekrwienie (np. fenylefryna, fenylopropanoloamina, pseudoefedryna), leków przeciwkaszlowych (np. dekstrometorfan, kodeina lub hydrokodon) lub leków przeciwbólowych (np. paracetamol, aspiryna). [0322] Odczulanie na alergeny to metoda leczenia, w której alergeny są wstrzykiwane do ciała pacjenta w celu zmniejszenia lub wyeliminowania odpowiedzi alergicznej. Jest ono także znane jako immunoterapia alergenem, odczulanie lub iniekcyjne leczenie alergii. Stosuje się je często w połączeniu z innymi metodami leczenia alergii, ale często jako podstawowe leczenie. Było ono z powodzeniem stosowane, gdy nie jest możliwe unikanie alergenu. Typowe leczenie odczulaniem na alergeny obejmuje podskórne wstrzykiwanie sterylnego alergenu w rosnących dawkach raz lub dwa razy w tygodniu aż do osiągnięcia dawki, która wytwarza przejściowy niewielki obszar lokalnego stanu zapalnego w miejscu wstrzyknięcia. Dawkę następnie podaje się w schemacie podtrzymania raz na 2-4 tygodni. Alergiczne odczulanie jest najczęściej używane w leczeniu astmy alergicznej i alergicznego nieżytu nosa, choć odniesiono sukces w leczeniu anafilaksji. Odczulanie również skutecznie
92 91- stosowano przez zastosowanie adiuwantów takich, jak adiuwant Freunda, który jest emulsją wodnego antygenu w oleju mineralnym. Efekt fizjologiczny tworzy nierozpuszczalny tłuszczowy depot, z którego stopniowo uwalniane są kropelki alergenu. Inną postacią odczulania alergenów jest polimeryzacja monomerycznych alergenów z aldehydem glutarowym w celu utworzenia cząsteczki o względnie niewielkiej alergenności (tj. dającej odpowiedź alergiczną), zachowując skuteczny poziom immunogenności. F. Dawkowanie farmaceutyczne: [0323] Dawki i pożądane stężenie leku w kompozycji farmaceutycznej według niniejszego wynalazku może zmieniać się w zależności od konkretnego zastosowania. Określenie odpowiedniego dawkowania i sposobu podawania pozostaje w zakresie umiejętności znawcy. Doświadczenia na zwierzętach dostarczają wiarygodnych wskazówek dla określania dawek skutecznych w terapii człowieka. Międzygatunkowe skalowanie skutecznych dawek można wykonać zgodnie z zasadami ustanowionymi przez Mordenti, J. i Chappell, W. The Use of Interspecies Scaling in Toxicokinetics, In Toxicokinetics and New Drug Development, Yacobi i wsp., Eds, Pergamon Press, New York 1989, strony [0324] Gdy stosowane jest tu podawanie in vivo opisanych polipeptydów lub przeciwciał, prawidłowe ilości dawkowania mogą się zmieniać od około 10 ng/kg do około 100 mg/kg masy ciała ssaka lub więcej na dzień, korzystnie około 1 mg/kg/dzień do 10 mg/kg/dzień, w zależności od drogi podania. Wskazówki co do konkretnych dawek i sposobów dostarczania są przedstawione w literaturze; patrz na przykład, US Pat. Nry 4,657,760; 5,206,344; lub 5,225,212. W zakresie wynalazku jest to, że różne formulacje będą skuteczne dla różnych leczeń i różnych zaburzeń, i że podawanie przeznaczone do leczenia konkretnego narządu lub tkanki może wymagać dostarczania w sposób inny niż w przypadku innego narządu lub tkanki. Ponadto, dawkowanie może być podawane w jednej lub większej liczbie oddzielnych podań lub poprzez wlew ciągły. Dla podawań powtarzanych w ciągu kilku dni lub dłużej, zależnie od stanu, leczenie kontynuuje się do wystąpienia pożądanego zahamowania objawów choroby. Jednakże, inne schematy dawkowania mogą być przydatne. Postęp tej terapii może być łatwo monitorowany za pomocą konwencjonalnych technik i oznaczeń. G. Podawanie Formulacji [0325] Formulacje według niniejszego wynalazku, w tym, bez ograniczania do odtwarzanych formulacji, podaje się ssakowi wymagającemu leczenia białkiem, korzystnie człowiekowi, w zgodzie ze znanymi sposobami takimi, jak podawanie dożylne, jako bolus lub przez ciągłą infuzję przez pewien okres czasu, domięśniowo, dootrzewnowo, podskórnie, wewnątrzmózgowo-rdzeniowego, wewnątrznosowo, domaziówkowo, dooponowo, doustnie, miejscowo lub przez inhalację. [0326] W korzystnych przykładach wykonania, kompozycje podaje się ssakowi przez podanie podskórne (tj. pod skórę). Dla tych celów, formulację można wstrzykiwać przy użyciu strzykawki. Jednakże, dostępne są inne urządzenia do podawania preparatu takie, jak urządzenia do iniekcji (np. urządzenia INJECT-EASE i GENJECT ); iniektory w
93 92- długopisie (jak GENPEN ); urządzenia autoiniektory, urządzenia bezigłowe (np. MEDIJECTOR i BIOJECTOR ); i podskórne układy dostarczania oparte na plastrach. [0327] W konkretnym przykładzie wykonania, niniejszy wynalazek dotyczy zestawów dla pojedynczej jednostki dawkowania do podawania. Takie zestawy obejmują pojemnik z wodną formulacją białka lub przeciwciała terapeutycznego, w tym jedno- lub wielokomorowe wstępnie napełnione ampułkostrzykawki. [0328] Odpowiednia dawka ( terapeutycznie skuteczna ilość ) białka będzie zależeć, na przykład, od stanu, który ma być leczony, ciężkości i przebiegu choroby, tego, czy białko jest podawane w celach zapobiegawczych, czy leczniczych, wcześniejszego leczenia, wywiadu klinicznego pacjenta i odpowiedzi na białko, rodzaju użytego białka oraz od uznania lekarza prowadzącego. Białko korzystnie podaje się pacjentowi jednorazowo lub jako seria leczeń i może być podawane pacjentowi w dowolnej chwili od rozpoznania dalej. Białko może być podawane jako jedyne leczenie lub w skojarzeniu z innymi lekami lub terapiami użytecznymi w leczeniu danego stanu. [0329] W przypadku, gdy białko jest przeciwciałem, początkową proponowaną dawką do podawania pacjentowi jest od około 0,1-20 mg/kg, czy to, na przykład, przez jedno lub więcej oddzielnych podań. Jednakże mogą być przydatne inne schematy dawkowania. Postęp tej terapii można łatwo monitorować za pomocą konwencjonalnych technik. [0330] Zastosowania formulacji anty-ige (np. rhumabe-25, rhmabe-26, Hu-901) obejmują na przykład leczenie lub profilaktykę chorób alergicznych mediowanych IgE, zakażenia pasożytnicze, śródmiąższowe zapalenie pęcherza moczowego i astmę. W zależności od choroby lub schorzenia, które ma być leczone, podaje się pacjentowi terapeutycznie skuteczną ilość (np. od około 1-15 mg/kg) przeciwciała anty-ige. H. Artykuły przemysłowe [0331] W innym przykładzie wykonania według wynalazku, zapewniony jest artykuł przemysłowy, który zawiera formulację i korzystnie zapewnia instrukcje dotyczące jego stosowania. Artykuł przemysłowy zawiera pojemnik. Odpowiednie pojemniki obejmują, na przykład, butelki, fiolki (np. fiolki dwukomorowe), strzykawki (jak pojedyncze lub podwójne strzykawki dwukomorowe) i probówki. Pojemnik może być wykonany z różnych materiałów takich, jak szkło lub tworzywo sztuczne. Pojemnik zawiera formulację. Etykieta, która znajduje się na albo jest dołączona do pojemnika może podawać wskazówki dla odtworzenia i/lub zastosowanie. Etykieta może ponadto wskazywać, że lek może lub powinien być podany podskórnie. Pojemnik zawierający formulację może być fiolką wielokrotnego użytku, która pozwala na wielokrotne podawanie (np. od 2-6 podań) odtworzonej formulacji. Artykuł przemysłowy może ponadto zawierać drugi pojemnik zawierający odpowiedni rozpuszczalnik (np. BWFI). Po zmieszaniu rozcieńczalnika i liofilizowanej formulacji, końcowe stężenie białka w odtworzonej formulacji będzie ogólnie wynosić co najmniej 50 mg/ml. Artykuł przemysłowy może ponadto zawierać inne materiały pożądane z handlowego lub użytkowego punktu widzenia, w tym inne bufory, rozcieńczalniki, filtry, igły, strzykawki i wkładki do opakowania z instrukcjami stosowania.
94 93- [0332] Wynalazek będzie pełniej zrozumiały przez odniesienie do poniższych przykładów. Nie powinny one jednak być interpretowane jako ograniczające zakres wynalazku. Wszystkie cytowania w niniejszym ujawnieniu są niniejszym wyraźnie włączone przez odniesienie. [0333] W innym przykładzie wykonania, wynalazek zapewnia artykuł przemysłowy zawierający formulacje opisane w niniejszym dokumencie do podawania w autoiniektorze. Autoiniektor można określić jako urządzenie do iniekcji, które w wyniku aktywacji, dostarczy jego zawartość bez dodatkowych niezbędnych działań ze strony pacjenta lub podającego. Są one szczególnie odpowiednie do samodzielnego podawania formulacji terapeutycznych, jeśli szybkość dostarczania musi być stała, a czas dostarczania jest większy niż kilka chwil. PRZYKŁAD 1 Izolacja IgE naczelnych [0334] Ludzką IgE klonowano z linii ludzkich komórek szpiczaka IgE U266 (ATCC, Manassas, VA, TIB #196). Sekwencje rezus i cyno IgE oznaczano przez klonowanie i sekwencjonowanie cdna z IgE pochodzącej z obwodowych komórek jednojądrzastych krwi rezus i cyno, które stymulowano w celu wytworzenia komórek przełączanych IgE (PBMC). Startery przedni i odwrotny (pokazane poniżej) zaprojektowane na podstawie sekwencji ludzkich IgE. Leżący wyżej starter przedni zaprojektowano tuż przed domeną CH2. Starter odwrotny zaprojektowano na końcu domeny transbłonowej. PBMC rezus i cyno otrzymano z California National Primate Research Center (Davis, CA). 5 x 10 6 PBMC hodowano z IL-4 (100 ng/ml) i CD40L (3 µg/ml) (R&D Systems, Minneapolis, odpowiednio MN, #204-IL i #617-CL) przez 4 dni. Komórki następnie zebrano, przygotowano RNA (RNeasy Mini Kit, #74106, Qiagen, Valencia, CA), i cdna wykonano stosując zestaw BD Sprint Powerscript oligo dt priming kit (BD Biosciences, San Jose, CA, #639558). Następnie przeprowadzono RT-PCR [94 C 2 minuty; 94 C 15 sekund; 60 C 30 sekund; 68 C 2 minuty; 30 cykli; 68 C 10 minut]. Dodano polimerazę Taq (10 minut 72oC) po zainicjowaniu cykli aby dodać zwisy A do klonowania TA (Invitrogen, Carlsbad, CA, # ). Produkty PCR klonowano przez klonowanie TOPO-TA i klony następnie poddano weryfikacji sekwencji (Genentech, Inc.). Sekwencje ludzkie, rezus i cyno dopasowano przy użyciu własnych programów do analizy (Genentech, Inc.). Poniższe homologie są dla sekwencji pomiędzy domenami CH2 i transbłonowymi. Między rezus (432 aminokwasy) i cyno (431 aminokwasy) było 6 różnic co stanowi około 98.6% homologii. Między człowiekiem i rezus było 53 różnic co równa się 87.7% homologii. Między człowiekiem a cyno było 56 różnic, co równa się 87% homologii (Patrz Figura 1). Startery zastosowane do klonowania: [0335] Startery przednie powyżej sekwencji CH2: 5 -ccgcccaccgtgaagatcttacagtc (SEQ ID NO:63) Starter odwrotny na końcu domeny transbłonowej: 5 -cggctcgagactaggcgtggggctggaggacgttg (SEQ ID NO:64) PRZYKŁAD 2
95 94- Izolacja Ab anty-ige/m1 Wytwarzanie białka fuzyjnego CH3-CH4-M1 /Fc [0336] Białko fuzyjne ludzka IgE CH3-CH4-M1 /Fc wygenerowano za pomocą amplifikacji PCR domen ludzkich IgE CH3-CH4-M1, oraz dodatkowych 15 aminokwasów bezpośrednio za domeną M1 z cdna otrzymanego z komórek U266 (ATTC Manassas, VA, TIB#196). Przygotowano RNA z komórek U266 (RNeasy Mini Kit, #74106, Qiagen, Valencia, CA) i cdna wytworzono stosując BD Sprint Powerscript oligo dt priming (BD Biosciences, San Jose, CA, #639558). Przeprowadzono RT-PCR i produkt PCR klonowano za pomocą klonowania TOPO-TA (Invitrogen, Carlsbad, CA, # ). N-końcową sekwencję sygnałową dodano przez mutagenezę PCR do ekspresji w komórkach ssaczych, i sekwencję sygnałowa +CH3+CH4+M1 +15 aminokwasów sklonowano do wektora ekspresyjnego (prk huigg1 Fc; Genentech) dla wygenerowania fuzji C-końcowej z regionem Fc ludzkiej IgG1. Sekwencja białka końcowego, włącznie z sekwencją sygnałową i Fc ludzkiej IgG1 jest następująca: [0337] Białko wytwarzano przez przejściową transfekcję komórek CHO (Genentech), i białko oczyszczono z supernatantu hodowli komórkowej przy użyciu techniki chromatograficznej z kolumną sefaroza Białko A. Immunizacja i tworzenie hybrydom [0338] Panel pan-specyficznych przeciwciał, które selektywnie wiążą część M1 ludzkiej IgE- M1 wytworzono stosując białko CH3-CH4-M1 /Fc. Do każdej poduszeczki tylnej łapy myszy BALB/c wstrzyknięto 1 µg CH3-CH4-M1 /Fc zawieszonego w adjuwancie monofosforylo-lipid A i dikorynomykolan trehalozy (MPL + TDM) (Corixa, Hamilton, MT) w odstępach 3- do 4-dniowych. Surowicę pobrano po 8 dawkach przypominających i oznaczano miano za pomocą testu immunoenzymatycznego (ELISA) i sortowanie komórek aktywowane fluorescencją i cytometrią przepływową (skrining FACS, patrz następny rozdział) w celu zapewnienia, że myszy wykazywały dobrą odpowiedź immunologiczną na CH3-CH4-M1 /Fc. Trzy dni po ostatniej dawce przypominającej, komórki węzłów podkolanowych poddano fuzji z linią komórek szpiczaka myszy P3X63Ag.U.1 (patrz, na przykład, Chuntharapai i wsp., 1997, Methods Enzymol. 288:15-27). Zfuzowane komórki hybrydoma wybrano z niezfuzjowanych komórek węzłów podkolanowych lub komórek szpiczaka stosując selekcję hypoksantyna-aminopteryna-tymidyna (HAT) w pożywce D z
96 95- zestawu ClonaCell hybridoma selection kit (StemCell Technologies, Inc., Vancouver, BC, Canada), uzyskując wygenerowane 4224 klony. Skrining dla pan-specyficznych przeciwciał anty-ludzki M1 [0339] Klony hybrydom, wytworzonych jak opisano poprzednio, zbadano pod kątem produkcji przeciwciał monoklonalnych wiążących do regionu M1 z białka CH3-CH4- M1 /Fc, regionm1 ludzkiego IgE-M1 ulegał stabilnej ekspresji na powierzchni linii komórkowej chłoniaka z komórek B myszy A20 (IgE-M1 /A20), i region M1 ludzkiej IgE- M1 ulegał stabilnej ekspresji na powierzchni ludzkiej linii komórkowej chłoniaka z komórek B BJAB (IgE-M1 /BJAB). Negatywne kontrole obejmowały ludzki CD-4/Fc, ludzką IgE ulegającą stabilnej ekspresji na powierzchni linii komórkowej chłoniaka z komórek B myszy A20 (IgE/A20), linię komórkową chłoniaka z komórek B myszy A20 (A20), ludzką IgE ulegającej stabilnej ekspresji na powierzchni ludzkiej linii komórkowej chłoniaka z komórek B BJAB (IgE/BJAB) i ludzką linię komórkową chłoniaka z komórek B BJAB (BJAB). Skrining ELISA [0340] Do przebadania 4224 klonów, wykonano test immunoenzymatyczny (ELISA) zasadniczo w sposób opisany w Baker i wsp., 2002, Trends Biotechnol., 20: Testy przeprowadzono w płytkach zarówno 384, jak i 96-dołkowych. W skrócie, 384- (lub 96-) dołkową płytkę pokryto 50 µl koziego przeciw-ludzkiego IgG Fc (MP Biomedicals, Irvine, CA) w stężeniu 2 µg/ml w buforze do powlekania (0.05 M bufor węglanowy, ph 9.6), zamknięto i przechowywano przez noc w temperaturze 4 C. Po usunięciu roztworu powlekającego, do każdego dołka dodano 80 µl (lub 200 µl dla płytki 96-dołkowej) roztworu testowego/blokującego zawierającego 0.5% albuminy surowicy bydlęcej i 0.05% Tween -20 w PBS (ph 7.4) (rozcieńczalnik ELISA) i płytki inkubowano w temperaturze pokojowej przez jedną godzinę z mieszaniem. Następnie dołki przemyto trzykrotnie 100 µl (lub 300 µl dla płytki 96-dołkowej) 0.05% Tween -20 w PBS (bufor do przemywania). [0341] Po etapie przemywania, dodano do każdego dołka 50 µl (lub 100 µl dla płytki 96- dołkowej) roztworu zawierającego antygen 0.4 µg/ml CH3-CH4-M1 /Fc lub ludzki CD-4/Fc w rozcieńczalniku do ELISA, i płytki inkubowano w temperaturze pokojowej przez jedną godzinę z mieszaniem. Dołki przemyto trzy razy buforem do przemywania jak poprzednio. Supernatant z pojedynczych klonów hybrydom dodano w taki sposób, że jeden dołek z CH3- CH4-M1 /Fc i jeden dołek z ludzkim CD-4/Fc otrzymały po 50 µl (lub 100 µl dla płytki 96- dołkowej) supernatantu z pojedynczego klonu hybrydomy. Płytki inkubowano w temperaturze pokojowej przez jedną godzinę z mieszaniem, i dołki przemyto trzy razy buforem do przemywania jak poprzednio. [0342] Po przemyciu, dodano do każdego dołka 50 µl (lub 100 µl dla płytki 96-dołkowej) rozcieńczenia 1:1000 owczej przeciw-mysiej IgG skoniugowanej do peroksydazy chrzanowej (brak reaktywności krzyżowej z ludzką IgG (MP Biomedicals)) w rozcieńczalniku do ELISA. Płytki inkubowano w temperaturze pokojowej przez jedną godzinę z mieszaniem, przemyto trzy razy buforem do przemywania jak poprzednio i osuszono przez poklepanie. Dołki wywołano dodając 50 µl (lub 100 µl dla płytki 96-dołkowej) mikrodołkowego substratu dla
97 96- peroksydazy tetrametylobenzydyny (TMB) (BioFX Laboratories, Owing Mills, MD, nr katalogowy TMBW ) do każdego dołka i inkubując w temperaturze pokojowej przez 5-10 minut lub aż do zaobserwowania dobrej zmiany koloru. Wywoływanie zatrzymano dodając 50 µl (lub 100 µl dla płytki 96-dołkowej) roztworu zatrzymującego TMB Stop Solution (BioFX Laboratories nr katalogowy BSTP ) do każdego dołka. Płytki analizowano za pomocą czytnika płytek Sunrise (Tecan US, Inc., Research Triangle Park, NC) przy 650 nm. [0343] Jako kontrole użyto krew z wstępnego pobierania i polisurowice. Próbki krwi z wstępnego pobierania zawierały mysie surowice przed immunizacją, a próbki polisurowic zawierały mysie anty-surowice otrzymane po 10 immunizacjach. Skrining FACS [0344] Aby przesiać 3221 klonów generowanych w Przykładzie 1, wykonano sortowanie komórek aktywowane fluorescencją (FACS) stosując linie komórkowe IgE-M1 /A20 i IgE- M1 /BJAB. Linie komórkowe IgE/A20, IgE/BJAB, A20 i BJAB służyły jako kontrole negatywne. Komórki ponownie zawieszono i wirowano 500g przez 5 minut w 4 C. Pożywkę odessano i komórki ponownie zawieszono w 4 C FACSFlow (BD Biosciences, San Jose, CA) bufor z dodatkiem 1% płodowej surowicy bydlęcej (bufor do barwienia komórek). Komórki odwirowano jak poprzednio, pożywkę odessano, a komórki ponownie zawieszono przy 2x10 6 komórek/ml buforu do barwienia komórek w 4 C. Komórki dodano do 96-dołkowych okrągłodennych płytek przy 50 µl/dołek (1x10 5 komórek/dołek) i do każdego dołka dodano 100 µl supernatantu z pojedynczych klonów hybrydom tak, że każdy supernatant hybrydomy inkubowano z jednym dołkiem zawierającym każdą linię komórkową. Płytkę inkubowano na lodzie przez 30 minut. Płytkę odwirowano przy 500g przez 5 minut w 4 C i supernatanty odessano. Każdy dołek ponownie zawieszono w 200 buforu do barwienia komórek w 4 C, i płytki odwirowano jak poprzednio. Bufor do barwienia komórek odessano. [0345] Po etapie przemywania, komórki w każdym dołku zawieszono ponownie w 100 µl rozcieńczenia 1:1000 z kozim anty-mysia IgG Fc sprzężonym z R-fikoerytryną (Jackson Immunoresearch, West Grove, PA) w buforze do barwienia komórek w 4 C, i płytki inkubowano w ciemności na lodzie przez 30 minut. Płytkę odwirowano przy 500g przez 5 minut w 4 C i supernatanty odessano. Każdy dołek zawieszono w 200 buforu do barwienia komórek w 4 C, i płytki odwirowano jak poprzednio. Bufor do barwienia komórek odessano. Komórki w każdym dołku ponownie zawieszono w 200 µl buforu do barwienia komórek w 4 C i przeniesiono do 1.2ml probówek do mikromiareczkowania (Quality Scientific Plastics, Petaluma, CA). FACS przeprowadzono na FACScan lub FACSCalibur (BD Biosciences). Sekwencjonowanie N-terminalne/Izolacja RNA/sekwencjonowanie i klonowanie [0346] Przeciwciała oczyszczono z supernatantu hybrydom anty-ige/m1 i analizowano dla sekwencjonowania N-końcowego w celu oznaczania klonalności w soli fizjologicznej buforowanej fosforanem. Każde przeciwciało rozdzielono na Precast 4-20% Tris HCl SDS- PAGE (Invitrogen, Carlsbad, CA) w warunkach redukujących. Każdy rozdzielony łańcuch ciężki (HC) i łańcuch lekki (LC) poddano sekwencjonowaniu N-końcowemu stosując N-
98 97- końcowej sekwencer Procise 494 (Applied Biosystems, Foster City, CA) stosując cykl 20 min, jak opisali Henzel i wsp. Analytical Biochemistry 267, (1999). Pierwszych 25 reszt zsekwencjonowano w celu ustalenia monoklonalności. Gdy łańcuch HC lub LC Ab zablokowano grupą piroglutamylową co sprawiało, że N-końcowy aminokwas stał się niedostępny do sekwencjonowania Edmana, przed sekwencjonowaniem przeprowadzono usunięcie grupy blokującej. Grupę piroglutamylową usunięto enzymem aminopeptydazą piroglutaminianową (PGAP, Sigma, St Louis, MO) stosując protokół opisany przez Pham i wsp. Electrophoresis 2005, [0347] Sekwencję łańcucha ciężkiego (HC) i łańcucha lekkiego (LC) przeciwciała M1 7A6, 1C11, 47H4, 26A11, 45C1 i 28E9 określono jako następującą: 7A6 HC QVQLQQSGAELVRPGASVTLSCKAS (SEQ ID NO:66) LC DIVMSQSPSSLTVSVGEKVTLSCKS (SEQ ID NO:67) 1C11 HC QVQLQQSGAELVRPGASVTLSCKAS (SEQ ID NO:68) LC DIVMSQSPSSLAVSVGEKVTMSCKS (SEQ ID NO:69) 47H4 HC EVKLVESGGGLVQPGGSRKLSCAAS (SEQ ID NO:70) LC DVVLTQTPLSLPVSLGDQASI (SEQ ID NO:71) 26A11 HC EVQLQQSGPELVKPGASVKMSCKAS (SEQ ID NO:72) LC DIQMTQTTSSLSASLGDRVTITCRS (SEQ ID NO:73) 45C1 HC QIQLVQSGPELKKPGETVK (SEQ ID NO:74) LC DVVMTQTPLTLSVTIGQPASISCK (SEQ ID NO:75) 28E9 HC EVKLVESGGGLVQPGGSLRLSCATS (SEQ ID NO:76) LC DIQMTQSPASLSVSVGETVTFTCR (SEQ ID NO:77)
99 98- W celu określenia sekwencji pełnej długości, zdegenerowane startery 5 zaprojektowano z sekwencji aminokwasowych na końcu N i znanych segmentów genów Ig i przeprowadzono PCR stosując zdegenerowany poniżej sekwencje konserwatywne łańcucha ciężkiego i lekkiego. 7A6 Startery przednie HC FOR.BsiWI 5 -tcgacgtacgctcaggttcagctgcagcaatctggggctgagctgg (SEQ ID NO:78) LC 1C11 FOR.EcoR V 5 -gatcgatatcgtgatgtcccagtctccctcctccctaac (SEQ ID NO:79) HC FOR.BsiWI 5 -tcgacgtacgctcaggttcaattgcagcagtctggggctgagctgg (SEQ ID NO:80) LC 47H4 FOR.EcoR V 5 -gatcgatatcgtaatgtctcagtctccttcctccctagc (SEQ ID NO:81) HC LC 26A11 HC LC 45C1 HC LC 28E9 HC LC 9.1HCF.Bsi WI 47H4LCF.E corv G7F7HCF. BsiWI 2B4LCF.Ec orv 2G6HCF.Bs iwi 9C10LCF.E corv 9.1HCFBsi WI 5E10.LCF. EcoRV 5 -tcgacgtacgctgaggtgaagttggtggagtctgggggaggcttag (SEQ ID NO:82) 5 -gatcgatatcgtgctgactcagactccactctccctgcc (SEQ ID NO:83) 5 -tcgacgtacgctgaggtccagctccagcagtctggacctgagc (SEQ ID NO:84) 5 -gatcgatatccagatgacccaaactacatcctccctg (SEQ ID NO:85) 5 -tcgacgtacgctcagatccagttggtgcagtctggacctgagctg (SEQ ID NO:86) 5 -gatcgatatcgtgatgacgcagactccactcactttgtcgg (SEQ ID NO:87) 5 -tcgacgtacgctgaggtgaagctggtggagtctgaaggaggcttgg (SEQ ID NO:88) 5 -gatcgatatccagatgcccagtctccagcctccctatc (SEQ ID NO:89)
100 99- Starter Łańcucha Ciężkiego (HCR) Starter Łańcucha Lekkiego (LCR) Startery odwrotne 5 -ctggacagggatccagagttccaggtcactgtcactggctcaggg (SEQ ID NO:90) 5 -ctgtaggtgctgtctttgctgtcctgatcagtccaactg (SEQ ID NO:91) [0348] RNA zbierano z hybrydom anty-ige/m1 (RNeasy Mini Kit, #74106, Qiagen, Valencia, CA) i cdna otrzymano stosując zestaw BD Sprint Powerscript oligo dt priming kit (BD Biosciences, San Jose, CA, #639558). PCR przeprowadzono przy użyciu powyższych starterów i produkty PCR sklonowano stosując zestaw TOPO-TA kit (Invitrogen, Carlsbad, CA, # ). PCR przeprowadzono stosując Platinum Pfx High Fidelity Polymerase (Invitrogen, Carlsbad, CA, # ) z warunkami PCR [94 C 2 minuty; 94 C 15 sekund; 60 C 30 sekund; 68 C 2 minuty; 30 cykli; 68oC 10 minut]. Klony przeszukiwano pod kątem wstawek i sekwencjonowano wiele klonów (Genentech, Inc.) aby zweryfikować oryginalną sekwencję genu łańcucha ciężkiego i lekkiego anty-ige /M1. [0349] Domeny zmienne przeciwciał anty-m1 subklonowano do wektorów ekspresyjnych w różnych regionach Fc w celu wygenerowania chimerycznych przeciwciał z różnych gatunków i/lub izotypowych Fc. Startery zaprojektowano aby klonować domenę zmienną (CDR + zrąb) na regionach Fc mysiej IgG2a, mysiej IgG2a-DANA i ludzkiej IgG1. [0350] Startery zaprojektowanego do wprowadzania miejsc restrykcyjnych w sekwencji łańcucha ciężkiego i lekkiego. Łańcuchy ciężkie sklonowano za pomocą BsiWI i Apal. Łańcuchy lekkie sklonowano za pomocą EcoRV i KpnI. Nowe produkty reakcji PCR były następnie trawione i ligowae do migg2a (LPG10), migg2a-dana (perk-e27dana), lub huigg1 dla łańcucha ciężkiego, lub wektorów ekspresyjnych mkappa (LPG2) bądź hukappa (Genentech, Inc.). W ostatecznych klonach zweryfikowano sekwencję (Genentech, Inc.). [0351] Startery 3 i kombinacje starterów przednich i odwrotnych do klonowania zmiennych sekwencji CDR są następujące: 7A6 Startery Odwrotne HC REV.ApaI 5 -accgatgggcccttggtggaggctgaagagactgtgag (SEQ ID NO:92) LC REV.KpnI 5 -ccttggtaccccctccgaacgtgtacggatagctataatattg (SEQ ID NO:93) 1C11 HC REV.ApaI 5 -gaccgatgggcccttggtggaggctgaggagactgtg (SEQ ID NO:94) LC REV.KpnI 5 -ccttggtaccccctccgaacgtgtacggatagctataa (SEQ ID NO:95) 47H4
101 100- HC LC 47H4HCR.ApaI 47H4LCR.KpnI 5 -tgggcccttggtggaggctgaggagacggtgactgag (SEQ ID NO:96) 5 -ttccaacttggtacctccacc (SEQ ID NO:97) 26A1 1 HC LC 7G8HCR. ApaI 47H4LCR.KpnI 5 - gaccgatgggcccttggtggargctgcagagacagtgaccagag (SEQ ID NO:98) 5 -ttccaacttggtacctccacc (SEQ ID NO:99) 45C1 HC LC 28E9 HC LC 45C1H CR.Apa I 5C2LC R.KpnI 28E9.H C.REV. ApaI 1D5.2L CR,Kpn I 5 -cgatgggcccttggtggargckgaggagacggtgagaatg (SEQ ID NO:100) 5 -agcttggtaccccctccg (SEQ ID NO:101) 5 -cgatgggcccttggtggaggctgaggagacggcgactgag (SEQ ID NO:102) 5 - ttccaacttggtacccgagccg (SEQ ID NO:103) Przedni Kombinacje starterów: Odwrotny 7A6: HC 7A6.FOR.BsiWI --- 7A6.REV.ApaI LC 7A6.FOR.EcoRV --- 7A6.REV.KpnI 1C11: HC 1C11.FOR.BsiWI --- 1C11.REV.ApaI LC 1C11.FOR.EcoRV --- 1C11.REV.KpnI 47H4: HC 9.1.HCF.BsiWI --- 7H4HCR.ApaI
102 101- LC* 47H4.LCF.EcoRV H4.LCR.KpnI 26A11 HC C7F7.HCF.BsiWI --- 7G8.HCR.ApaI LC 2B4.LCF.EcoRV H4.LCR.KpnI 45C1 HC 2G6.HCF.BsiWI C1.HCR.ApaI LC 9C10.LCF.EcoRV --- 5C2.LCR.KpnI 28E9 HC 9.1.HCF.BsiWI E9.HCR.ApaI LC 5E10.LCF.EcoRV -- 1D52.LCR.KpnI [0352] Łańcuch lekki 47H4 miał wewnętrzne miejsce KpnI, które wykluczało bezpośrednie klonowanie do wektorów ekspresyjnych mkappa i hukappa. Wewnętrzne miejsce KpnI zmutowano stosując zestaw Quick Change XL Site Directed Mutagenesis Kit (Stratagene, Cedar Creek, Texas, # ) [95 C 1minuta; 95 C 50 sekund; 60 C 50 sekund; 68 C 4.5 minuty; 68 C 7minut]. Startery zaprojektowano zgodnie z wymaganiami zestawu do mutacji pojedynczego nukleotydu w miejscu KpnI, który nie zmienia sekwencji aminokwasowej. TAC zmutowano do TAT, co zachowało tyrozynę (Y) na tej pozycji. Zweryfikowano sekwencję w ostatecznych klonach (Genentech, Inc.). [0353] Starterami użytymi do mutagenezy były: FOR 5 -cc tat tta cat tgg tat ctg cag aag cca ggc c (SEQ ID NO:104) REV 5 -ggc ctg gct tct gca gat acc aat gta aat agg (SEQ ID NO:105) PRZYKŁAD 2A Humanizacja Przeciwciał Anty-IgE/M1 [0354] Przykład ten opisuje humanizację mysich przeciwciał anty-ige/m1 26A11, 7A6 i 47H4. Materiały i metody [0355] Domenę M1 ludzkiej IgE zasocjowanej z błoną eksprymowano w fuzji Fc w komórkach CHO i oczyszczono konwencjonalnymi środkami. Hybrydomy z ekspresją przeciwciała 26A11, 7A6 i 47H4 otrzymano przez immunizację myszy rekombinowanym białkiem fuzyjnym Fc-M1 i zidentyfikowano za pomocą testu ELISA stosując szalki powlekane M1 -Fc. Funkcjonalne przeciwciała zidentyfikowano na podstawie ich zdolności do wiązania się do komórek eksprymujących M1 i promowania apoptozy. [0356] Klonowanie mysich domen zmiennych 26A11, 7A6 i 47H4 - Całkowity RNA ekstrahowano z komórek hybrydomy wytwarzających 26A11, 7A6 lub 47H4 stosując standardowe metody. Domeny zmienną lekką (VL) i zmienną ciężką (VH) amplifikowano za
103 102- pomocą RT-PCR ze starterami zdegenerowanymi do łańcuchów ciężkich i lekkich. Te startery są specyficzne dla N-terminalnej sekwencji aminokwasowej regionów VL i VH. Odpowiednio, startery LC i HC zostały zaprojektowane tak, aby hybrydyzowały z odpowiednim regionem w lekkiej stałej (CL) i stałej domenie ciężkiej 1 (CH1), które są wysoce konserwatywne u wszystkich gatunków. Sekwencję polinukleotydową wstawek oznaczano przy zastosowaniu rutynowych metod sekwencjonowania. Sekwencje aminokwasowe VL oraz VH 26A11, 7A6 i 47H4 przedstawiono na odpowiedni Figurach 6A i 6D, 6B i 6E, i 6C i 6F. [0357] Bezpośrednie przeszczepy regionów hiperzmiennych na akceptorowy ludzki konsensusowy zrąb - Fagemid stosowany w tej pracy jest wektorem do prezentacji jednowartościowego Fab-g3 i składa się z 2 otwartych ramek odczytu, pod kontrolą pojedynczego promotora phoa. Pierwsza otwarta ramka odczytu składa się z sekwencji sygnałowej stii zfuzjowanej z domenami VL i CH1 akceptorowego łańcucha lekkiego, a druga składa się z sekwencji sygnałowej stii zfuzowanej z domenami VH i CH1 łańcucha ciężkiego akceptorowego, a następnie mniejszego białka P3 płaszcza faga. [0358] Domeny VL i VH z mysich przeciwciał 26A11, 7A6 lub 47H4 (mu26a11, mu7a6 lub mu47h4) dopasowano z sekwencjami konsensusowymi ludzkiego VL kappa I (huki) i ludzkiego VH podgrupy III (huiii). Aby dokonać przeszczepy CDR, regiony hiperzmienne z mu26a11, mu7a6 lub mu47h4 szczepiono do konsensusowych zrębów akceptorowych hukj i huiii aby wygenerować bezpośrednie przeszczepy CDR (26A11.v1, 7A6.v1 lub 47H4.v1) (Figury 6A-6F). W domenie VL następujące regiony przeszczepiono do ludzkiego konsensusowego akceptora: pozycje (L1), (L2) i (L3). W domenie VH przeszczepiono pozycje (H1), (H2) i (H3). MacCallum i wsp. [MacCallum i wsp. J. Mol. Biol. 262: (1996))] zbadali struktury krystaliczne kompleksu przeciwciała z antygenem i okazało się, że pozycja 49 i 94 łańcucha ciężkiego jest częścią regionu kontaktowego, tym samym uzasadnione wydaje się aby zawrzeć te pozycje w definicji CDR-H2 i CDR-H3 przy humanizowaniu przeciwciał. [0359] Humanizowane przeciwciała anty-ige/m1 26A11.v1, 7A6.v1 i 47H4.v1 wytworzono jak przeciwciała IgG za pomocą mutagenezy Kunkela wektorów ekspresyjnych LC i HC stosując oligonukleotydy osobno dla każdego regionu hiperzmiennego. Zmiany aminokwasów w celu zwiększenia stabilności również wykonano za pomocą mutagenezy Kunkela. Kunkel i wsp., J. Biol. Chem. 263(29): (1988). Właściwe klony zostały zidentyfikowane przez sekwencjonowanie DNA. [0360] Wywtarzanie IgG - Do celów przesiewowych, warianty IgG początkowo wytwarzano w komórkach 293. Wektory kodujące VL i VH (25 µg) były transfekowane do komórek 293 przy użyciu systemu Fugene. 500 µl FuGene zmieszano z 4.5 ml pożywki DMEM nie zawierającej FBS i inkubowano w temperaturze pokojowej przez 5 min. Każdy łańcuch (25 µg) dodano do tej mieszaniny i inkubowano w temperaturze pokojowej przez 20 min, a następnie przeniesiono do pięciu butelek T-150 do transfekcji przez noc w temperaturze 37 C w 5% CO 2. Następnego dnia pożywkę zawierającą mieszaninę transfekcyjną usunięto i zastąpiono 23 ml pożywki PS04 z 0.1 ml/l pierwiastków śladowych (A0934) i 10 mg/l
104 103- insuliny (A0940). Komórki inkubowano przez kolejne 5 dni, po czym pożywkę zebrano przy 1000 rpm przez 5 min i jałowo przesączono z wykorzystaniem sączka o niskim wiązaniu białka 0,22 um. Próbki mogą być przechowywane w 4 C po dodaniu 2.5 ml 0.1% PMSF na każde 125 ml pożywki. [0361] Oznaczenia powinowactwa - Oznaczenia powinowactwa wykonywano analizą Scatcharda, ELISA wiązania komórek i powierzchniowym rezonansem plazmonowym z wykorzystaniem BIAcore or A100. [0362] Analiza Scatcharda komórki Daudi IgE/M1 ludzkie, rezus i cyno przygotowano do wiązania w zimnym buforze wiążącym złożonym z zasadowej pożywki, z 10mM HEPES ph 7.4, i 2% FBS oraz 40ug/ml ludzkiej IgG. Komórki rozcieńczono do stężenia 1.7 x 10 6 komórek/ml, i trzymano na lodzie do momentu ich dodania do oznaczenia. Białka/przeciwciała joduje się metodą Iodogen. Różne stężenia zimnego białka wytwarza się w trzech powtórzeniach, stosując rozcieńczenie 1:2 poczynając od stężenia nasycenia i kończąc na stężeniu zera, w sumie 14 stężeniach. Gorące białko w pojedynczym stężeniu dodano do wszystkich roztworów dla konkurowania z zimnym białkiem. Na koniec, komórki są dodawane do mieszaniny gorącego i zimnego białka, stosowano 250,000 komórek na próbkę. Test utrzymuje się w 4 C przez 4 godziny, wytrząsając. Każdą próbkę następnie zbiera się i przesącza przez błonę, przemywa co najmniej 3-krotnie buforem do wiązania, pozostawia do wyschnięcia, a następnie zlicza przez 1 minutę, stosując Perkin Elmer Wizard 1470 Auto Gamma Counter. [0363] Konkurencyjny elektrochemilluminescencyjny test wiązania do komórek - Względne powinowactwo wiązania humanizowanych wariantów mierzono stosując test konkurencyjnego elektrochemiluminescencyjnego wiązania do komórek: Transfekowane komórki BJAB eksprymujące IgE-M1 (BJAB-IgE/M1 )(długi) lub kontrolne transfekowane komórki nie eksprymujące M1 (BJAB-IgE) (krótki) przemyto solą fizjologiczną buforowaną fosforanem (PBS) i wysiano przy 25,000 komórek/dołek w 25 µl PBS na 96-dołkowych płytkach MULTI-ARRAY High Bind (Meso Scale Discovery). Komórki inkubowano na płytkach w ciągu jednej godziny w temperaturze pokojowej, aby umożliwić przylgnięcie komórek do dołków. W celu zablokowania niespecyficznego wiązania, dodano 25 µl 30% FBS w PBS do każdego dołka. Płytki następnie inkubowano w temperaturze pokojowej przez 30 minut z delikatnym wytrząsaniem. Roztwór blokujący zdekantowano i dodano rozcieńczenia seryjne humanizowanych wariantów ( nm) uzupełnione stałą ilością przeciwciała biotynylowanego anty-ige M1 (33.3 nm 26A11 lub 47H4) w 25 µl PBS zawierającego 2% FBS (bufor do testu). Po jednej godzinie inkubacji w temperaturze pokojowej z łagodnym mieszaniem, płytki przemyto trzy razy PBS (300 µl/dołek), z użyciem płuczki do płytek mikrotitracyjnych (ELx405 Select, Bio-Tek Instruments, Inc., Winooski, VT). Związane przeciwciała wykryto przez dodanie 25 µl 0.25 µg/ml znakowanej rutenem streptawidyny do buforu do testu. Po jednej godzinie inkubacji w temperaturze pokojowej, płytki przemyto i dodano oparty na Tris Read Buffer T (1x, bez środka powierzchniowo czynnego) (Meso Scale Discovery) (150 µl/dołek). Sygnały elektrochemiluminescencyjne rejestrowano stosując czytnik Sector Imager 6000 (Meso Scale Discovery).
105 104- [0364] Biacore Wykorzystano dwa kierunki; M1 -Fc albo określony wariant przeciwciała immobilizowano (około 100 RU, w 10 mm Octan Sodu ph 4.8 na płytce czujnikowej CM5) i odpowiedni ligand (odpowiednio wariant przeciwciała lub M1 Fc) służył jako analit (nastrzyknięty z szybkością przepływu 30 µl/min, stosując 2-krotne seryjne rozcieńczenie 0.5 do 1000 nm w PBST). Każdą próbkę analizowano z 3 minutami asocjacji i 10 minutami dysocjacji. Po każdym nastrzyku płytkę regenerowano stosując 10 mm glicynę ph 1.7. [0365] Biacore A-100 -M1 - immobilizowano (około 100 RU, w 10 mm Octanie Sodu ph 4.8 na płytce czujnikowej CM-5) i wariant przeciwciała służył jako analit (nastrzyknięty z szybkością przepływu 30 µl/min, stosując 2-krotne seryjne rozcieńczenie 0.5 do 1000 nm w PBST). Każdą próbkę analizowano z 5 minutami asocjacji i 5 minutami dysocjacji. Po każdym nastrzyku płytkę regenerowano stosując 10 mm glicynę ph 1.7. [0366] Odpowiedź wiązania skorygowano przez odjęcie kontrolnej komórki przepływowej z komórek przepływowych wariantu IgG 18F7. Do analizy kinetyki wykorzystano model Langmuira 1:1 równoczesnego dopasowywania k on i k off. Wyniki i dyskusja [0367] Przeszczepy CDR 26A11, 7A6 i 47H4 - Ludzki zrąb akceptorowy stosowany do humanizacji jest oparty na ludzkiej konsensusowej domenie kappa I VL i konsensusowej ludzkiej domenie VH podgrupy III. Domeny VL i VH mu26a11, mu7a6 i mu47h4 dopasowano z ludzkimi domenami kappa I i podgrupy III; każdy region determinujący komplementarność (CDR) identyfikowano i przeszczepiano do ludzkiego zrębu akceptorowego w celu wygenerowania przeszczepu CDR, który może ulegać ekspresji jako IgG (Figury 6A-6F). [0368] Ocena Powinowactwa - 26A11.v1, 7A6.v1 i 47H4.v1 oceniano za pomocą testu ELISA wiązania do komórek i powierzchniowego rezonansu plazmonowego (Tabela 1). Testy te wykazały, że przeszczepy CDR miały bardzo podobne powinowactwo do swoich odpowiednich przeciwciał hybrydom. [0369] Eliminacja miejsc potencjalnej deamidacji i tworzenia kwasu izo-asparaginowego w CDR-L1 z 26A11 i 47H4 - Aby uniknąć potencjalnych problemów produkcyjnych, wyeliminowano miejsca tworzenia kwasu izo-asparaginowego Asn 28 -Gly 29 w CDR-L1z 47H4.v1 i Asn 31 -Ser 32 w CDR-L1 z 26A11.v1 przez próbkowanie reszt znajdujących się w innych przeciwciał w tych pozycjach (Figura 6A, 6C, Tabela I). Zmiany Gly 29 na Ala 29 w CDR-L1 z 47H4.v1 (47H4.v2 i 47H4.v5), oraz Ser 32 na Tyr 32 w CDR-L1 z 26A11 (26A11.v3 i 26A11.v6) lub Ala 32 (26A11.v2 lub 26A.11.v5) okazały się być odpowiednimi zamiennikami. Ponadto, zmiana Asn 31 na albo Ser 31 (26A11.v13 lub 26A11.v15), albo Gln 31 (26A11.v14 lub 26A11.v16) służyła również do wyeliminowania potencjalnej deamidacji i tworzenia kwasu izo-asparaginowego i dostarczała kandydatów o powinowactwach podobnych do odpowiednich przeciwciał hybrydomy. [0370] Potencjalne utlenianie Met 34 w CDR-H1 z albo 26A11.v1, albo 47H4.v1 unikano zmieniając tą resztę na Ile 34 w obu przeciwciałach bez wpływu na wiązanie M1.
106 105- PRZYKŁAD 3 Specyficzność Ab anty-m1 [0371] Specyficzność klonów przeciwciał anty-ige/m1 testowano na linii ludzkich komórek B BJAB (ATCC Manassas, VA, #HB-136) transfekowanych IgE z sekwencją M1 ( postać długa ) lub IgE bez sekwencji M1 ( postać krótka ). Linie komórkowe IgE BJAB wytworzono przez retrowirusową transdukcję komórek BJAB stosując wektor ekspresyjny pmscv (Washington University, St. Louis, MO). cdna pełnej długości ciężkich i lekkich łańcuchów ludzkiego receptora komórek B IgE uzyskano z ludzkiej linii komórkowej szpiczaka IgE U266 (ATCC, Manassas, VA, TIB#196) i subklonowano do wektora retrowirusowego w połączeniu z IRES (wewnętrzne miejsce wejścia rybosomu) aby umożliwić bicistronową kotransfekcję i koekspresję łańcuchów ciężkich i lekkich przeciwciał. Dalszy opis metody transdukcji retrowirusowej jest przedstawiony poniżej. [0372] Komórki pakujące PG13 (ATCC CRL-10686) wysiano na 10 cm płytce do hodowli tkankowych przy 2 x 10 6 komórek na płytkę (DMEM o wysokiej zawartości glukozy, 10% FBS, penicylina, streptomycyna, 2 mm L-glutaminy) na 24 godziny. Komórki transfekowano konstruktami pmscv DNA stosując FuGENE 6 i hodowano przez 2 dni w temperaturze 37 C, 5% CO2. Supernatant hodowli komórkowej zawierający cząstki retrowirusowe zbierano i przesączono przez filtr 0.4 mikrona. Jałowy siarczan protaminy dodano do końcowego stężenia 10 µg/ml, i 4 ml supernatantu wykorzystano do infekowania w przybliżeniu 1 x 10 6 komórek BJAB infekcją przez wirowanie w temperaturze 32 C w ciągu 90 minut, a następnie dalej hodowano w supernatancie retrowirusowym przez 3-4 godzin w temperaturze 37 C w 5% CO 2. Zakażone komórki BJAB odzyskano, przeniesiono do pożywki do hodowli komórkowych RPMI (RPMI, 10% FBS (Hyclone, Logun, UT, #SH ), penicylina, streptomycyna, 2 mm L-glutaminy) i ekspandowano dla sortowania. Pozytywnie transfekowane komórki zidentyfikowano metodą cytometrii przepływowej przy użyciu przeciwciała anty-ludzka IgE MAE11 (Genentech) do wykrywania powierzchniowego IgE. [0373] Przeciwciała anty-ige/m1 są specyficzne dla postaci długiej i nie są specyficzne dla postaci krótkiej błonowej IgE, co określono za pomocą barwienia w cytometrii przepływowej linii komórkowej z postacią długą i postacią krótką IgE BJAB. Przeciwciało anty-ludzka IgE (Mae11) (Genentech, Inc.) zastosowano jako pozytywna kontrola dla zdolności wiązania zarówno postaci długiej, jak i krótkiej IgE. Przeciwciała anty-gp120 migg1 lub antyambrozja migg2a zastosowano jako kontrole izotypowe (Genentech, Inc). [0374] Komórki BJAB hoduje się w RPMI 10% FBS (Hyclone, Logun, UT, #SH ), Penicylina/Streptomycyna, 2 mm L-glutaminy (Genentech, Inc.). 0.5 x 10 6 Komórek z długim BJAB lub krótkim BJAB blokowano mysią (10 µl) (VWR, #RLD ) i ludzką surowicą (10 µl) (Sigma, St. Louis, MO, #S-2257) przez 15 minut na lodzie w 100 µl buforu do przemywania FACs (2% FBS w 1X PBS (Genentech, Inc.). Komórki następnie barwiono 1 µg/ml każdego przeciwciała anty-m1 przez kolejne 20 minut na lodzie i następnie przemyto 1 ml buforu do przemywania FACs i odwirowano przy 1200 rpm przez 5 minut. Komórki
107 106- ponownie zawieszono w 100 µl buforu do przemywania FACs, a następnie barwiono kozią anty-mysia IgG-PE (5 µl) (Caltag/Invitrogen, Carlsbad, CA, #M ) na lodzie w ciągu 20 minut. Komórki płukano jak poprzednio i ponownie zawieszono w 1ml buforu do przemywania FACs do analizy na urządzeniu FACs Calibur (BD, Inc, Franklin Lakes, NJ). Wszystkie testowane przeciwciała były specyficzne dla postaci długiej IgE i nie wiązały komórek krótkie BJAB-IgE powyżej kontroli izotypowej. Zaobserwowano szeroki zakres względnego powinowactwa oparty na intensywności barwienia: [0375] Również ocenialiśmy przeciwciała anty-m1 wiążące się do linii komórkowej U266 (ATCC, Manassas, VA, #TIB196), które naturalnie eksprymują niski poziom ludzkiej IgE zawierającej M1 na powierzchni komórki. Komórki U266 utrzymuje się w wysokiej gęstości (1-2x106/ml) w pożywce hybrydomy [RPMI 1640, 15% płodowej surowicy bydlęcej, 2 mm L-glutaminy, 10 mm HEPES, 4.5 g/l glukozy, 1.5 g/l wodorowęglanu]. Komórki U266 barwiono przeciwciałami anty-m1 przy 1µg/ml. 47H4, 47G4 i 42A5 barwiły U266 na poziomach zbliżonych do Mae11. Przeciwciała 42H4, 45C1 i 28E9 barwiły U266 na bardzo niskich poziomach. 7A6, 1C11, 26A11, 51D2 i 26B11 nie barwiły U266. (Figura 2C) PRZYKŁAD 3A Specyficzność Wiązania Mysiego Anty-IgE/M1 [0376] Figury 2D-F przedstawiają wiązanie mysich przeciwciał anty-ige/m1 47H4, 26A11 i 7A6 do zbioru linii komórkowych, które eksprymują ludzki rezus lub cyno IgE/M1. [0377] Specyficzność klonów przeciwciał anty-ige/m1 testowano na linii ludzkich komórek B BJAB (ATCC Manassas, VA, #HB-136) i Daudi (ATCC #CCL-213) zainfekowanych przez retrowirusy do ekspresji postaci długiej IgE z sekwencją M1 lub postacią krótką IgE bez sekwencji M1. Komórki BJAB hoduje się w RPMI 10% FBS (Hyclone, Logun, UT, #SH ), Penicylina/Streptomycyna, 2 mm L-glutaminy (Genentech, Inc.). 0.5x106 Komórki z długim BJAB lub krótkim BJAB blokowano mysią (10 µl) (VWR, #RLD ) i ludzką surowicą (10µl) (Sigma, St. Louis, MO, #S-2257) w ciągu 15 minut na lodzie w 100µl buforu do przemywania FACs (2% FBS w 1X PBS (Genentech, Inc.)). Komórki następnie barwiono 1µg/ml każdego przeciwciała anty-m1 przez kolejne 20 minut na lodzie i następnie przemyto 1 ml buforu do przemywania FACs i odwirowano przy 1200 rpm przez 5 minut. Komórki ponownie zawieszono w 100µl buforu do przemywania FACs, a następnie barwiono kozim anty-mysia IgG-PE (5µl) (Caltag/Invitrogen, Carlsbad, CA, #M ) na lodzie w ciągu 20 minut. Komórki płukano jak poprzednio i ponownie zawieszono w 1 ml buforu do przemywania FACs do analizy na urządzeniu FACs Calibur (BD, Inc, Franklin Lakes, NJ). Wszystkie testowane przeciwciała były specyficzne dla postaci długiej IgE i nie wiązały komórek BJAB-IgE/krótki powyżej kontroli izotypowej. Zaobserwowano szeroki zakres względnego powinowactwa oparty na intensywności barwienia. [0378] Specyficzność klonów przeciwciał anty-ige/m1 testowano na linii ludzkich komórek B Daudi zainfekowanych przez retrowirusy do ekspresji błonowej IgE z sekwencją M1 (postać długa) lub błonowej IgE bez sekwencji M1 (postać krótka). Przeciwciała anty- IgE/M1 (47H4, 26A11, 7A6) są specyficzne dla postaci długiej i nie są specyficzne dla
108 107- postaci krótkiej (Figura 2D). Przeciwciało anty-gp120 migg1 zastosowano jako kontrolę izotopową (Genentech, Inc). [0379] Linię ludzkich komórek B Daudi transdukowano retrowirusem zawierającym kasetę ludzka IgE/naczelny M1 dla każdego z rezus lub cyno lub ludzkiego IgE/M1. Po transdukcji komórki eksprymujące ludzki, rezus, lub cyno IgE/M1 posortowano sorterem FACS do >98% czystość w ciągu 3-4 sortowań. [0380] Figura 2F przedstawia zdolność przeciwciała anty-ige/m1 do wiązania rezus i cyno M1. 47H4 i 7A6 wiążą zarówno rezus, jak i cyno M1, natomiast 26A11 wiąże wyłącznie rezus M1. [0381] Oprócz transfekowanych linii komórek Daudi, oceniliśmy wiązanie przeciwciała anty- IgE/M1 do U266, ludzkiej linii komórkowej szpiczaka IgE (ATCC, Manassas, VA, #TIB196). Komórki U266 utrzymuje się przy wysokim zagęszczeniu (1-2x106/ml) w pożywce hybrydom [RPMI 1640, 15% płodowa surowica bydlęca, 2 mm L-glutaminy, 10mM HEPES, 4.5 g/l glukozy, 1.5g/L wodorowęglanu]. Komórki U266 barwiono przeciwciałami anty-m1 przy 1 µg/ml. Przeciwciało anty-ige/m1 47H4 wiązało U266 i nie wiązało 26A11 oraz 7A6 (Figura 2E). PRZYKŁAD 3B Specyficzność Wiązania Humanizowanego anty-ige/m1 [0382] Przykład 3B przedstawia wiązanie humanizowanego przeciwciała anty-ige/m1 do zbioru linii komórkowych, które eksprymują ludzki, rezus lub cyno IgE/M1. [0383] Stosując taką samą linii komórek ludzkich B Daudi lub BJAB z nadmierną ekspresją ludzkiej, rezus i cyno M1 omówioną w Przykładzie 3A, specyficzność humanizowanych klonów przeciwciał anty-ige/m1 testowano na linii ludzkich komórek B Daudi zainfekowanych przez retrowirusy do ekspresji postaci długiej IgE z sekwencją M1 lub postacią krótką IgE bez sekwencji M1. Humanizowane przeciwciała anty-ige/m1 (47H4v5, 26A11v6, 7A6v1) są specyficzne dla postaci długiej i nie są specyficzne dla postaci krótkiej (bez M1 ) [Figura 2G]. HERCEPTIN mab anty-her2 huigg1 było stosowane jako kontrola izotypu (Genentech, Inc.). Dodatkowo, humanizowane przeciwciała anty-ige/m1 (47H4v5 i 26A11v6) wiążą się z liniami komórkowymi U266 (Figura 2H). [0384] Figura 2I wykazuje zdolność humanizowanego przeciwciała anty-ige/m1 do wiązania rezus, jak i cyno M1. Przeciwciała 47H4v5 i 7A6v1 wiążą zarówno rezus, jak i cyno M1, natomiast 26A11v6 wyłącznie wiąże tylko rezus M1. PRZYKŁAD 4 Względne Powinowactwa Wiązania przeciwciał anty-m1 [0385] Zaobserwowaliśmy szeroki zakres intensywności wiązania przeciwciał anty-ige/m1 przy 1 µg/m). W celu oceny względnego powinowactwa przeciwciał dla długiego receptora IgE, barwiliśmy komórki BJAB długie seriami rozcieńczeń przeciwciała anty-m1 (1, 0.1, 0.01, µg/ml). Komórki BJAB długie blokowano mysią i ludzką surowicą (10 µl) przez
109 minut na lodzie. Komórki barwiono odpowiednią ilością przeciwciała anty-m1, lub kontrolami izotypowymi anty-gp 120 migg1 lub anty-ambrozja migg2a przez kolejne 20 minut. Komórki następnie przemyto w buforze do przemywania FACs (1 ml), odwirowano (1200 rpm przez 5 minut) i następnie ponownie zawieszono w 100 µl buforu do przemywania FACs z przeciwciałem kozim anty-mysia IgG-PE (5 µl) (CALTAG/Invitrogen, Carlsbad, CA #M ) dla wykrywania. Komórki płukano ponownie, jak wyżej, a następnie analizowano na urządzeniu FACs Calibur (BD, Inc, Franklin Lakes, NJ). Ustawienia napięcia oparto na µg/ml kontroli izotypowej. Na podstawie tych wyników uszeregowano kandydatów przeciwciał anty-ige/m1 według względnego powinowactwa wiązania: 42H4 > 7A6, 47H4, 47G4 > 42A5, 26A11, 45C1 > 51D2, 26B11 > 1C11 > 28E9 [0386] Dodatkowo w stosunku do oznaczeń powinowactwa, przez miareczkowanie i wykrywanie FACS przeciwciał, określiliśmy także powinowactwa wybranych przeciwciał anty-m1 do linii komórek BJAB eksprymujących IgE postać długą za pomocą analizy Scatcharda. Komórki BJAB-z długim przygotowano dla wiązania w zimnym buforze wiążącym składający się z pożywki podstawowej, z 10mM HEPES ph 7.4, i 2% FBS oraz 40 µg/ml ludzkiej IgG. Komórki rozcieńczano do stężenia 1.7 x 10 6 komórek/ml, i trzymano na lodzie do momentu dodania do testu. Przeciwciała jodowano metodą Iodogen. Różne stężenia nieznakowanego przeciwciała przygotowano w trzech powtórzeniach, stosując rozcieńczenie 1:2 począwszy od stężenia nasycenia i kończące się na stężeniu zero, w sumie 14 stężeniach. Jodowane przeciwciało w pojedynczym stężeniu dodano do wszystkich rozcieńczeń nieznakowanego przeciwciała konkurencyjnego. Na koniec do każdej mieszanki jodowanego i nieznakowanego przeciwciała dodano 250,000 komórek BJAB-z długim. Próbki inkubowano w 4 C w ciągu 4 godzin, mieszając. Każdą próbkę następnie zebrano i przesączono przez błonę, przemyto co najmniej 3 razy buforem wiążącym, pozostawiono do wyschnięcia i następnie mierzono w ciągu 1 minuty stosując Perkin Elmer Wizard 1470 Auto Gamma Counter. PRZYKŁAD 4A Powinowactwo Scatcharda mysich przeciwciał anty-ige/m1 [0387] Komórki BJAB i Daudi z ekspresją różnych postaci IgE z lub bez M1 (Ludzki, Rezus, Cyno) przygotowano dla wiązania w zimnym buforze wiążącym składającym się z pożywki podstawowej, z 10mM HEPES ph 7.4 i 2% FBS oraz 40ug/ml ludzkiej IgG. Komórki rozcieńcza się do stężenia 1.7 x 10 6 komórek/ml, i utrzymuje na lodzie do momentu dodania do testu. Białka/przeciwciała joduje się metodą Iodogen. Różne stężenia białka zimnego wytwarza się w trzech powtórzeniach, stosując rozcieńczenie 1:2 począwszy od stężenia nasycenia i kończące się na stężeniu zero, w sumie 14 stężeniach. Gorące białko w pojedynczym stężeniu dodaje się do każdego rozcieńczenia w celu konkurencji z zimnym białkiem. Na koniec, komórki dodaje się do mieszaniny białek gorącego i zimnego, zastosowano 250,000 komórek na próbkę. Test utrzymuje się w 4 C w ciągu 4 godzin, mieszając. Każdą próbkę zbiera się i sączy przez błonę, przemywa co najmniej 3 razy
110 109- buforem wiążącym, pozostawia do wyschnięcia i następnie zlicza przez 1 minutę, stosując Perkin Elmer Wizard 1470 Auto Gamma Counter. [0388] Figura 3O podsumowuje powinowactwa mysich przeciwciał anty-m1 mierzonych metodą analizy Scatcharda. Mysie przeciwciało 47H4 migg1 wiązało ludzkie, rezus, oraz cyno M1 z powinowactwami odpowiednio 0.79, 0.77 i 0.97 nm. Mysie przeciwciała migg1 26A11 i migg1 7A6 wiązały ludzki M1 z powinowactwami odpowiednio 2.3 i 0.64 nm. [0389] Figura 3P podsumowuje powinowactwa wiązania humanizowanego przeciwciała anty- IgE/M1 do ludzkiego, rezus i cyno M1. Humanizowane przeciwciało huigg1 47H4v5 wiązało ludzkie, rezus, oraz cyno M1 z powinowactwami odpowiednio 1.5, 1.3 i 2.5 nm. Humanizowane przeciwciała 47H4 v2, 47H4 v1, 26A11 v6, 26A11 v14 i 26A11 v1 wiązały ludzki M1 z powinowactwami odpowiednio 0.54, 0.37, 1.85, 1.5 i PRZYKŁAD 5 Badania Blokowania/Wiązania Epitopu Anty-IgE/M1 [0390] Badania blokowania przeprowadzano między kandydatami przeciwciał anty-ige/m1 w celu określenia nakładania się lub rozróżnienia epitopów wiążących. Gdy przeciwciało blokujące i wykrywające były różnego izotypu (tj. IgG1 vs. IgG2a), te różnice izotypów były wykorzystane do wyznaczenia zakresu blokowania przez wykrywanie przeciwciałem drugorzędowym specyficznym dla izotypu (CALTAG/Invitrogen, Carlsbad, CA, kozia antymysia IgG1-PE #32004, lub kozia anty-mysia IgG2a #M32204). Kiedy przeciwciała przeciwciała blokującego i wiążącego były tego samego izotypu, przeciwciała koniugowano z biotyną (Pierce, Rockford, IL, EZ-łącznik-sulfo-NHS-LC-Biotyna #CAS ) i wykrywano streptawidyną-pe (BD #554061), lub koniugowano z Alexa-647 (Molecular Probes/Invitrogen, Carlsbad, CA, #A20173). Przeciwciała blokujące początkowo i wykrywające zastosowano w stosunku 1:1 (10 µg/ml) (Figury 4A-4B). Kombinacje przeciwciał, które początkowo nie dały blokowania lub tylko częściowe blokowanie testowano dalej za pomocą stosunku przeciwciało blokujące do wykrywającego 10:1 1. Komórki BJAB-długi blokowano mysią i ludzką surowicą (jak opisano powyżej), a następnie barwiono przeciwciałem blokującym(10 µg/ml) w buforze do przemywania FACs (2% FBS/1X PBS) przez 20 minut na lodzie. Komórki płukano w buforze do przemywania FACs, a następnie barwiono przeciwciałem do wykrywania przy stosunku 1:1 (10 µg/ml) lub 10:1 (1 µg/ml) przez 20 minut na lodzie. Komórki płukano ponownie, jak wyżej, a następnie analizowano na urządzeniu FACs Calibur (BD, Inc, Franklin Lakes, NJ). Barwione komórki porównano z przeciwciałem kontrolnym dla izotypu albo migg1 anty-gp120 (Genentech, Inc.) albo migg2a anty-ambrozja (Genentech, Inc.) (Fig. 4C). Dla porównania całkowite zablokowanie oznaczano przez barwienie tym samym nieskoniugowanym przeciwciałem, jeśli to możliwe. [0391] Badania blokowania ujawniły trzy główne grupy wiązania A i B i C. Grupę A podzielono dalej na dwie grupy A1 (47H4, 47G4, 42H4, 42A5), oraz A2 (45C1, 51D2, 26A11) na podstawie częściowego nakładania się epitopów (Fig. 4D). Grupa C (26B11) miała bardzo niewiele wspólnego z innymi kandydatami przeciwciał. Trzech kandydatów nakładało
111 110- się bardziej szeroko niż inni. 1C11 wydawał się mieć nakładające się wiązanie z grupą A1 i A2, i był tylko częściowo blokowany przez grupę B (28E9). 7A6 należy przede wszystkim do grupy A1, ale miało częściowe oddziaływanie z kandydatami grupy A2. 28E9 należy do grupy wszystkich do siebie. 28E9 wykazywał tylko częściowe oddziaływanie z kandydatami grupy A2. PRZYKŁAD 5A Mapowanie epitopów [0392] Figury 5A-E przedstawiają badania wiązania epitopowego wykonane przy użyciu przeciwciała anty-ige/m1. Mysie przeciwciała 47H4, 7A6 i 26A11 przedstawione są na Figurach 5B i 5D, natomiast humanizowane warianty 47H4v5, 7A6v1 i 26A11v6 przedstawione są na Figurach 5C i 5E. Badania mapowania epitopów: Mysi Kandydaci [0393] Wygenerowano 15 peptydów obejmujących sekwencję otaczającą i zawierająca ludzki M1 (Clifford Quan) (Figura 5A). Peptydy ponownie zawieszono w albo w dh20 albo 50% DMSO. Peptydy powleczono na 96-dołkowych płytkach (płytki NUNC Maxisorp high protein-binding capacity ELISA plates # ) przy 1 µg/ml w buforze do powlekania (0.05M Węglan/wodorowęglan (ph9.6) (100 µl na dołek). Białko fuzyjne M1 -Fc stosowano jako pozytywną kontrolę dla wiązania. Negatywne kontrole obejmowały dołki powlekane ludzką IgE (U266) i dołki niepowlekane. Peptydy pozostawiono na noc do powlekania w 4 C. Następnego dnia rano Płytki przemyto 3 razy buforem do przemywania (PBS/ 0.05% Tween- 20 (ph 7.4)). Płytki następnie zblokowano w buforze do blokowania (PBS, 0.5% BSA (ph 7.4)) przez 1 godzinę z łagodnym mieszaniem. Rozcieńczenia przeciwciał (40 ng/ml i Ing/ml) do testu (47H4 migg1, 47H4v5 huigg1, 26A11 migg1, 26A11v6 huigg1, 7A6 migg1, 7A6v1 huigg1) wykonano w Magic Buffer [PBS (ph7.4), 0.5% BSA. 0.05% Tween-20, 10 ppm Proclin., 0.2% BgG, 0.25% Chaps, 5 mm EDTA, 0.35M NaCl]. Przeciwciało antygp120 migg1 i huigg1 HERCEPTIN zastosowano jako kontrole odpowiednio dla mysiego i ludzkiego przeciwciała anty-ige/m1. Po wystarczającym zablokowaniu płytek (0.5% BSA/1X PBS), płytki przemyto 3 razy i następnie do płytki w trzech powtórzeniach dodano przeciwciała (100 µl na dołek). Płytki inkubowano następnie przez 2 godziny w temperaturze pokojowej. Płytki z mysim przeciwciałem płukano 3 razy, a następnie dodano anty-mysia IgG1-Biotyna (BD Biosciences (A85-1) #553441) (1:10,000, 100 µl na dołek) i inkubowano przez 1 godzinę w temperaturze pokojowej. Po 3 przemyciach dodano SA-HRP (BD Biosciences #554066) (1:20,000, 100 µl na dołek) i inkubowano przez 1 godzinę. Płytki z ludzkim przeciwciałem IgG1 przemyto jak poprzednio i dodano anty-huigg.fc-hrp (Jackson ImmunoResearch (# )) (1:10,000, 100 µl na dołek) i inkubowano przez 1 godzinę w RT. Po zakończeniu drugorzędowych inkubacji, pyłki przemyto 6 razy i dodano substrat TMB (BD OptEIA #555214) (100 µl na dołek). Reakcję substrat-hrp zatrzymano 1M H3PO4 po -4 minutach. Następnie płytki odczytano przy 450/650. Dane przedstawiono jako względne OD (OD450 / CD650).
112 111- [0394] Figura 5B i 5D ilustruje wiązanie mysich kandydatów 47H4, 26A11 i 7A6 anty- IgE/M1 do peptydów M1. Wiązanie tych kandydatów koreluje z pogrupowaniem epitopów określonym przez badania blokowania przeciwciała. migg1 47H4 wiązało peptyd 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8). migg1 7A6 wiązało peptydy 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8) i 5 (RAPDRVLCHSGQQQG) (SEQ ID NO:9). migg1 26A11 wiązało peptydy 7 (GQQQGLPRAAGGSVP) (SEQ ID NO:11) i 8 (PRAAGGSVPHPRCH) (SEQ ID NO:12). [0395] Figury 5C i 5E ilustrują wiązanie humanizowanych przeciwciał anty-ige/m1 do peptydów M1. Mapowanie epitopów przeprowadzono jak opisano powyżej dla odpowiednich mysich przeciwciał, z tym, że użyto innego przeciwciała drugorzędowego. Płytki z ludzkim przeciwciałem IgG1 przemyto jak poprzednio i dodano przeciwciało drugorzędowe anty-igg.fc-hrp (Jackson ImmunoResearch (# ) (1:10,000, 100 µl na dołek) i inkubowano przez 1 godzinę w RT. Wiązanie epitopów odpowiadało temu dla macierzystych przeciwciał mysich. 47H4 v5 wiąże peptyd 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8). 7A6 v1 wiąże peptydy 4 (SAQSQRAPDRVLCHS) (SEQ ID NO:8) i 5 (RAPDRVLCHSGQQQG) (SEQ ID NO:9). migg1 26A11 wiąże peptydy 7 (GQQQGLPRAAGGSVP) (SEQ ID NO:11) i 8 (PRAAGGSVPHPRCH) (SEQ ID NO:12). PRZYKŁAD 6 Indukcja Apoptozy przez Mysie Anty-IgE/M1 w Komórkach Daudi-IgE/Długa [0396] Komórki Daudi-Długa (IgE/M1 ) użyto do testowania wpływu powierzchniowego sieciowania IgE na indukcję apoptozy. Komórki Daudi (ATCC, Manassas, VA, #CCL-213) to linia ludzkich komórek chłoniaka Burkitta, o której wiadomo, że jest podatna na apoptozę w odpowiedzi na sieciowanie BCR. Komórki Daudi eksprymujące postać długą IgE wytworzono przez retrowirusową transdukcję ciężkich i lekkich łańcuchów U266 IgE z M1, jak opisano powyżej do wytwarzania komórek BJAB z długą IgE. Analiza metodą cytometrii przepływowej komórek Daudi-IgE/Długa z zastosowaniem przeciwciał przeciwko endogennym IgM i transfekowane receptorem komórek B IgE wskazują na większą ekspresję IgM niż IgE. Apoptozę komórek Daudi-IgE/Długa badano w komórkach hodowanych z gęstością 0,2 x 10 6 / 1.5ml w pożywce hodowlanej [RPMI, 10% FBS (Hyclone, Logun, UT, #SH ), Penicylina/Streptomycyna (Gibco/Invitrogen, Carlsbad, CA # ), 2 mm Glutamina (Genentech, Inc.), 10 mm HEPES (7.2) (Genentech, Inc.), 1 mm pirogronianu sodu (Gibco/Invitrogen, Carlsbad, CA #11360), 1.59 g/l roztworu wodorowęglanu sodu (Gibco/Invitrogen, Carlsbad, CA # )]. Przed rozpoczęciem testu apoptozy, martwe komórki usunięto na gradiencie Ficoll (GE Healthcare # ) w celu zmniejszenia poziomu tła śmierci komórek w teście. 0.2 x 10 6 komórek hodowano w trzech powtórzeniach z lub bez przeciwciał anty-m1 lub przeciwciał kontrolnych w roztworze w ciągu 72 godzin. Komórki następnie zebrano i analizowano pod kątem apoptozy stosując zestaw Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA #556547). Komórki płukano dwukrotnie w zimnym PBS (Genentech, Inc.) i następnie ponownie zawieszono w 100 µl 1X buforu do wiązania (0.1 M Hepes / NaOH (ph7.4), 1.4 M NaCl, 25 mm CaCl 2 ) (BD Biosciences, San Jose, CA, # E). Komórki następnie
113 112- barwiono 2.5 µl przeciwciałem aneksyna V-FITC (BD Biosciences, San Jose, CA # X) i 5 µl jodku propidiowego (PI) (BD Biosciences, San Jose, CA # E) w ciemności. Po 15 minutach, do każdej probówki dodano 400 µl 1X buforu do wiązania i komórki analizowano na urządzeniu do cytometrii przepływowej FACS Calibur (BD, Inc. Franklin Lakes, NJ). Dla każdej próbki zebrano około 10-20,000 zdarzeń. Umierające komórki są określane jako aneksyna V dodatnie i ujemne PI. Martwe komórki są pozytywne zarówno dla aneksyny V, jak i PI. Procent każdej populacji (martwych i umierających) obliczono stosując oprogramowanie do analizy FACS FlowJo (Tree Star, Inc., Ashland, Oregon). Dane w trzech powtórzeniach uśredniono i obliczono odchylenia standardowe. Procent apoptozy obliczono jak sumę komórek martwych i umierających i wykreślono wykres za pomocą Excela (Microsoft, Inc.). [0397] Kamptotecynę (Sigma-Aldrich, St. Louis, MO, #C mg) i przeciwciało anty- IgM (JacksonImmuno Research, West Grove, PA, # ) użyto jako kontrolę pozytywną do indukcji apoptozy w linii komórkowej Daudi-IgE/Długa. Przeciwciała migg1 anty-gp120 lub migg1 anty-ambrozja (Genentech, Inc.) użyto jako kontrole ujemne w tym teście. Komórki Daudi-IgE/Długie hodowano z szeregiem stężeń przeciwciał anty-ige/m1 lub kontroli izotypowych (25, 10, 1, 0.1, 0.01, µg/ml). Przeciwciałami anty-ige/m1 poddanymi badaniu były 7A6, 47H4, 47G4, 42A5, 42H4, 1C11, 26A11, 51D2, 45C1, 26B11 i 28E9 (Genentech, Inc.). Również dla porównania użyto przeciwciało anty-ludzka IgE (Mae11-mIgG1). [0398] Przeciwciała kontroli izotypowej anty-gp120 i anty-ambrozja nie indukowały apoptozy powyżej komórek traktowanych. Poziomy tła apoptozy były w zakresie 10-15% w każdym doświadczeniu. Przeciwciała anty-ige/m1 7A6 (>10 µg/ml), 47H4 (>10 µg/ml), i 47G4 (25 µg/ml) indukowały apoptozę komórek Daudi IgE Długi. 26A11 wywoływało podobne wielkości apoptozy (~30-50%), ale w znacznie niższych stężeniach (>0.1 µg/ml). 42A5, 51D2, 45C1, 26B11 i 28E9 nie indukowały apoptozy powyżej poziomu tła. Mae11 również nie indukował apoptozy powyżej poziomu tła. [0399] Przeprowadziliśmy także test apoptozy w obecności drugorzędowego przeciwciała F(ab ) 2 koziego anty-mysia IgG (ab ) 2 (Jackson ImmunoResearch, West Grove, PA # ) do supersieciowania receptora komórek B IgE na linii komórkowej Daudi. Doświadczenia sieciowania wykonywano w obecności koziego anty-mysia przeciwciała F(ab ) 2 IgG (30 µg) (Jackson ImmunoResearch, West Grove, PA, # ). Za wyjątkiem 26B11 (30%), wszystkie przeciwciała indukowały maksymalne poziomy apoptozy 70-80%, ale przy różnych stężeniach. Przeciwciała te mogą być podzielone na dwie grupy. 7A6, 1C11, 26A11, 51 D2, 45C1 i 28E9 indukowały maksymalną apoptozę przy najwyższych stężeniach. Inna grupa 47H4, 47G4, 42A5, i 42H4 i MAE11 wykazywała zmniejszenie apoptozy w wyższych stężeniach. PRZYKŁAD 6A Indukcja Apoptozy Humanizowanych Przeciwciał anty-ige/m1 w komórkach Daud/IgEdługi
114 113- [0400] Komórki Daudi-Długa (IgE/M1 ) użyto do testowania wpływu powierzchniowego sieciowania IgE na indukcję apoptozy w tej linii komórkowej. Komórki Daudi (ATCC, Manassas, VA, #CCL-213) to linia ludzkich komórek chłoniaka Burkitta, o której wiadomo, że są podatne na apoptozę w odpowiedzi na sieciowanie BCR. Komórki Daudi-IgE/Długa hodowano z gęstością 0.2x10 6 / 1.5 ml w pożywce hodowlanej [RPMI, 10% FBS (Hyclone, Logun, UT, #SH ), Penicylina/Streptomycyna (Gibco/Invitrogen, Carlsbad, CA # ), 2mM Glutamina (Genentech, Inc.), 10mM HEPES (7.2) (Genentech, Inc.), 1mM Pirogronian Na (Gibco/Invitrogen, Carlsbad, CA #11360), 1.59 g/l roztworu wodorowęglanu sodu (Gibco/Invitrogen, Carlsbad, CA # )] w nocy poprzedzającej, a następnie martwe komórki usunięto na gradiencie Ficoll (GE Healthcare # ) następnego dnia. To obniżyło poziomy tła śmierci komórek w teście. 0.2x10 6 komórek hodowano w trzech powtórzeniach z lub bez przeciwciał anty-m1 lub przeciwciał kontrolnych w roztworze w ciągu 72 godzin. Komórki następnie zebrano i analizowano pod kątem apoptozy stosując zestaw Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA #556547). Komórki płukano dwukrotnie w zimnym PBS (Genentech, Inc.) i następnie ponownie zawieszono w 100µl 1X buforu do wiązania (0.1 M Hepes / NaOH (ph7.4), 1.4 M NaCl, 25 mm CaCl 2 ) (BD Biosciences, San Jose, CA, # E). Komórki następnie barwiono 2.5µl przeciwciałem aneksyna V-FITC(BD Biosciences, San Jose, CA # X) i 5µl jodku propidiowego (PI) (BD Biosciences, San Jose, CA # E) w ciemności. Po 15 minutach dodano do każdej probówki 400µl 1X buforu do wiązania i komórki analizowano na urządzeniu do cytometrii przepływowej FACS Calibur (BD, Inc. Franklin Lakes, NJ). Dla każdej próbki zebrano około 10-20,000 zdarzeń. Umierające komórki są określane jako aneksyna V dodatnie i ujemne PI. Martwe komórki są pozytywne zarówno dla aneksyny V, jak i PI. Procent każdej populacji (martwych i umierających) obliczono stosując oprogramowanie do analizy FACS FlowJo (Tree Star, Inc., Ashland, Oregon). Dane w trzech powtórzeniach uśredniono i obliczono odchylenia standardowe. Procent apoptozy obliczono jak sumę komórek martwych i umierających i wykreślono wykres za pomocą Excela (Microsoft, Inc.). [0401] Przeciwciało Anty-IgR4 (JacksonImmuno Research, West Grove, PA, # ) stosowano jako kontrolę pozytywną do indukcji apoptozy w linii komórkowej Daudi- IgE/Długie. huigg1 HERCEPTIN (Genentech, Inc.) zastosowano jako kontrolę negatywną w tym teście. Komórki Daudi-IgE/Długa hodowano z szeregiem stężeń przeciwciał anty- IgE/M1 lub kontroli izotypowej (25, 10, 1, 0.1, 0.01, µg/ml). Doświadczenia wtórnego sieciowania przeprowadzono w obecności koziego przeciwciała F(ab ) 2 anty-ludzka IgG (50µg) (Biosource #AHI1301). Zbadanymi przeciwciałami anty-ige/m1 były huigg147h4v5, 26A11 v6, 7A6v1. [0402] Kontrola izotypowa huigg1 HERCEPTIN nie indukowała apoptozy powyżej nietraktowanych komórek. Poziomy tła apoptozy były w zakresie 10-15% w każdym doświadczeniu. Przeciwciała anty-ige/m1 47H4 v5, 26A11v6 i 7A6v1 indukowały apoptozę w zakresie 30-40% (Figura 8A).
115 114- [0403] Powtórzyliśmy te same oznaczenia w obecności przeciwciała drugorzędowego F(ab )2 koziego anty-ludzka IgG (Biosource #AHI1301) aby sieciować receptor IgE na linii komórkowej Daudi. Wszystkie przeciwciała indukowały maksymalne poziomy apoptotyczne 70-90%, z pewnym spadkiem aktywności apoptotycznej w wysokich stężeniach pierwszorzędowego przeciwciała anty-ige/m1 (Figura 8B). 47H4v5 typu dzikiego vs. afukozylowane w indukowaniu apoptozy bez wtórnego sieciowania. [0404] Afukozylowaną (AF) wersję 47H4 v5 wytworzono przy użyciu linii komórkowych BIOWA (Genentech, Inc.). Wersje zarówno WT, jak i AF 47H4 v5 były zdolne do indukowania apoptozy na podobnym poziomie (Figura 8C). Tabela 1 Powinowactwa Wiązania Humanizowanych Anty-IgE/M1 mu26 A11 26A1 1.v1 26A1 1.v2 26A1 1.v3 26A1 1.v4 26A1 1.v5 26A1 1.v6 26A1 1.v7 L1-31/ 32 NS NS Zmiany w Zekwencji NA NY H1-34/ 35 M M M M M M M M Scatc hard KD (nm) 1.6 nm BJAB-IgE wiązanie komórek IC50 (nm) Biacore A100 immobili zowane M1 Biacore 2000 immobili zowane M1 immobili zowana IgG IC50 (nm) KD (nm) KD (nm) KD (nm) 12 25/ NS IM NA IM NY IM NS IH 890 bardzo słabe
116 115-26A1 1.v10 26A1 1.v11 26A1 1.v12 26A1 1.v13 26A1 1.v14 26A1 1.v15 26A1 1.v16 mu47 H4 47H4. v1 47H4. v2 47H4. v3 47H4. v4 47H4. v5 47H4. v6 NS IN ~ NA IN NY IN SS QS M M M M SS IM QS IM L1-27e/ 28 H1-34 H3-10 0k NG M M 0.4 nm NG M M NA M M NG I L NA I L >130 0 NA I M NA M L H1-34 H3-10 0k 69 56
117 116- mu7a 6 7A6.v 1 7A6.v 2 PRZYKŁAD 7 M M 0.6 nm M M I L Indukcja Przepływu Wapnia [0405] Zbadano zdolność przeciwciał anty-m1 do indukcji przepływu wapnia w komórkach Daudi-IgE/Długa jako dowód, że przeciwciała te mogą indukować sygnalizację komórkową dalszego receptora IgE komórek B. Komórki Daudi (ATCC, Manassas, VA, #CCL-213), które wykazują nadekspresję postaci długiej IgE/M1 hodowano przez noc w niewielkiej gęstości 0.2 x 10 6 /ml. Następnego dnia rano martwe komórki usunięto na gradiencie Ficoll (GE Healthcare # ). Komórki obciążano indo-1 w masie, aby zapewnić równomierne obciążenie pomiędzy wszystkimi bodźcami. Komórki ponownie zawieszono w stężeniu 5 x 10 6 /ml w pożywce hodowlanej (RPMI-10% FBS (Hyclone), Penicylina/Streptomycyna, 2 mm L-Glutamina) i w połączeniu z barwnikiem wykrywania wapnia Indo-1 (5 µm) (Molecular probes/invitrogen, Carlsbad, CA, #11223). Komórki następnie przemyto w pożywce hodowlanej i ponownie zawieszono w 10 6 /ml komórek stosowano na bodziec. Próbki ogrzewano w temperaturze 37 C w łaźni wodnej przez 5 minut przed analizą na urządzeniu FACs Vantage (BD, Inc, Franklin Lakes, NJ). Próbki były początkowo analizowane przez 1 minutę w celu wytworzenia linii podstawowej, a następnie próbki były stymulowane 20 µg każdego przeciwciała, z wyjątkiem anty-igm (10 µg). Dane zebrano dla 8-10 minut. Analizę kinetyczną przeprowadzono na oprogramowaniu do analizy FACS FlowJo (Tree Star, Inc., Ashland, Oregon). Dane podaje się jako stosunek Indo Związany/ Indo Wolny. Wszystkie przeciwciała anty-m1 porównano z kontrolą izotypową migg1 anty-gp120 (20 µg). Anty-ludzka IgM (Jackson Immuno-Research, West Grove, PA, # ) i anty-ige (Mae11, Genentech, Inc.) użyto jako kontrole pozytywne. 7A6, 47H4, 47G4, 26A11 i 1C11 indukowały przepływ wapnia w komórkach Daudi IgE długi. 28E9 i 42A5 indukowały przepływ wapnia na niskim poziomie. 45C1, 26B11 i 51D2 nie indukowały przepływu wapnia. PRZYKŁAD 8 Indukcja ADCC za pomocą 47H4 v5 Anty-IgE/M1 wt i afukozylowanego [0406] Cytotoksyczność komórkowa zależna od przeciwciał (ADCC) umożliwia komórkom cytotoksycznym na wiązanie (przez przeciwciało) do antygenu komórki docelowej i następnie zabijanie komórki docelowej cytotoksyną. Przeciwciała anty-ige/m1 posiadające aktywności ADCC lub zwiększoną aktywność ADCC mogą zwiększyć wartość terapeutyczną w leczeniu zaburzeń mediowanych IgE.
118 117- [0407] Stwierdzono, że przeciwciała wytwarzane w komórkach ssaczych, które są afukozylowane mają zwiększoną aktywność ADCC. Następujące doświadczenie opisuje wytwarzanie przeciwciał afukozylowanych anty-ige/m1. [0408] Komórki NK izolowano ze 100 ml pełnej krwi (RosetteSep #15065, Stem Cell Technologies). Czystość komórek NK określono metodą barwienia anty-ludzkie CD56. Komórki NK o czystości >70% CD56+ użyto w każdym teście. Przeciwciała anty-ige/m1 i kontrolę izotypową huigg1 Mab anty-her2 HERCEPTIN miareczkowano seryjnie. Przeciwciała te (50 µl) inkubowano z komórkami BJAB z nadekspresją ludzkiej IgE/M1 na powierzchni komórek w ciągu 30 minut w temperaturze pokojowej w RPMI-1640 (bez czerwieni fenolowej) (BioWhitaker #12-918F) z 1% FBS (Linię komórkową wytworzono jak opisano powyżej). Komórki NK (50µl) następnie dodano do tej linii komórkowej przy stosunku 15:1 (150,000 komórek NK na 10,000 celów (BJAB-Długi)). Oznaczenia przeprowadzono w trzech powtórzeniach. BJAB-huDługa, przeciwciała i komórki NK inkubowano następnie przez 4 godziny w temperaturze 37 C. Po hodowli, płytki 96 dołkowe z dnem w kształcie litery U zwirowywano i zbierano supernatanty (100µl). Supernatanty testowano następnie pod względem uwalniania LDH przy użyciu testu reakcji LDH (Roche # ). Sam cel i zlizowany cel wykorzystano do obliczenia procentowej cytotoksyczności. Supernatanty były inkubowane w równej objętości z mieszaniną reakcyjną LDH przedstawioną przez producenta przez minut. Następnie płytki odczytano przy 490 nm. % cytotoksyczności obliczono następująco: = (Wartość dośw. Sam cel) / Zlizowany cel Sam cel). Dane wykreślono stosując Kaleidagraph i najlepsze dopasowanie krzywych stosowano do generowania wartości ED50. [0409] Na Figurze 10, przeciwciało kontroli izotypowej huigg1 HERCEPTIN indukowało niski poziom aktywności. Przeciwciała anty-ige/m1 indukowały specyficzną cytotoksyczność. Postacie typu dzikiego i afukozylowane 47H4v5 Anty-IgE/M1 indukowały podobną maksymalną % cytotoksyczność (~ 70-80%). Afukozylowane 47H4v5 wykazywało silniejsze działanie niż postać typu dzikiego (EC50 afukozylowanego 47H4v5 wyniosło ~0.83 nm; EC50 47H4v5 typu dzikiego wyniosło ~6.6 nm). PRZYKŁAD 9 Działanie mysich przeciwciał anty-ige/m1 w Atopowej Myszy hu-scid [0410] Figury 12A i 13A przedstawiają zdolność przeciwciała anty-ige/m1 (odpowiednio zarówno mysiego, jak i humanizowanego) do hamowania wytwarzania IgE w surowicy i komórek plazmatycznych produkujących IgE w atopowym modelu hu-scid. Doświadczenie 1 (# ) badało mysich kandydatów anty-ige/m1 i doświadczenie 2 (# ) badało humanizowanych kandydatów anty-ige/m1. [0411] Dawców przeszukiwano dla poziomów IgE w surowicy z naszego wewnętrznego programu dawców krwi Leukopack. Dwoje dawców, wybranych dla dostarczenia komórek dla atopowego modelu hu-scid, samych zidentyfikowało się jako mający alergie i mieli poziomy IgE w surowicy 1696 i 1152 ng/ml. Zdrowi dawcy mają zazwyczaj IgE od <100 do 300 ng/ml.
119 118- [0412] Doświadczenia z hu-scid przedstawione są na Figurach 12A i 13A. PBMC wyodrębniono przez leukoforezę i gradient gęstości Ficoll (GE Healthcare # ) i zliczono komórek PBMC (10 8 na 100µl) wstrzykiwano dootrzewnowo napromieniowanym myszom SCID-beige dniu zero. Aby nakierować reakcję w kierunku wytwarzania IgE, myszy traktowano anty-ifny (BD Biosciences, San Jose, CA, #554698) i przeciwciałami anty-il12 (100µg/dawkę) (BD Biosciences, San Jose, CA, #554659) w dniach 0 i 3; i rhil-4 (100ng/ dawkę) (R&D Systems, Minneapolis, MN, #204-IL-010) w dniach 2,3,4. Myszy traktowano przeciwciałami doświadczalnymi (tj. anty-ige/m1 ) trzy razy w tygodniu (mniej więcej co trzy dni) zaczynając od dnia 0 do końca badania. Ludzkie komórki pozostawiono do ekspansji pomiędzy 7 a 14 dniem, po którym myszy poddano eutanazji. Krwawienia pobrano w dniu 7 i na końcu badania. Ostatniego dnia eksperymentu myszy uśmiercano i izolowano splenocyty. Zawiesiny pojedynczych komórek użyto do oznaczenia IgE Elispot i FACs w celu określenia stopnia rozpuszczenia komórek ludzkich i zubożenia komórek plazmatycznych IgE. [0413] Dane są wyrażone jako średnia ± odchylenie standardowe. Wartości p obliczono stosując oprogramowanie statystyczne JMP. Test Dunnetta porównuje średnie grup, gdzie wszystkie grupy testowe są testowane wobec grupy referencyjnej. Każda para t Studenta porównuje każdą parę grup stosując test t Studenta. Następnie stosowana jest korekta Bonferroniego w celu dostosowania wartości p z każdej pary t Studenta dla uniknięcia porównań parami. Progiem dla wszystkich wartości p jest Procent zmiany w danych podanych na Figurach 12A-I i 13A-H obliczono między grupą leczoną i grupą kontrolną bez normalizacji do wartości wyjściowych. [0414] Wolne poziomy IgE oznaczano za pomocą procedury testu ludzkiego ELISA, w którym płytki 384-dołkowe ELISA Maxisorp (Nalge Nunc International, Rochester, NY) powleczono 1 µ g/ml przeciwciała monoklonalnego MAE11 w 50 mm buforze węglanowym, ph 9.6, w 4 C przez noc i blokowano 0.5% albuminy surowicy bydlęcej, w PBS w temperaturze pokojowej przez 1 godzinę. Wzorce ( ng/ml) i trójkrotne rozcieńczenia próbek (minimalne rozcieńczenie 1: 10 w celu uniknięcia efektu surowicy) w PBS zawierającej 0.5% bydlęcej albuminy surowicy, 0.05% polisorbatu 20, 10 części na milion Proclin 300 (Supelco, Bellefonte, PA), 0.25% CHAPS (Sigma, St. Louis, MO), 0.2% bydlęcych globulin g (Biocell, Rancho Dominguez, CA) i 5 mm EDTA inkubowano na płytkach w temperaturze pokojowej przez 1 godzinę. Związaną IgE wykrywano przez inkubację znakowanego peroksydazą koziego przeciwciała anty-ludzkiego IgE (Kirkegaard & Perry Laboratories, Gaithersburg, MD) w dołkach przez 1 godzinę. Dodano substrat 3,3,5,5 - tetrametylobenzydynę (Kirkegaard & Perry Laboratories, Gaithersburg, MD) i reakcję zatrzymano przez dodanie 1 M kwasu fosforowego. Płytki przemyto między etapami. Absorbancję odczytywano przy 450 nm w czytniku do układacza Titertek (ICN, Costa Mesa, CA). Krzywą standardową dopasowuje się przy użyciu programu do dopasowania do czteroparametrowej krzywej regresji nieliniowej (opracowany przez Genentech). Punkty danych wpadające w liniowy zakres krzywej standardowej stosowano do obliczenia stężenia IgE w próbkach.
120 119- [0415] Panel izotypowej immunoglobuliny ludzkiej wykonano przez wzięcie mysiego przeciwciała monoklonalnego (mab) do ludzkiej IgG1 (klon 2C11. Abcam Inc., Cambridge, MA) rozcieńczono do 4 ug/ml w 0.05M buforze węglanu sodu o ph 9.6, i powleczono na płytkach ELISA (384-dołkowe, płytki o wysokim wiązaniu, Greiner Bio One, Monroe, NC) podczas całonocnej inkubacji w 4 C. Po przemyciu 3 razy buforem do przemywania (PBS / 0.05% Tween-20), płytki blokowano PBS / 0.5% bydlęcej albuminy surowicy (BSA) od 1 do 2 godzin. Te i wszystkie inne inkubacje przeprowadzono w temperaturze pokojowej na wytrząsarce orbitalnej. Próbki surowicy myszy rozcieńczono 1:100, a następnie wykonano seryjnie rozcieńczenia 1:3 stosując bufor do testu (PBS / 0.5% BSA / 0.05% Tween 20 / 0.25% CHAPS / 5mM EDTA / 15PPM Proclin). Przy użyciu tego samego buforu przygotowano seryjne rozcieńczenia IgG1 na podstawie krzywej standardowej ( ng/ml). Rozmrożono wstępnie rozcieńczone kontrole przy wysokich, średnich i niskich regionach krzywej standardowej. Po etapie blokowania, płytki przemyto i próbki, standardy i kontrole dodano do 384 dołkowych płytek i inkubowano przez 2 godziny. Płytki przemyto i biotynylowane mysie mab do ludzkiej IgG1 (Zymed Laboratories, South San Francisco, CA) rozcieńczono 1:500 w buforze do testu i dodano do przemytych płytek na 1 godzinę inkubacji. Streptawidynę-peroksydazę chrzanową (SA-HRP) (GE Healthcare, Piscataway, NJ) rozcieńczono 1:40,000 w buforze do testu i dodawano do płytek po przemyciu. Po inkubacji przez 30 minut, po ostatnim etapie przemywania, dodano tetrametylobenzydynę (TMB) (Kirkegaard & Perry Laboratories, Gaithersburg, MD) i barwę wywoływano przez 10 do 15 minut. Reakcję zatrzymano przez dodanie 1M kwasu fosforowego. Gęstości optyczną uzyskano za pomocą czytnika mikropłytek (450 nm, 650 nm długości fali odniesienia), i stężenia próbek obliczono z 4-parametrowych dopasowań krzywych standardowych. Minimalne kwantyfikowalne stężenie ludzkiej IgG1 w próbkach surowicy myszy wynosiło 1.22 ug/ml. [0416] Tę ogólną metodę ELISA opisaną powyżej zastosowano również do badania innych izotypów Ig. IgG2 [0417] Mysie mab anty-ludzka IgG2 (specyficzne dla łańcucha γ 2 ) (Southern Biotech, Birmingham, AL) powleczono na płytkach ELISA przy 4 ug/ml. Zakres krzywej standardowej ludzkiej IgG2 wynosił ng/ml. Biotynylowane Mysie mab antyludzka IgG2 (specyficzne dla łańcucha γ 2 ) (Southern Biotech) rozcieńczono do 0.5 µg/ml i stosowano jako przeciwciało wykrywające. SA-HRP dodano do 384 dołkowych płytek przy rozcieńczeniu 1:10,000. Minimalne kwantyfikowalne stężenie ludzkiej IgG2 w próbkach surowicy myszy wynosiło 1.22 µg/ml. IgG3 [0418] Mysie mab anty-ludzka IgG2 (Zymed Laboratories) rozcieńczono do 1 µg/ml i powleczono na płytkach ELISA. Zakres krzywej standardowej ludzkiej IgG3 wynosił ng/ml. Biotynylowane mysie mab anty-ludzka IgG3 (specyficzne dla łańcucha γ 3 ) (Southern Biotech) rozcieńczono do 0.1 µg/ml i stosowano jako przeciwciało wykrywające.
121 120- SA-HRP dodano do 384 dołkowych płytek przy rozcieńczeniu 1:20,000. Minimalne kwantyfikowalne stężenie ludzkiej IgG3 w próbkach surowicy myszy wynosiło 78.1 ng/ml. IgG4 [0419] Oczyszczone mysie mab anty-ludzka IgG4 (BD Pharmingen, San Jose, CA) rozcieńczono do 0.25 ug/ml i powleczono na płytkach ELISA. Zakres krzywej standardowej ludzkiej IgG4 wynosił ng/ml. Skoniugowane z biotyną mysie mab anty-ludzka IgG4 (BD Pharmingen) rozcieńczono do 0.25 µg/ml i stosowano jako przeciwciało wykrywające. SA-HRP dodano do 384 dołkowych płytek przy rozcieńczeniu 1:20,000. Minimalne kwantyfikowalne stężenie ludzkiej IgG4 w próbkach surowicy myszy wynosiło 39.1 ng/ml. IgA [0420] Oczyszczone mysie mab anty-ludzka IgA 1 /A 2 (BD Pharmingen) rozcieńczono do 2 µg/ml i powleczono na płytkach ELISA. Zakres krzywej standardowej ludzkiej IgA wynosił ng/ml. Skoniugowane z biotyną mysie mab anty-ludzka IgA 1 /A 2 (BD Pharmingen) rozcieńczono do 1 µg/ml i stosowano jako przeciwciało wykrywające. SA-HRP dodano do 384 dołkowych płytek przy rozcieńczeniu 1:80,000. Minimalne kwantyfikowalne stężenie ludzkiej IgA w próbkach surowicy myszy wynosiło 39.1 ng/ml. IgM [0421] Oczyszczone mysie mab anty-ludzka IgM (BD Pharmingen) rozcieńczono do 0.5 ug/ml i powleczono na płytkach ELISA. Zakres krzywej standardowej ludzkiej IgM wynosił ng/ml. Skoniugowane z biotyną mysie mab anty-ludzka IgM (BD Pharmingen) rozcieńczono do 0.5 µg/ml i stosowano jako przeciwciało wykrywające. SA-HRP dodano do 384 dołkowych płytek przy rozcieńczeniu 1:40,000. Minimalne kwantyfikowalne stężenie ludzkiej IgM w próbkach surowicy myszy wynosiło 156 ng/ml. [0422] Splenocyty hu-scid, splenocyty myszy lub komórki węzłów chłonnych były liczone przy użyciu mikrosfer Fluoresbrite YG (Polysciences, Inc. #18862) na FACs Calibur Flow Cytometer (BD (San Jose, CA)). [0423] Testy Elispot zastosowano do określenia częstość komórek produkujących IgE w osoczu u myszy hu-scid po leczeniu przeciwciałami rhuil-4, anty-ifnγ i anty-il-12 przez 15 dni. Płytki Elispot (Millipore, Billerica, MA, #MSIPS4510, przeźroczyste) powleczono rozcieńczeniem 1:500 pierwszorzędowego nieskoniugowanego przeciwciała anty-ludzka IgE (AXXORA, San Diego, CA, #BET-A80-108A) w buforze do powlekania [Genentech, Inc. (A3323)] przy 100µl/dołek przez noc w 4 C. Roztwór zawierający przeciwciało pierwszorzędowe zdekantowano i płytki przemyto dwa razy buforem do przemywania (1X PBS, 0.05% Tween-20). Błony blokowano 200µl/dołek 0.5% BSA w 1X PBS przez 0,5-1 godziny w temperaturze 37 C. Roztwór blokujący usunięto i do płytki dodano pozytywne komórki kontrolne lub splenocyty SCID-hu. Linię komórkową ludzkiego szpiczaka U266 produkującą IgE (ATCC, Manassas, VA, #TIB196) zastosowano jako dodatnią kontrolę testu. Komórki U266 dodano do płytki rozpoczynając od stężenia 3000 komórek/0.1ml (=0.032x10 6 /ml) podłoża hybrydom [RPMI 1640, 15% płodowa surowica bydlęca, 2 mm L-
122 121- glutaminy, 10mM HEPES, 4.5 g/l glukozy, 1.5g/L wodorowęglanu], a następnie wykonano siedem seryjnych rozcieńczeń (1:2) z. Zawiesiny pojedynczych komórek z całej śledziony ponownie zawieszono w 1ml pożywki. 100µl dodano do pierwszego dołka. Następnie wykonano siedem 1:2 seryjnych rozcieńczeń. Komórki hybrydomy U266 i śledziony inkubowano przez noc w 37 C. Następnego dnia płytki przemyto trzy razy buforem do przemywania (1XPBS, 0.05% Tween-20) i następnie 100µl/dołek drugorzędowego przeciwciała anty-ludzka IgE-skoniugowana z fosfatazą alkaliczną (Sigma, St. Louis, MO, A- 3525), dodano przy 1:2000 w 0.5% BSA / 1X PBS do każdego dołka i inkubowano w 37 C przez 2 godziny. Płytki przemyto trzy razy i następnie spłukano raz ddh 2 0 (Genentech, Inc.). Do każdego dołka dodano 100 µl/dołek roztworu BCIP/NBT (R&D Systems, Minneapolis, MN, Elispot Blue Color Module, #SEL002) i inkubowano w ciemności przez 30 minut. Płytki następnie płukano raz ddh20 i płytki pozostawiono do wyschnięcia w temperaturze pokojowej. Płyty zostały wysłane do zliczenia BD Biosciences (BD, Inc., Franklin Lakes, NJ). Ilość komórek plazmatycznych obliczono stosując liczbę punktów wykrytych, krotność rozcieńczenia (wkład komórkowy) i całkowitą liczbę komórek śledziony. [0424] Ludzkie komórki plazmatyczne wytworzono przez usunięcie śledziony w dniu 15 z myszy SCID-hu leczonej przeciwciałem. Zawiesiny pojedynczych komórek przygotowywano i przesączono. 5x10 6 komórek blokowano ludzkimi (Sigma, St. Louis, MO, #S-2257) i mysimi surowicami (VWR, #RLD ), a następnie barwiono przeciwciałami antyludzkim CD38 PE (BD Biosciences, San Jose, CA, #555460) i anty-ludzkim FITC PE (Dakocytomation, Glostrup, Denmark, #K2311) w buforze do przemywania FACs (2% FBS, 1X PBS) na lodzie w ciągu 20 minut. Komórki następnie przemyto i analizowano na urządzeniu FACs Calibur (BD, Inc., Franklin Lakes, NJ). Ekspresję ludzkich CD38 zastosowano jako kontrolę pozytywną dla pomyślnego przekazania ludzkiego PBMC. Komórki plazmatyczne określano jako CD38 wysokie, PC+. [0425] W doświadczeniu # (Figura 12A), trzy przeciwciała anty-ige/m1 (47H4, 26A11 i 7A6) testowano wobec kontroli izotypowej anti-gp120-muigg1. Myszy leczone kontrolą izotypową generowały wysokie poziomy ludzkiej IgE w dniu 9. Leczenie kandydatami anty-ige/m1 zmniejszało poziomy IgE o 65-84% (Figury 12 B-C). Leczenie anty-ige/m1 także zmniejszało wytwarzanie IgE przez komórki in vivo 19-69% (Figura 12D-E). Inne Ig w surowicy nie miały istotnego wpływu na leczenie anty-ige/m1 (zmniejszenie o ~30% zaobserwowano dla kilku przeciwciał anty-m1 dla huig1, IgG3 i IgG4 (Figura 12F-G). Nie zaobserwowano zmniejszenia odsetka całkowitej ilości komórek plazmatycznych śledziony z leczeniem anty-ige/m1 (Figura 12H-I). [0426] W doświadczeniu # (Figura 13A), humanizowane anty-ige/m1 (47H4v5) testowano wobec izotypowo kontrolnego przeciwciała huigg1 HERCEPTIN. Myszy leczone kontrolą izotypową generowały wysokie poziomy IgE w surowicy w dniu 8. Leczenie huigg1 anty-ige/m1 zmniejszało poziomy IgE o 79% (Figura 13B-D) i komórek plazmatycznych produkujących IgE o 75% (Figura 13C-D). Nie obserwowano zmniejszenia w innych Ig surowicy (IgG1, IgG2, IgG3, IgG4, IgM, IgA) (Figura 13E-F) lub procentu wszystkich komórek plazmatycznych w śledzionie (Figura 13G-H). Badania te wykazują, że
123 122- przeciwciała anty-ige/m1 specyficznie zmniejszają IgE w surowicy i komórkach wytwarzających IgE, ale nie immunoglobulin innych izotypów. PRZYKŁAD 10 Transgeniczne Myszy z Ludzkim M1 Generowanie myszy transgenicznych z knockin hum1 [0427] Ponieważ myszy normalnie nie wyrażają domeny M1 podobnej do ludzi, wytworzyliśmy myszy z ludzką domeną M1 z knockin do mysiego locus IgE (Figura 14A). Ludzka sekwencja M1 wykorzystuje mysie splicingowe miejsce akceptorowe M1 powyżej i następnie jest poddana fuzji z mysią sekwencją M1 poniżej (Figura 14B). Kasetę IRES-EGFPpA - Neo również wpasowano w 3 mysiej sekwencji M2. Ten knockin pozwoli na ekspresję mysiej IgE z domeną ludzkiego M1 na powierzchni komórek B IgE+. Sekwencja M1 nie będzie ulegać ekspresji na wydzielanej IgE. Przesiewanie linii z knockin M1 (PCR) [0428] Myszy z knockin ludzkiego M1 genotypowano za pomocą PCR z użyciem starterów specyficznych dla mysiego locus IgE i ludzkiej sekwencji M1. Startery stosowane były następujące: WT FOR 5 -GGCCAAAGACCCTAAGACAGTC (SEQ ID NO:106) hum1 FOR 5 -GGGCTGGCTGGCGGCTCCGC (SEQ ID NO:107) WT REV 5 -CTATGCCCTGGTCTGGAAGATG (SEQ ID NO:108) [0429] Oczyszczony genomowy DNA analizowano z powyższych starterów, stosując 32 cykli o następującym programie [94 C 4 min; 94 C 1 min; 60 C 30 s; 72 C 1 min (30 cykli); 72 C 10 min]. Produkty PCR następnie analizowano na 2% żelu agarozowym 0,5 x TBE. Genotyp PCR dzikiego typu dał produkt 668 bp, a knockin hu-m1 dał produkt 457 bp (Figura 14C). Przesiewanie linii z knockin M1 (Southern) [0430] Komórki ES z knockin hum1 i końcowe linie mysie przesiewano i weryfikowano za pomocą Southern blot. 10 µg oczyszczonej komórki ES lub DNA ogona genomowego trawiono Hindlll dla lewego ramienia lub BamHI dla prawego ramienia przez noc. Strawiony DNA następnie analizowano na żelu z 0.8% Agarozą 1X TAE. DNA przeniesiono na błonę nylonową (Roche # ) stosując bufor do denaturacji (1.5M NaCl; 0.5M NaOH) przez noc. Błonę przepłukano, usieciowano UV, a następnie zanurzono w roztworze DIG Easy Hyb (Roche # ) na 4 godziny z obracaniem w 46 C. Sondy wytworzono przez PCR przy użyciu zestawu PCR DIG probe Synthesis, zgodnie z zaleceniami producenta (Roche # ). [0431] Startery dla lewego ramienia były następujące: FOR 5 -TGTCTGGTGGTGGACCTGGAAAGCG (SEQ ID NO:109) Rev 5 -TCCTCGCTCTCCTGCTCTGGTGGTG (SEQ ID NO:110)
124 123- [0432] Startery dla prawego ramienia były następujące: FOR 5 -CCATGCAACCTAGTATCCTATTCTC (SEQ ID NO:111) Rev 5 -CTTTATACAGGAGAACCTAGCCCAG (SEQ ID NO:112) [0433] Sondy były testowane wobec nieznakowanego produktu PCR aby zapewnić zwiększoną wielkość i dobre znakowanie DIG. Bloty były następnie sondowane przez noc z zagotowaną sondą przez noc w 46 C z obracaniem. Kolejnego dnia bloty następnie przemyto i wywołano przeciwciałem anty-dig zgodne ze wskazówkami producenta. Bloty poddano ekspozycji na błonę przez minut. Figura 14D przedstawia wyniki southerna. Myszy typu dzikiego wygenerowały fragment 7.4 kb HindIII lewego ramienia, który staje się 3kB fragmentem w alleli knockin hu-m1. Myszy typu dzikiego wygenerowały fragment 14.1 kb BamHI prawego ramienia, który staje się 18.1 kb fragmentem w alleli knockin hum1. Przedstawiono myszy heterozygotyczne i typu dzikiego (Figura 14D). PRZYKŁAD 11 Transgeniczny Model Hu-M1 : Immunizacja TNP-OVA [0434] Przykład ten ilustruje zdolność kandydatów anty-ige/m1 do zapobiegania wytwarzaniu przeciwciał IgE uzyskanych przez stymulowanie TNP-OVA zarówno w pierwotnej odpowiedzi immunologicznej (Dośw. # F; Figury 15A-I) lub pamięciowej odpowiedzi immunologicznej (Dośw. # B; Figury 16A-K) na TNP-OVA. TNP-OVA lub trinitrofenylo-owalbumina jest silnym immunogenem często wykorzystywanym do generowania silnych odpowiedzi immunologicznych przeciwciał. [0435] Dane są wyrażone jako średnia ± odchylenie standardowe. Wartości p obliczono stosując oprogramowanie statystyczne JMP. Test Dunnetta porównuje średnie grup, gdzie wszystkie grupy testowe są testowane wobec grupy referencyjnej. Każda para t Studenta porównuje każdą parę grup stosując test t Studenta. Następnie stosowana jest korekta Bonferroniego w celu dostosowania wartości p z każdej pary t Studenta dla uniknięcia porównań parami. Progiem dla wszystkich wartości p jest Procent zmiany w danych podanych dla odpowiedzi pierwotnej (Figury 15A-I) i pamięciowej odpowiedzi immunologicznej (Figury A-K) obliczono przez odjęcie średniej wartości dla grupy niezakażonej lub nieimmunizowanej od każdej z wartości dla myszy, średnie dla grup przeliczono ponownie i następnie obliczono procentową zmianę w stosunku do grupy kontrolnej dla każdego punktu czasowego. [0436] Immunizacja TNP-OVA myszy z knockin ludzkiej IgE/M1 (C57BL/6) indukuje wyważoną odpowiedź Th1/Th2 in vivo. Poziomy IgE specyficznej antygenowo po pierwotnej immunizacji i prowokacji osiągają poziomy ~ ng/ml. W doświadczeniu # F, myszy z knockin hum1 (C57B1/6) immunizowano TNP-OVA (Biosearch Technologies, Novato, CA) (100 µg w 2 mg ałunu na mysz i.p.) w dniu 0 (Figura 15A). Myszy były następnie traktowane 0.1 mg/kg przeciwciał trzy razy w tygodniu pomiędzy dniami 0 a 28 (Figura 15A). W doświadczeniu # B, mysz z knockin hum1 (C57B1/6) immunizowano TNP-OVA/Ałun w dniu 0 (jak poprzednio) i następnie prowokowano TNP-
125 124- OVA (100 µg na mysz i.p.) w dniu 28 (Figura 16A). Myszy były leczone 10 mg/kg przeciwciał między dniami 28 i 49 (Figura 16A). Od myszy pobrano krew w trakcie odpowiedzi immunologicznej w celu monitorowania IgE specyficznej antygenowo w surowicy i poziomów IgG1. [0437] IgG1 TNP-OVA specyficzną dla IgE mierzono przy użyciu płytek do ELISA (384 dołkowa z powierzchnią MaxiSorp, Nunc, Neptune, NJ) powleczona przez godzin w 2-8 C 25 µl/dołek TNP-OVA (TNP:OVA stosunek 13:1) (Biosearch Technologies, Novato, CA) rozcieńczoną 0.5 µg/ml w 50mM węglan/wodorowęglan sodu, ph 9.6. Płytki następnie zlano i osuszono. Bufor do blokowania (PBS, 0.5% BSA, ph 7.2, 50 µl/dołek) dodano do płytki na 1-2 godziny. Te i wszystkie następne inkubacje przeprowadzono w temperaturze pokojowej z łagodnym mieszaniem. Próbki rozcieńczono 1/50 w rozcieńczalniku do testu (PBS, 0.5% BSA, 0.05% Tween-20, 0.01% Proclin 300), a następnie rozcieńczono seryjnie 1/3 na jedenaście punktów. Standardy były seryjnie rozcieńczano dwukrotnie w rozcieńczalniku do testu na jedenaście punktów, począwszy od 1ng/ml dla IgG1 Anty-TNP OVA i 3ng/ml dla IgE Anty-TNP OVA. Płytki przemyto trzy razy buforem do przemywania (PBS, 0.05% Tween-20, ph 7.2) i wzorce, próbki i kontrole dodano (25µL/dołek) do płytek w dwóch powtórzeniach w celu oddzielnego wykrywania izotypów IgG1 i IgE. Po godzinach inkubacji, płytki przemyto sześć razy buforem do przemywania. Biotynylowane szczurze m Ab anty-mysia IgG1 i IgE (BD Biosciences, San Diego, CA) rozcieńczono do 0.5 µg/ml dla IgG1 lub 1.0 µg/ml dla IgE w rozcieńczalniku do testu i dodano do odpowiednich płytek (25 µl/dołek). Płytki inkubowano przez godz. i przemyto sześć razy buforem do przemywania. Koniugat streptawidyna-peroksydaza chrzanowa (AMDEX, GE Healthcare, Piscataway, NJ) rozcieńczono 1/80,000 dla IgG1 lub 1/5,000 dla IgE i dodano do płytki (25 µl/dołek) na 30 min. Po przemyciu sześć razy buforem do przemywania, płytki wywołano dodając 25µL/dołek tetrametylobenzydyny (Kirkegaard & Perry Laboratories, Gaithersburg, MD) i inkubując przez 15 min dla IgG1 lub 25 min dla IgE. Reakcję przerwano przez dodanie 25 µl/dołek 1M H 3 PO 4, i odczytywano absorbancję przy 450 nm, z odniesieniem 620 nm. Wyniki dla nieznanych próbek interpolowano z dopasowań 4-parametrowych krzywych standardowych. [0438] Komórki plazmatyczne myszy ze śledziony lub węzłów chłonnych były najpierw identyfikowane przez oddzielenie komórek CD138+. Te komórki plazmatyczne produkujące IgE oznaczono ilościowo metodą Elispot. Dla wszystkich komórek produkujących IgE, przeciwciało anty-ige (od BD OptEIA mige zestaw #555248) powleczono na płytkach MultiScreen HTS (Millpore #MS1PS4W10) w rozcieńczeniu 1:2 przy użyciu buforu do powlekania ELISA użyciu w 50 µl na dołek. Powlekanie przeprowadzono w 4 C przez noc. Kolejnego ranka płytki przemyto 5 razy 200µl jałowego PBS-tween 20 (0.05%) za pomocą pipety wielokanałowej pod wyciągiem do hodowli tkankowej. Płytki następnie zblokowano w 300 µl PBS / 5% BSA lub pożywce do hodowli komórkowej (RPMI-1640, 10% FBS) przez dwie godziny w temperaturze pokojowej. Zawiesiny pojedynczych komórek przygotowywano ze śledziony lub węzłów chłonnych, i komórki zliczano FACS (jak opisano powyżej). Komórki przygotowano do wysiania, począwszy od 10 7 komórek, a następnie 3-4 krotne rozcieńczenia dla modeli immunizacji TNP-OVA, lub 4x10 6 komórek, a następnie 2
126 125- krotne rozcieńczenia dla modeli zakażenia Nippostrongylus. Mysią linię komórek B A20 stosowano jako kontrolę ujemną, a linię komórek TIB-141 użyto jako kontrolę dodatnią. Rozcieńczenia komórek przygotowano w pożywce do hodowli komórkowej (RPMI % FBS). Po etapie blokowania, płytki przemyto raz pożywką do hodowli komórkowej i odessano. Komórki wysiano w 100µl na dołek. Płytki następnie inkubowano przez noc w 37 C, 5% CO 2. Następnego dnia płytki przemyto 6 razy stosując SkanWasher 300 (Molecular Devices (Sunnyvale, CA)) za pomocą PBS-tween 20 (0.05%). Biotynylowane drugorzędowe przeciwciało z zestawu mige BD OptEIA (#555248) stosowano następnie w stężeniu odnotowanym przez producenta, zwykle pomiędzy 1:250-1:500 w 100µl/dołek. Przeciwciało drugorzędowe przygotowano w PBS / 1% BSA. Płytki następnie inkubowano przez 1 godzinę w temperaturze pokojowej i następnie przemyto 6 razy stosując SkanWasher300 za pomocą PBS-tween 20 (0.05%). Streptawidynę AP (R&D #SEL002) dodano następnie w rozcieńczeniu 1:60 w 100 µl na dołek, ponownie przygotowanym w PBS / 1% BSA, i inkubowano przez 1 godzinę w temperaturze pokojowej. Płytki następnie przemyto jak poprzednio 3 razy i następnie płukano dwa razy wodą destylowaną. Pozostałą ciecz usunięto na papierowym ręczniku. Następnie dodano reagent do wywoływania BCIP/NBT (100 µl/dołek), i płytki inkubowano przez 30 minut w temperaturze pokojowej w ciemności. Substrat zdekantowano, a płytki przemyto dwukrotnie wodą dejonizowaną. Płytki odwrócono w celu usunięcia nadmiaru wody i pozostawiono do wyschnięcia. Plamki oznaczono ilościowo stosując mikroskop dysekcyjny. [0439] Doświadczenie # F badało zdolność anty-ige/m1 do zapobiegania odpowiedzi IgE na pierwotną immunizację TNP-OVA. Myszy leczone kontrolą izotypową anty-gp120 wytwarzały IgE w surowicy, osiągając maksymalne poziomy od dnia 8 do 14 (~25 ng/ml) (Figura 15B). Leczenie anty-ige/m1 zapobiegało wzrostowi IgE w surowicy w dniu 8 (98%) i 14 (99%) (Figura 15C-D). Zmniejszenie to było znacząco różne dla zwierząt leczonych izotypowo i nie różniły się istotnie od myszy nieimmunizowanych (Figury 15C-E). Anty-IgE/M1 wpływał na poziomy IgG1 specyficznej antygenowo tylko nieznacznie przez 28 dni doświadczenia (Figury 15F-I). [0440] Doświadczenie # B badało zdolność anty-ige/m1 do zapobiegania wtórnej/pamięciowej odpowiedzi IgE na TNP-OVA. Wtórna odpowiedź IgE na dawkę przypominającą TNP-OVA w dniu 28 jest szybsza, osiągając szczytową wartość po 4 dniach, a nie 8-9 dniach, jak w pierwotnej odpowiedzi (Figura 16B). Leczenie kandydatami anty- IgE/M1 począwszy od dawki przypominającej w dniu 28 zmniejszyło wzrost IgE między dniem 28 a 35 (Figura 16B). Poziomy IgE leczonej anty-ige/m1 były znacznie zmniejszone w porównaniu z kontrolą dla izotypu, 59-75% w dniu 32 i 90-93% w dniu 35 (Figury 16C-D). W dniach poziomy IgE nie różniły się istotnie od myszy nieimmunizowanych (Figura 16E). Analiza powierzchni pod krzywą dla tych grup leczonych między dniami 28 i 49 ponadto wskazuje, że anty-ige/m1 zmniejszało poziomy IgE w surowicy o 74-84%, i średni poziom dzienny IgE specyficznej antygenowo również zmniejszało o 74-83% (Figury 16F- H). Brak znaczącego zmniejszenia IgG1 specyficznej antygenowo zaobserwowano po leczeniu anty-ige/m1 (Figury 16I-K).
127 126- PRZYKŁAD 12 Model Transgeniczny Hu-M1 : Model zakażenia Nippostrongylus brasiliensis [0441] Przykład ten ilustruje zdolność przeciwciała anty-ige/m1 do zapobiegania produkcji IgE (Figury 17A-D), oraz terapeutycznego zmniejszania wytwarzanie IgE w wywołanym uprzednio zakażeniu Nippostrongylus brasiliensis. Zmniejszenie wytwarzania IgE oceniano przez podawanie przeciwciała anty-ige/m1 zarówno przy szczytowych poziomach IgE odpowiedzi immunologicznej (Figury 18A-I), jak i gdy stosuje je późno w odpowiedzi immunologicznej (Figury 19 [0442] A-G). [0443] Nippostrongylus brasiliensis jest nicieniem przewodu pokarmowego, który zakaża szczury i myszy. Jego jaja wylęgają się w kale i dojrzewają do zakaźnej larwy w stadium L3. Larwy L3 przechodzą przez skórę gospodarza i migrują do naczyń krwionośnych, a następnie podróżują do płuc po jednym lub dwóch dniach. Po rozwinięciu do stadium larwy L4 w płucach, podróżują wzdłuż tchawicy gospodarza i przełyku i docierają do jelita czczego w dniu 2 do 3. Larwy dojrzewają do dorosłych robaków i parują się i produkują jaja około dnia 5. Jaja N. brasiliensis są wydalane z gospodarzy wraz z kałem. [0444] Myszy zakażone N. brasiliensis wykazują początkową wrodzoną odpowiedź immunologiczną, a następnie silną odporność typu 2 w celu usunięcia zakażenia. Depozycja dopełniacza i fibronektyny występuje we wcześniejszym stadium zakażenia, co ułatwia rekrutację leukocytów i przyczepność. Komórki efektorowe takie, jak granulocyty eozynochłonne, leukocyty zasadochłonne i komórki T CD4 + Th2, są rekrutowane do płuc do walki z infekcją. Cytokiny Th2, IL-4 i IL-13, odgrywają istotną rolę w utrzymaniu odpowiedzi immunologicznej w płucach i są wymagane dla oczyszczenia z robaków w jelitach. Reakcje typu 2 charakteryzują się wysokim poziomem produkcji przeciwciał, w tym przeciwciał IgE. [0445] Mysz z knockin ludzkiej IgE/M1 (G57BL/6) generują silne odpowiedzi IgE na zakażenie N. brasiliensis osiągając maksimum w dniu (Figury 17B, 18B, 19B). Poziomy IgE w surowicy spadają następnie do niższego, ale trwale podwyższonego poziomu. Myszy zakażono Nippostrongylus brasiliensis w dniu 0. Wszystkie trzy doświadczenia zakażania N. brasiliensis leczono 10 mg/kg przeciwciał migg1 anty-ige/m1 lub kontroli izotypowej migg1 anty-gp120. W badaniu prewencyjnym (Figura 17A), myszy z knockin hum1 leczono trzy razy na tydzień, począwszy od dnia 0 do dnia 21. W badaniu produkcji szczytowej (Figura 18A), myszy z knockin hum1 traktowano trzy razy w tygodniu pomiędzy dniami 11 a 21. W badaniu późnej interwencji (Figura 19A), myszy z knockin hum1 traktowano trzy razy w tygodniu pomiędzy dniami 41 i 62. [0446] Dane są wyrażone jako średnia ± odchylenie standardowe. Wartości p obliczono stosując oprogramowanie statystyczne JMP. Test Dunnetta porównuje średnie grup, gdzie wszystkie grupy testowe są testowane wobec grupy referencyjnej. Każda para t Studenta porównuje każdą parę grup stosując test t Studenta. Następnie stosowana jest korekta Bonferroniego w celu dostosowania wartości p z każdej pary t Studenta dla uniknięcia
128 127- porównań parami. Progiem dla wszystkich wartości p jest Procent zmiany w danych podanych w badaniach prewencyjnym i produkcji szczytowej obliczono przez odjęcie średniej wartości grup niezakażonych lub nieimmunizowanych od każdej z wartości dla myszy, średnie dla grup przeliczono ponownie i następnie obliczono procentową zmianę w stosunku do grupy kontrolnej dla każdego punktu czasowego. W badaniu późnej interwencji, zmiany procentowe obliczono w każdej grupie leczenia porównując dzień 48 i 55 do dnia 41. [0447] Komórki plazmatyczne wytwarzające IgE określono i oceniono metodą Elispot, jak opisano poprzednio w Przykładzie 12. [0448] Prewencyjne badanie testowało zdolność anty-ige/m1 w celu zapobiegania wzrostowi IgE w odpowiedzi na pierwotne zakażenie Nippostrongylus. Myszy kontrolne (myszy leczone izotypowo) osiągnęły wysoki poziom produkcji IgE w dniu 15 po infekcji (~3000 ng/ml). Myszy leczone anty-ige/m1 wykazywały zmniejszoną produkcję IgE po zakażeniu (zmniejszenie 93-94% w porównaniu do migg1 anty-gp120), która nie różniła się istotnie od myszy niezakażonych (Figury 17B-D). [0449] Badanie produkcji szczytowej testowało zdolność anty-ige/m1 do zmniejszania poziomów IgE terapeutycznie w szczycie odpowiedzi IgE na zakażenie N. brasiliensis. Wszystkie badane grupy osiągnęły wysoki poziom IgE (~2000 ng/ml) w dniu 11 po zakażeniu Nippostrongylus (Figura 18B). Leczenie anty-ige/m1 rozpoczęto w dniu 11 w szczycie tej odpowiedzi. Kandydaci migg1 anty-ige/m1 zmniejszali poziomy IgE w surowicy o 82-89% w ciągu czterech dni od leczenia (Figura 18C-D). W dniu 21 poziomy IgE u myszy leczonych anty-ige/m1 uległy zmniejszeniu o 97-98%, osiągając poziomy, które nie są statystycznie różne od grupy kontrolnej niezakażonej (Figura 18E). Komórki plazmatyczne wytwarzające IgE oznaczono ilościowo za pomocą Elispot anty-ige. Zakażenie N. brasiliensis wywoływało znaczną liczbę komórek produkujących IgE w zarówno węzłach chłonnych krezkowych (~30 komórek na 4x10 6 ; Figura 18F), jak i śledzionie (~10 komórek na 4x10 6 ; Figura 18G). W dniu 21 leczenie anty-ige/m1 zmniejszyło komórki wytwarzające IgE w węzłach chłonnych krezkowych o 88-94% oraz w śledzionie o 57-66%. Częstotliwość wszystkich komórek plazmatycznych (komórek CD138+) w krezkowych węzłach chłonnych i śledzionie zwiększyła się we wszystkich grupach leczonych w porównaniu do myszy niezakażonych, i nie występowała znacząca zmiana w ogólnej częstotliwości komórek plazmatycznych w każdym narządzie w związku z leczeniem anty-ige/m1 (Figura 18H-I). Wyniki te przedstawiają zdolność przeciwciał anty-ige/m1 do zmniejszenia IgE w surowicy do poziomów nominalnych przez zubożenie komórek produkujących IgE in vivo, nawet wtedy, gdy są stosowane w poziomie szczytowym produkcji IgE. [0450] Badanie późnej interwencji testowało zdolność anty-ige/m1 do zmniejszania niskiego poziomu trwałej IgE, który osiąga się późno w cyklu zakażenia. Wszystkie grupy leczone osiągały szczyt produkcji IgE około dnia 15. Po inicjacji leczenia anty-ige/m1 w dniu 41, były pewne różnice w poziomach IgE w surowicy między grupami leczonymi (Figury 19B-C), które znormalizowano przez odniesienie do poziomów dnia 41 jako 100% (Figura 19D). Leczenie anty-ige/m1 istotnie zmniejszało poziomy IgE w surowicy w porównaniu do kontroli izotypowej anty-gp120 migg1 między dniami 48 i 55 (Figura 19E-
129 128- G). W dniu 48 i 55 poziomy IgE zmniejszyły się o odpowiednio 64-75% i 75-84%, w porównaniu z kontrolą izotypową (miggi anty-gp120), która spadła o odpowiednio 20% i 37% (Figury 19F-G). Różnica średnich poziomów IgE między leczonymi grupami nie była znacząco różna od grupy kontrolnej izotypowej (gp120) przed rozpoczęciem terapii anty- IgE/M1 (Figura 19E). Jednakże leczenie przeciwciałami anty-ige/m1 pokazało bardziej dramatyczny i istotny spadek poziomów IgE w surowicy niż obserwowany w grupie kontrolnej w dniu 48 (Figura 19F) i dniu 55 (Figura 19G). Zmianę procentową i wartość p (d41) obliczono porównując każdy dzień leczonych grup 48 lub 55 do tej samej grupy wartości początkowej w dniu 41 przed leczeniem. PRZYKŁAD 13 Ekspresja przeciwciała anty-ige/m1 w komórkach ssaków [0451] Przykład ten ilustruje wytwarzanie potencjalnie glikozylowanych postaci pożądanego białka lub przeciwciała poprzez rekombinacyjną ekspresję w komórkach ssaczych. [0452] Wektor prk5 (patrz EP 307,247, opublikowany 15 marca 1989), jest stosowany jako wektor ekspresyjny. Opcjonalnie, DNA kodujące łańcuch lekki i/lub łańcuch ciężki przeciwciała anty-ige/m1 poddaje się ligacji do prk5 wybranymi enzymami restrykcyjnymi w celu umożliwienia wstawienia DNA za pomocą sposobów ligacji, jak opisano w Sambrook i wsp., supra. [0453] W jednym przykładzie wykonania, wybranymi komórkami gospodarza mogą być komórki 293. Ludzkie komórki 293 (ATCC CCL 1573) hoduje się do konfluencji w płytkach do hodowli tkankowych w pożywce takiej, jak DMEM uzupełnionej płodową surowicą cielęcą i ewentualnie składnikami odżywczymi i/lub antybiotykami. Około 10 µg DNA kodującego przeciwciało anty-ige/m1 zligowane do prk5 miesza się z około 1 µg DNA kodującego gen VA RNA [Thimmappaya i wsp., Cell, 31:543 (1982)] i rozpuszcza w 500 µl 1 mm Tris-HCl, 0.1 mm EDTA, M CaCl 2. Do tej mieszaniny dodano kroplami 500 µl 50 mm HEPES (ph 7.35), 280 mm NaCl, 1.5 mm NaPO 4, i osad pozostawia się do utworzenia przez 10 minut w 25 C. Osad zawiesza się i dodaje do komórek 293 i pozostawia do osadzenia przez około cztery godziny w 37 C. Pożywkę hodowlaną odsysa się i dodaje 2 ml 20% glicerolu w PBS na 30 sekund. Komórki 293 następnie przemywa się pożywką wolną od surowicy, dodaje świeżą pożywkę i komórki inkubuje przez około 5 dni. [0454] Około 24 godzin po transfekcji, pożywkę hodowlaną usuwa się i zastępuje pożywką (samą) lub pożywką hodowlaną zawierającą- 200 µci/ml 35 S-cysteiny i 200 µci/ml 35 S- metioniny. Po 12 godzinach inkubacji, zbiera się kondycjonowaną pożywkę, zatęża na filtrze przez wirowanie i wprowadza na 15% żel z SDS. Przetwarzany żel można wysuszyć i podać ekspozycji na błonę przez wybrany okres czasu, do ujawnienia obecności przeciwciała anty- IgE/M1. Hodowle zawierające transfekowane komórki można poddać dalszej inkubacji (w pożywce wolnej od surowicy) i pożywkę bada się w wybranych testach biologicznych. [0455] W alternatywnej technice, przeciwciało anty-ige/m1 można wprowadzać do komórek 293 przejściowo, z zastosowaniem metody z siarczanem dekstranu opisanym przez Somparyrac i wsp., Proc. Natl. Acad. Sci., 12:7575 (1981). Komórki 293 hoduje się do
130 129- maksymalnej gęstości w kolbie obrotowej i dodaje 700 µg DNA kodującego przeciwciało anty-ige/m1 zligowane do prk5. Komórki są najpierw zatężane z kolby obrotowej przez wirowanie i płukane PBS. Osad DNA-dekstran inkubuje się w peletce komórek przez cztery godziny. Następnie komórki traktuje się 20% glicerolu przez 90 sekund, przemywa pożywką do hodowli tkankowej i ponownie wprowadza do kolby obrotowej zawierającej pożywkę do hodowli tkankowych, 5 µg/ml insuliny bydlęcej i 0.1 µg/ml transferryny bydlęcej. Po około czterech dniach, kondycjonowaną pożywkę odwirowuje się i przesącza w celu usunięcia komórek i resztek. Próbkę zawierającą wyeksprymowane przeciwciało anty-ige/m1 można następnie zatężyć i oczyścić jakimkolwiek wybranym sposobem, takim, jak dializa i/lub chromatografia na kolumnie. [0456] W innym przykładzie wykonania, przeciwciało anty-ige/m1 można eksprymować w komórkach CHO. DNA kodujący przeciwciało anty-ige/m1 zligowany z prk5 można transfekować do komórek CHO stosując znane reagenty takie, jak CaPO 4 lub DEAEdekstran. Jak opisano powyżej, hodowle komórkowe można inkubować, a pożywkę zastąpić pożywką hodowlaną (samą) lub pożywką zawierającą znacznik radioaktywny taki, jak 35 S- metionina. Po ustaleniu obecności przeciwciała anty-ige/m1, pożywkę hodowlaną można zastąpić pożywką wolną od surowicy. Korzystnie, hodowle inkubuje się przez około 6 dni, a następnie zbiera kondycjonowaną pożywkę. Pożywkę zawierającą wyekpsrymowane przeciwciała anty-ige/m1 można następnie zatężać i oczyszczać dowolnym wybranym sposobem. [0457] Warianty tagowane epitopem przeciwciała anty-ige/m1 można również eksprymować w komórkach gospodarza CHO. DNA kodujący przeciwciało anty-ige/m1 zligowany z prk5 można subklonować do wektora prk55. Wstawka subklonu może ulegać PCR aby ulec fuzji w ramce z wybranym tagiem epitopowym takim, jak poli-his w bakulowirusowym wektorze ekspresyjnym. Poli-his tagowany DNA kodujący wstawkę znakowanego przeciwciała anty-ige/m1 można następnie subklonować do wektora SV40, zawierającego marker selekcyjny taki, jak DHFR do selekcji stabilnych klonów. W końcu, komórki CHO można transfekować (jak opisano powyżej) wektorem prowadzonym SV40. Znakowanie można przeprowadzić, jak opisano powyżej, dla potwierdzenia ekspresji. Pożywkę hodowlaną zawierającą wyeksprymowane, poli-his tagowane przeciwciało anty- IgE/M1 można następnie zatężać i oczyszczać dowolnym wybranym sposobem takim, jak przez chromatografię powinowactwa chelatu Ni 2+. [0458] Przeciwciało anty-ige/m1 można również eksprymować w komórkach CHO i/lub COS procedurą przejściowej ekspresji lub w komórkach CHO inną procedurą stabilnej ekspresji. [0459] Stabilną ekspresję w komórkach CHO wykonuje się stosując następującą procedurę. Białka eksprymuje się jako konstrukt IgG (immunoadhezyny), w którym sekwencje kodujące dla postaci rozpuszczalnych (np. domen zewnątrzkomórkowych) odpowiednich białek są poddane fuzji z sekwencją regionu stałego TgG1 zawierającą zawias, CH2 i domeny CH2 i/lub jest postacią poli-his tagowaną.
131 130- [0460] Po amplifikacji PCR odpowiednie DNA są subklonowane do wektora ekspresyjnego CHO przy zastosowaniu standardowych technik, jak opisano w Ausubel i wsp., Current Protocols of Molecular Biology, Unit 3.16, John Wiley and Sons (1997). Wektory ekspresyjne CHO konstruuje się tak, aby posiadały miejsca restrykcyjne 5 i 3 DNA będącego przedmiotem zainteresowania, aby umożliwić dogodne podmienianie cdna. Wektor użyty do ekspresji w komórkach CHO jest taki, jak opisano w Lucas i wsp., Nucl. Acids Res. 24:9 ( (1996), i wykorzystuje wczesny promotor SV40/enhancer do kierowania ekspresją cdna będącego przedmiotem zainteresowania i reduktazy dihydrofolianowej (DHFR). Ekspresja DHFR pozwala na selekcję dla stabilnego utrzymywania transfekcji poniższego plazmidu. [0461] Dwanaście mikrogramów pożądanego plazmidowego DNA wprowadza się do mniej więcej 10 milionów komórek CHO, przy użyciu dostępnych w handlu reagentów do transfekcji SUPERFECT (Quiagen), DOSPER lub FUGENE (Boehringer Mannheim). Komórki są hodowane jak opisano w Lucas i wsp., supra. Około 3 x 10-7 komórek zamraża się w ampułce do dalszego wzrostu i produkcji jak opisano poniżej. [0462] Ampułki zawierające plazmidowy DNA są rozmrażane przez umieszczenie ich w łaźni wodnej i mieszane przez worteksowanie. Zawartość pipetuje się do probówki do wirowania, zawierającej 10 ml pożywki i odwirowuje przy 1000 rpm przez 5 minut. Supernatant odsysa się i komórki ponownie zawiesza w 10 ml pożywki selekcyjnej (0.2 µm filtrowanej PS20 z 5% 0.2 µm diafiltrowanej płodowej surowicy bydlęcej). Komórki następnie dzieli się na równe objętości do 100 ml kolb obrotowych zawierających 90 ml pożywki selektywnej. Po 1-2 dniach, komórki są przenoszone do 250 ml kolby obrotowej wypełnionej 150 ml selektywnej pożywki wzrostowej i inkubuje w temperaturze 37 C. Po kolejnych 2-3 dniach, 250 ml, 500 ml i 2000 ml kolby obrotowe zaszczepia się 3 x 10 5 komórek/ml. Pożywkę dla komórek wymienia się na świeżą pożywkę przez wirowanie i ponowne zawieszanie w pożywce produkcyjnej. Chociaż można stosować dowolną odpowiednią pożywkę CHO, rzeczywiście stosowana może być pożywka opisana w patencie US nr , wydanym 16 czerwca L produkcyjną kolbę obrotową zaszczepia się przy 1.2 x 10 6 komórek/ml. W dniu 0 określa się liczbę komórek i ph. W dniu 1, próbkuje się kolbę obrotową i rozpoczyna przepłukiwanie filtrowanym powietrzem. W dniu 2, próbkuje się kolbę obrotową, temperaturę podnosi się do 33 C, i dodaje 30 ml 500 g/l glukozy i 0.6 ml 10% środka przeciwpiennego (np. 35% emulsja polidimetylosiloksanu, Dow Coming 365 Medical Grade Emulsion). Podczas wytwarzania, ph jest regulowane tak, aby utrzymać je przy około 7.2. Po upływie 10 dni, lub do momentu, gdy żywotność spada poniżej 70%, hodowlę komórkową zbiera się wirowanie i filtrowanie przez filtr 0.22 um. Przesącz albo przechowywano w 4 C lub bezpośrednio naniesiono na kolumny do oczyszczania. [0463] Dla poli-his tagowanych konstruktów, białka oczyszcza się za pomocą kolumny Ni- NTA (Qiagen). Przed oczyszczaniem, dodaje się imidazol do kondycjonowanej pożywki do stężenia 5 mm. Kondycjonowaną pożywkę pompuje się na 6 ml kolumnę Ni-NTA równowagowaną w 4 C, w 20 mm Hepes, ph 7.4, bufor zawierający 0.3 M NaCl i 5 mm imidazolu z szybkością przepływu 4-5 ml/min. Po załadowaniu, kolumnę przemywa się
132 131- dodatkowym buforem równowagującym, a białko wymywa się buforem równowagującym zawierającym 0.25 M imidazol. Wysoce oczyszczone białko jest następnie odsalane w buforze do przechowywania, zawierającym 10 mm Hepes, 0.14 M NaCl i 4% mannitol, ph 6.8, za pomocą kolumny 25 ml G25 Superfine (Pharmacia) i przechowywane w -80 C. [0464] Konstrukty immunoadhezyny (zawierające Fc) oczyszcza się z kondycjonowanej pożywki w następujący sposób. Kondycjonowaną pożywkę pompuje się na 5 ml kolumnę z Białkiem A (Pharmacia), którą równowagowano w 20 mm buforu fosforanu Na, ph 6.8. Po załadowaniu, kolumnę płukano buforem do równowagowania przed elucją 100 mm kwasem cytrynowym, ph 3.5. Eluowane białko natychmiast zobojętnia się przez zbieranie 1 ml frakcji do probówek zawierających 275 ul 1 M buforu Tris, ph 9. Wysoce oczyszczone białko jest następnie odsalane w buforze do przechowywania jak opisano powyżej dla białek tagowanych poli-his. Homogeniczność ocenia się w żelach poliakryloamidowych z SDS i przez sekwencjonowanie N-końcowego aminokwasu przez degradację Edmana. Depozyt Materiału [0465] Następujące materiały zdeponowano w American Type Culture Collection, University Blvd., Manassas, VA , USA (ATCC): Materiał Nr Dep. ATCC Data Dokonania Depozytu 7A6.18 PTA marca C PTA marca G4.6.2 PTA marca H PTA marca H4.6.9 PTA marca A PTA marca A PTA marca D PTA marca C PTA marca B PTA marca E PTA marca 2007 [0466] Depozyty te zostały wykonane zgodnie z postanowieniami Traktatu Budapesztańskiego o Międzynarodowym Uznawaniu Depozytu Drobnoustrojów dla celów Postępowania Patentowego oraz Regulacji pod tym traktatem (Traktatem Budapesztańskim). Zapewnia to utrzymanie realnej hodowli depozytów do trzydziestu (30) lat od daty dokonania depozytów i co najmniej pięć (5) lat po ostatnim wniosku o udostępnienie próbki. Depozyty będą udostępniane przez ATCC zgodnie z warunkami Traktatu Budapesztańskiego i na podstawie porozumienia osiągniętego między Genentech, Inc i ATCC, co zapewnia stałą i nieograniczoną dostępność do potomstwa hodowli depozytu do publicznej wiadomości w
133 132- momencie wydania Patentu US lub po upublicznieniu zgłoszenia patentowego US lub obcego, w zależności od tego, co nastąpi wcześniej i zapewnia dostępność potomstwa osobie określonej przez Komisarza ds. Patentów i Znaków Towarowych USA, który ma do tego prawo zgodnie z 35 USC 122 i przepisy Komisarza na tej podstawie (w tym 37 CFR 1.14 ze szczególnym odniesieniem do 886 OG 638). [0467] Cesjonariusz niniejszego zgłoszenia zgodził się, że jeśli hodowla materiałów dotyczących depozytu umrze lub zostanie utracona lub zniszczona podczas hodowli w odpowiednich warunkach, materiały zostaną niezwłocznie zastąpione takim samym innym po zawiadomieniu. Dostępność zdeponowanego materiału nie należy interpretować jako licencję na wykonanie tego wynalazku z naruszeniem praw przyznanych na mocy władzy jakiegokolwiek rządu, zgodnie z jego prawami patentowymi. [0468] Powyższy pisemny opis jest uważany za wystarczający, aby umożliwić znawcy w tej dziedzinie techniki realizację wynalazku w praktyce. Niniejszy wynalazek nie ma być ograniczany w swoim zakresie przez przedstawione tu przykłady. W istocie, różne modyfikacje wynalazku oprócz tych tu pokazanych i opisanych, staną się oczywiste dla znawców w tej dziedzinie na podstawie powyższego opisu. Lista Sekwencji [0469] <110> Genentech, Inc. Wu, Lawren Balazs, Mercedesz Brightbill, Hans Chan, Andrew C. Chen, Yvonne Man-yee Chuntharapai, Anan Dennis, Mark Wong, Terence <120> Apoptotyczne Przeciwciała Anty-IgE <130> P2474R1 WO <141> <150> US 60/ 896, 339 <151> <160> 112 <210> 1 <211> 431 <212> PRT <213> Homo sapiens
134 133- <220> <221> Inne... <223> ludzka IgE <400> 1
135 134- <210> 2 <211> 432 <212> PRT <213> Macaca mulatta <220> <221> inne... <223> rezus IgE <400> 2
136 135-
137 136- <210> 3 <211> 431 <212> PRT <213> Macaca fascicularis <220> <221> Inne... <223> Cyno IgE <400> 3
138 137- <210> 4 <211> 104 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki Fragment IgE <400> 4
139 138- <210> 5 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 1 <400> 5 <210> 6 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 2 <400> 6 <210> 7 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 3 <400> 7 <210> 8 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 4 <400> 8 <210> 9 <211> 15
140 139- <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 5 <400> 9 <210> 10 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1 peptyd 6 <400> 10 <210> 11 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' pept y d 7 <400> 11 <210> 12 <211> 14 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 8 <400> 12 <210> 13 <211> 15 <212> PRT <213> Sztuczna sekwencja
141 140- <220> <223> M1' peptyd 9 <400> 13 <210> 14 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 10 <400> 14 <210> 15 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 11 <400> 15 <210> 16 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 12 <400> 16 <210> 17 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 13 <400> 17
142 141- <210> 18 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 14 <400> 18 <210> 19 <211> 15 <212> PRT <213> Sztuczna sekwencja <220> <223> M1' peptyd 15 <400> 19 <210> 20 <211> 108 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Kappa I ludzki łańcuch lekki przeciwciała <400> 20
143 142- <210> 21 <211> 108 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Marine 26A11 łańcuch lekki przeciwciała <400> 21 <210> 22 <211> 108 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 26A11 v 1, 4 łańcuch lekki przeciwciała <400> 22
144 143- <210> 23 <211> 108 <212> PRT <213> Sztuczna sekwencja <220> <223> humanizowane 26A11 v2, 5 łańcuch lekki przeciwciała <400> 23 <210> 24 <211> 108 <212> PRT <213> Sztuczna sekwencja <220> <223> humanizowane 26A11 V3, 6 łańcuch lekki przeciwciała <400> 24
145 144- <210> 25 <211> 108 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 26A11 V13, 15 łańcuch lekki przeciwciała <400> 25 <210> 26 <211> 108 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 26A11 V14, 16 łańcuch lekki przeciwciała <400> 26
146 145- <210> 27 <211> 114 <212> PRT <213> Mus msculus <220> <221> Inne... <223> Mysie 7A6 łańcuch lekki przeciwciała <400> 27 <210> 28 <211> 114 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 7A6 V. 1 łańcuch lekki przeciwciała <400> 28
147 146- <210> 29 <211> 113 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Humanizowane 47H4 łańcuch lekki przeciwciała <400> 29 <210> 30 <211> 113 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 47H4 V. 1, 3 łańcuch lekki przeciwciała <400> 30
148 147- <210> 31 <211> 113 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 47H4 V łańcuch lekki przeciwciała <400> 31 <210> 32 <211> 113 <212> PRT <213> Homo sapi ens <220> <221> Inne... <223> Łańcuch ciężki ludzkiego przeciwciała III <400> 32
149 148- <210> 33 <211> 112 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 26A11 łańcuch ciężki przeciwciała <400> 33 <210> 34 <211> 112 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 26A11 V. 1-3, 13, 14 łańcuch ciężki przeciwciała
150 149- <400> 34 <210> 35 <211> 112 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 26A11 V. 4-6, 15, 16 łańcuch ciężki przeciwciała <400> 35 <210> 36 <211> 122 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 7A6 łańcuch ciężki przeciwciała
151 150- <400> 36 <210> 37 <211> 122 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 7A6 V. łańcuch ciężki przeciwciała <400> 37 <210> 38 <211> 117 <212> PRT <213> Mus musculus
152 151- <220> <221> Inne... <223> Mysie 47H4 łańcuch ciężki przeciwciała <400> 38 <210> 39 <211> 117 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 47H4 V. 1,2 łańcuch ciężki przeciwciała <400> 39 <210> 40 <211> 117 <212> PRT <213> Sztuczna sekwencja
153 152- <220> <223> Humanizowane 47H4 V.3,4 łańcuch ciężki przeciwciała <400> 40 <210> 41 <211> 117 <212> PRT <213> Sztuczna sekwencja <220> <223> Humanizowane 47H4 V. 5 łańcuch ciężki przeciwciała <400> 41 <210> 42 <211> 117 <212> PRT <213> Sztuczna sekwencja
154 153- <220> <223> Humanizowane 47H4 V.6 łańcuch ciężki przeciwciała <400> 42 <211> 446 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 7A6 łańcuch ciężki przeciwciała pełnej długości <400> 43
155 154-
156 155- <210> 44 <211> 220 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 7A6 łańcuch lekki przeciwciała pełnej długości <400> 44
157 156- <210> 45 <211> 442 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 47H4 łańcuch ciężki przeciwciała pełnej długości <400> 45
158 157- <210> 46 <211> 219 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 47H4 łańcuch lekki pełnej długości <400> 46
159 158- <210> 47 <211> 436 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 26A11 łańcuch ciężki przeciwciała pełnej długości <400> 47
160 <210> 48 <211> 214 <212> PRT <213> Mus musculus 159-
161 160- <220> <221> Inne... <223> Mysie 26A11 łańcuch lekki przeciwciała pełnej długości <400> 48 <210> 49 <211> 449 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 45C1 łańcuch ciężki przeciwciała pełnej długości <400> 49
162 161-
163 162- <210> 50 <211> 219 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 45C1 łańcuch lekki przeciwciała pełnej długości <400> 50
164 163- <210> 51 <211> 446 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 28E9 łańcuch ciężki przeciwciała pełnej długości <400> 51
165 164- <210> 52 <211> 214 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 28E9 łańcuch lekki przeciwciała pełnej długości <400> 52
166 165- <210> 53 <211> 442 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 1C11 łańcuch ciężki przeciwciała pełnej długości <400> 53
167 166- <210> 54 <211> 220 <212> PRT <213> Mus musculus <220> <221> Inne... <223> Mysie 1C11 łańcuch lekki przeciwciała pełnej długości <400> 54
168 167- <210> 55 <211> 25 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VH - H1 <400> 55 <210> 56 <211> 14 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VH - H2
169 168- <400> 56 <210> 57 <211> 32 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VH - H3 <400> 57 <210> 58 <211> 11 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VH - H4 <400> 58 <210> 59 <211> 23 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VL - L1 <400> 59 <210> 60 <211> 15
170 169- <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VL - L2 <400> 60 <210> 61 <211> 32 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VL - L3 <400> 61 <210> 62 <211> 11 <212> PRT <213> Homo sapiens <220> <221> Inne... <223> Ludzki zrąb konsensusowy VL - L4 <400> 62 <210> 63 <211> 26 <212> DNA <213> Sztuczna sekwencja <220> <223> Przedni starter do klonowania stosowany w przykładzie 1
171 170- <400> 63 ccgcccaccg tgaagatcttacagtc 26 <210> 64 <211> 35 <212> DNA <213> Sztuczna sekwencja <220> <223> Odwrotny starter do klonowania stosowany w przykładzie 1 <400> 64 cggct cgaga ct aggcgt gg ggctggagga cgt t g 35 <210> 65 <211> 542 <212> PRT <213> Sztuczna sekwencja <220> <223> fuzja IgE Ch3-Ch4-M-Fc <400> 65
172 171-
173 172- <210> 66 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 7AG <400> 66 <210> 67 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 7AG <400> 67
174 173- <210> 68 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 1C11 <400> 68 <210> 69 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 1C11 <400> 69 <210> 70 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 47H4 <400> 70 <210> 71 <211> 21
175 174- <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 47H4 <400> 71 <210> 72 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 26A11 <400> 72 <210> 73 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 26A11 <400> 73 <210> 74 <211> 19 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 45C1
176 175- <400> 74 <210> 75 <211> 24 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 45C1 <400> 75 <210> 76 <211> 25 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch ciężki 28E9 <400> 76 <210> 77 <211> 24 <212> PRT <213> Mus musculus <220> <221> Inne... <223> N-terminalna sekwencja, łańcuch lekki 28E9 <400> 77
177 176- <210> 78 <211> 46 <212> DNA <213> Sztuczna sekwencja <220> <223> FOR. Bsi W 7AG HC starter przedni <400> 78 tcgacgtacg ct caggt t ca gct gcagcaa t ct ggggct g agctgg 46 <210> 79 <211> 39 <212> DNA <213> Sztuczna sekwencja <220> <223> FOR. EcoRV 7A6 LC starter przedni <400> 79 gatcgatat cgtgatgtccc agt ct ccct c ct ccct aac 39 <210> 80 <211> 46 <212> DNA <213> Sztuczna sekwencja <220> <223> FOR. Bsi W 1C11 HC starter przedni <400> 80 t cgacgt acg ct caggt t ca at t gcagcag t ct ggggct g agctgg 46 <210> 81 <211> 39 <212> DNA <213> Sztuczna sekwencja <220> <223> FOR. EcoRV 1C11 LC starter przedni <400> 81 gat cgat at c gtaatgtctc agtctccttc ctccctagc 39 <210> 82 <211> 46 <212> DNA <213> Sztuczna sekwencja
178 177- <220> <223> 9. 1 HCF. Bsi W 47H4 HC starter przedni <400> 82 t cgacgt acg ctgaggtgaa gt t ggt ggag t ct gggggag gcttag 46 <210> 83 <211> 39 <212> DNA <213> Sztuczna sekwencja <220> <223> 47H4LCF. EcoRV 47H4 LC starter przedni <400> 83 gat cgat at c gt gct gact c agactccact ctccctgcc 39 <210> 84 <211> 43 <212> DNA <213> Sztuczna sekwencja <220> <223> C7F7HCF. Bsi W 26A11 HC starter przedni <400> 84 t cgacgt acg ct gaggt cca gct ccagcag t ct ggacct g agc 43 <210> 85 <211> 37 <212> DNA <213> Sztuczna sekwencja <220> <223> 2B4LCF. EcoRV 26A11 LC starter przedni <400> 85 gat cgat at c cagat gaccc aaact acat c ct ccct g 37 <210> 86 <211> 45 <212> DNA <213> Sztuczna sekwencja <220> <223> 2G6HCF. Bsi W 45C1 HC starter przedni <400> 86 t cgacgt acg ct cagat cca gttggtgcag t ct ggacct g agct g 45
179 178- <210> 87 <211> 41 <212> DNA <213> Sztuczna sekwencja <220> <223> 9C10LCF. EcoRV 45C1 LC starter przedni <400> 87 gatcgatatc gtgatgacgc agact ccact cactttgtcgg 41 <210> 88 <211> 46 <212> DNA <213> Sztuczna sekwencja <220> <223> 9.1HCF BsiW 28E9 HC starter przedni <400> 88 t cgacgt acg ct gaggt gaa gct ggt ggag t ct gaaggag gct t gg 46 <210> 89 <211> 39 <212> DNA <213> Sztuczna sekwencja <220> <223> 5E10. LCF. EcoRV 28E9 LC starter przedni <400> 89 gat cgat at cagatgaccc agt ct ccagc ct ccct at c 39 <210> 90 <211> 45 <212> DNA <213> Sztuczna sekwencja <220> <223> Starter Łańcucha Ciężkiego (HCR) <400> 90 ct ggacaggg atccagagtt t ccaggt cact gt cact ggct caggg 45 <210> 91 <211> 39 <212> DNA <213> Sztuczna sekwencja
180 179- <220> <223> Starter Łańcucha Lekkiego (LCR) <400> 91 ct gt aggt gc t gt ct t t gct gt cct gat ca gtccaactg 39 <210> 92 <211> 38 <212> DNA <213> Sztuczna sekwencja <220> <223> REV. Apal 7A6 HC starter odwrotny <400> 92 accgatgggc ccttggtgga ggctgaagag act gt gag 38 <210> 93 <211> 43 <212> DNA <213> Sztuczna sekwencja <220> <223> REV. Kpnl 7A6 LC starter odwrotny <400> 93 ccttggtacc ccct ccgaac gtgtacggat agctataata ttgg 43 <210> 94 <211> 37 <212> DNA <213> Sztuczna sekwencja <220> <223> REV. Apal 1C11 HC starter odwrotny <400> 94 gaccgatggg cccttggtgg aggctgagga gactgtg 37 <210> 95 <211> 38 <212> DNA <213> Sztuczna sekwencja <220> <223> REV. Kpnl 1C11 LC starter odwrotny <400> 95 ccttggtacc ccct ccgaac gt gt acggat agct at aa 38
181 180- <210> 96 <211> 37 <212> DNA <213> Sztuczna sekwencja <220> <223> 47H4HCR. Apal 47H4 HC starter odwrotny <400> 96 t gggccct t g gtggaggctgg aggagacggt gactgag 37 <210> 97 <211> 21 <212> DNA <213> Sztuczna sekwencja <220> <223> 47H4LCR. Kpnl 47H4 LC starter odwrotny <400> 97 ttccaacttg gtacctccac c 21 <210> 98 <211> 44 <212> DNA <213> Sztuczna sekwencja <220> <223> 7G8HCR. Apal 26A11 HC starter odwrotny <400> 98 gaccgatggg cccttggtgg argctgcaga gacagtgacc agag 44 <210> 99 <211> 21 <212> DNA <213> Sztuczna sekwencja <220> <223> 47H4LCR. Kpnl 26A11 LC starter odwrotny <400> 99 ttccaacttg gt acct ccac c 21 <210> 100 <211> 40 <212> DNA <213> Sztuczna sekwencja
182 181- <220> <223> 45C1.HCR. Apal 45C1 HC starter odwrotny <400> 100 cgatgggccc ttggtggarg ckgaggagac ggtgagaatg 40 <210> 101 <211> 18 <212> DNA <213> Sztuczna sekwencja <220> <223> 5C2LCR. Kpnl 45C1 LC starter odwrotny <400> 101 agcttggtac cccctccg 18 <210> 102 <211> 40 <212> DNA <213> Sztuczna sekwencja <220> <223> 28E9. HC. REV. Apal 28E9 HC starter odwrotny <400> 102 cgatgggccc ttggtggagg ctgaggagac ggcgactgag 40 <210> 103 <211> 22 <212> DNA <213> Sztuczna sekwencja <220> <223> 1D5. 2LCR, Kpnl 28E9 LC starter odwrotny <400> 103 ttccaacttg gtacccgagc cg 22 <210> 104 <211> 33 <212> DNA <213> Mus musculus <400> 104 cctatttaca ttggtatctg cagaagccag gcc 33 <210> 105 <211> 33
183 182- <212> DNA <213> Sztuczna sekwencja <220> <223> Starter odwrotny mutagenezy <400> 105 ggcctggctt ctgcagatac caat gt aaat agg 33 <210> 106 <211> 22 <212> DNA <213> Sztuczna sekwencja <220> <223> M1' przesiewanie WT starter przedni <400> 106 ggccaaagac cctaagacag t c 22 <210> 107 <211> 20 <212> DNA <213> Sztuczna sekwencja <220> <223> M1' przesiewanie hum1' starter przedni <400> 107 gggctggctg gcggctccgc 20 <210> 108 <211> 22 <212> DNA <213> Sztuczna sekwencja <220> <223> M1' przesiewanie WT starter odwrotny <400> 108 ctatgccctg gtctggaaga tg 22 <210> 109 <211> 25 <212> DNA <213> Sztuczna sekwencja <220> <223> starter przedni lewego ramienia
184 183- <400> 109 tgtctggtgg tggacctgga aagcg 25 <210> 110 <211> 25 <212> DNA <213> Sztuczna sekwencja <220> <223> starter odwrotny przedni lewego ramienia <400> 110 t cct cgct ct cct cct ct gg tggtg g 25 <210> 111 <211> 25 <212> DNA <213> Sztuczna sekwencja <220> <223> starter przedni prawego ramienia <400> 111 ccat gcaacc t agt at cct a ttctc c 25 <210> 112 <211> 25 <212> DNA <213> Sztuczna sekwencja <220> <223> starter odwrotny prawego ramienia <400> 112 ctttatacag gagaacctag cccag 25 Dorota Rzążewska Rzecznik patentowy
185 184- Zastrzeżenia patentowe 1. Przeciwciało anty-ige/m1', które specyficznie wiąże epitop w IgE w odcinku M1' określonym przez reszty 317 do 351 z SEQ ID NO:1 i, które indukuje apoptozę w komórkach B eksprymujących IgE. 2. Przeciwciało według zastrz. 1, przy czym przeciwciało jest zdolne do wiązania IgE, która pochodzi od człowieka, małpy rezus lub małpy cynomolgus. 3. Przeciwciało według zastrz. 1 albo 2, przy czym przeciwciało specyficznie zubaża komórki B produkujące IgE, gdy podaje się jego terapeutycznie skuteczną ilość in vivo ssakowi i ewentualnie zmniejsza całkowitą IgE w surowicy lub zmniejsza wolną IgE w surowicy; przy czym ewentualnie IgE jest specyficzna dla alergenu. 4. Przeciwciało według zastrz. 3, które jest chimeryczne, humanizowane lub ludzkie. 5. Przeciwciało według dowolnego z poprzednich zastrzeżeń, które specyficznie wiąże się do tego samego epitopu, co wiązany przez przeciwciało wybrane z grupy składającej się z: 47H4 (Nr Dep. ATTC PTA-8270), 7A6 (Nr Dep. ATTC PTA-8268), 26A11 (Nr Dep. ATTC PTA- 8262), 47H4v5 zawierające zmienny łańcuch lekki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:31 (Fig. 6C) i zmienny łańcuch ciężki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:41 (Fig. 6F), 7A6v1 zawierające zmienny łańcuch lekki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:28 (Fig. 6B) i zmienny łańcuch ciężki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:37 (Fig. 6E), i 26A11 v6 zawierające zmienny łańcuch lekki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:24 (Fig. 6A) i zmienny łańcuch ciężki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:35 (Fig. 6D). 6. Przeciwciało według zastrz. 5, przy czym epitop odpowiada peptydowi wybranemu z grupy składającej się z: peptyd 4 (SEQ ID NO:8), peptyd 5 (SEQ ID NO:9), peptyd 7 (SEQ ID NO:11) lub peptyd 8 (SEQ ID NO:12) i korzystnie, gdy epitop odpowiada peptydowi 4 (SEQ ID NO:8). 7. Przeciwciało według dowolnego z poprzednich zastrzeżeń, które wiąże się z powinowactwem wiązania Scatcharda, które jest równoważne temu dla mysiego przeciwciała anty-ige/m1' 47H4 (Nr Dep. ATTC PTA-8270), i ewentualnie przy czym powinowactwo wynosi między 0.30 i 0.83 nm. 8. Przeciwciało według dowolnego z poprzednich zastrzeżeń, które specyficznie wiąże się z odcinkiem M1' IgE z powinowactwem wiązania Scatcharda, które jest równoważne temu dla humanizowanego przeciwciała anty-ige/m1' 47H4v5 zawierającego zmienny łańcuch lekki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:31 (Fig. 6C) i zmienny łańcuch ciężki o sekwencji aminokwasowej przedstawionej jako SEQ ID NO:41 (Fig. 6F), i ewentualnie przy czym powinowactwo wynosi około 1.5 nm.
186 Przeciwciało według dowolnego z poprzednich zastrzeżeń zawierające HVR łańcucha ciężkiego i łańcucha lekkiego przeciwciała lub jego fragmentu wiążącego antygen wybranego z grupy składającej się z: 26A11, jak przedstawiono w SEQ ID NO: 21 (Fig. 6A) i SEQ ID NO: 33 (Fig. 6D), 26A11v.1-16, jak przedstawiono w SEQ ID NO: (Fig. 6A) i SEQ ID NO: 34 i 35 (Fig. 6D), 7A6, jak przedstawiono w SEQ ID NO: 27 (Fig. 6B) i SEQ ID NO: 36 (Fig. 6E), 7A6v1, jak przedstawiono w SEQ ID NO: 28 (Fig. 6B) i SEQ ID NO: 37 (Fig. 6E), 47H4, jak przedstawiono w SEQ ID NO: 29 (Fig. 6C) i SEQ ID NO: 38 (Fig. 6F), i 47H4v1-6, jak przedstawiono w SEQ ID NO: 30 i 31 (Fig. 6C) i SEQ ID NO: (Fig. 6F), i korzystnie zawierające HVR ciężkiego i lekkiego łańcucha lub przeciwciało lub jego fragment wiążący antygen 47H4v1-6, jak przedstawiono w SEQ ID NO: 30 i 31 (Fig. 6C) i SEQ ID NO: (Fig. 6F). 10. Przeciwciało według zastrz. 9 zawierające zmienne regiony łańcuchów ciężkich i lekkich przeciwciała lub jego fragmentu wiążącego antygen wybranego z grupy składającej się z: 26A11, jak przedstawiono w SEQ ID NO: 21 (Fig. 6A) i SEQ ID NO: 33 (Fig. 6D), 26A11v.1-16, jak przedstawiono w SEQ ID NO: (Fig. 6A) i SEQ ID NO: 34 i 35 (Fig. 6D), 7A6, jak przedstawiono w SEQ ID NO: 27 (Fig. 68) i SEQ ID NO: 36 (Fig. 6E), 7A6v1, jak przedstawiono w SEQ ID NO: 28 (Fig. 6B) i SEQ ID NO: 37 (Fig. 6E), 47H4, jak przedstawiono w SEQ ID NO: 29 (Fig. 6C) i SEQ ID NO: 38 (Fig. 6F), i 47H4v1-6, jak przedstawiono w SEQ ID NO: 30 i 31 (Fig. 6C) i SEQ ID NO: (Fig. 6F). 11. Przeciwciało według zastrz. 10, które jest afukozylowane. 12. Peptyd wybrany z grupy składającej się z: peptyd 4 (SEQ ID NO:8), peptyd 5 (SEQ ID NO:9), peptyd 7 (SEQ ID NO:11) lub peptyd 8 (SEQ ID NO:12). 13. Kompozycja zawierająca przeciwciało określone dowolnym z zastrzeżeń 1-11 w kombinacji z co najmniej jednym farmaceutycznie dopuszczalnym nośnikiem. 14. Kompozycja według zastrz. 13 zawierająca ponadto jeden lub więcej leków wybranych z grupy składającej się z: przeciwciało anty-ige, lek przeciwhistaminowy, lek rozszerzający oskrzela, glikokortykoid, NSAID, antagonista TNF, antagonista integryny, środek immunosupresyjny, antagonista IL-4, antagonista IL-13, podwójny antagonista IL-4/IL-13, DMARD, przeciwciało, które wiąże się z markerem powierzchniowym komórki B i antagonista BAFF. 15. Wyizolowany kwas nukleinowy, który koduje przeciwciało lub jego fragment wiążący antygen zawierający HVR łańcucha ciężkiego i lekkiego apoptotycznego przeciwciała anty- IgE/M1' lub jego fragmentu wiążącego antygen wybranego z grupy składającej się z: 26A11, jak przedstawiono w SEQ ID NO: 21 (Fig. 6A) i SEQ ID NO: 33 (Fig. 6D), 26A11 v1-16, jak przedstawiono w SEQ ID NO: (Fig. 6A) i SEQ ID NO: 34 i 35 (Fig. 6D), 7A6, jak przedstawiono w SEQ ID NO: 27 (Fig. 6B) i SEQ ID NO: 36 (Fig. 6E), 7A6v1, jak przedstawiono w SEQ ID NO: 28 (Fig. 6B) i SEQ ID NO: 37 (Fig. 6E), 47H4, jak przedstawiono w SEQ ID NO: 29 (Fig. 6C) i SEQ ID NO: 38 (Fig. 6F), i 47H4v1-6, jak przedstawiono w SEQ ID NO: 30 i 31 (Fig. 6C) i SEQ ID NO: (Fig. 6F).
187 Kwas nukleinowy według zastrz. 15, zawierający ponadto kwas nukleinowy kodujący regiony zmienne łańcuchów ciężkich i lekkich sekwencji przeciwciała wybranego z grupy składającej się z: 26A11, jak przedstawiono w SEQ ID NO: 21 (Fig. 6A) i SEQ ID NO: 33 (Fig. 6D), 26A11v1-16, jak przedstawiono w SEQ ID NO: (Fig. 6A) i SEQ ID NO: 34 i 35 (Fig. 6D), 7A6, jak przedstawiono w SEQ ID NO: 27 (Fig. 6B) i SEQ ID NO: 36 (Fig. 6E), 7A6v1, jak przedstawiono w SEQ ID NO: 28 (Fig. 6B) i SEQ ID NO: 37 (Fig. 6E), 47H4, jak przedstawiono w SEQ ID NO: 29 (Fig. 6C) i SEQ ID NO: 38 (Fig. 6F), i 47H4v1-6, jak przedstawiono w SEQ ID NO: 30 i 31 (Fig. 6C) i SEQ ID NO: (Fig. 6F). 17. Kwas nukleinowy według zastrz. 16, przy czym kodowane przeciwciało jest afukozylowane. 18. Wektor, w którym kwas nukleinowy określony dowolnym z zastrzeżeń 15 do 17, jest funkcjonalnie połączony. 19. Komórka gospodarza zawierająca wektor określony zastrzeżeniem Komórka gospodarza według zastrz. 19, która jest ssacza, a korzystnie jest to komórka jajnika chomika chińskiego. 21. Sposób wytwarzania apoptotycznego przeciwciała anty-ige/m1' lub funkcjonalnego fragmentu obejmujący hodowanie komórki gospodarza określonej zastrzeżeniem 19 albo 20 w warunkach odpowiednich do ekspresji przeciwciała lub fragmentu i odzyskiwanie przeciwciała lub fragmentu. 22. Artykuł przemysłowy zawierający kompozycję określoną zastrzeżeniem 13 albo 14 i ulotkę informacyjną wykazującą zastosowanie do leczenia zaburzenia mediowanego przez IgE. 23. Artykuł według zastrz. 22, który jest (i) fiolką, lub (ii) ampułko-strzykawką, która ewentualnie zawiera ponadto urządzenie wstrzykujące takie, jak autoiniektor. 24. Przeciwciało określone dowolnym z zastrzeżeń 1 do 11 do zastosowania w sposobie specyficznego zubażania komórek B produkujących IgE, przy czym sposób ten obejmuje podawanie ssakowi terapeutycznie skutecznej ilości przeciwciała anty-ige/m1' i ewentualnie przy czym przeciwciało wykazuje aktywność ADCC. 25. Przeciwciało określone dowolnym z zastrzeżeń 1 do 11 do zastosowania w sposobie leczenia zaburzenia mediowanego przez IgE, przy czym sposób ten obejmuje podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1'. 26. Przeciwciało do zastosowania w sposobie leczenia według zastrz. 25, przy czym: (i) zaburzenie mediowane IgE jest wybrane z grupy składającej się z: alergiczny nieżyt nosa, astma alergiczna, astma niealergiczna, atopowe zapalenie skóry, alergiczna gastroenteropatia, anafilaksja, pokrzywka, alergie pokarmowe, alergiczna aspergiloza oskrzelowopłucna, choroby pasożytnicze, śródmiąższowe zapalenie pęcherza moczowego, zespół hiper-ige, ataksja-teleangiektazja, zespół Wiskotta-
188 187- Aldricha, atymiczna limfoplazja, szpiczak IgE, reakcja przeszczep przeciwko gospodarzowi i plamica alergiczna i/lub (ii) przeciwciało podaje się w kombinacji z terapeutycznie skuteczną ilością co najmniej jednego leku wybranego z grupy składającej się z: przeciwciało anty-ige, lek przeciwhistaminowy, lek rozszerzający oskrzela, glikokortykoid, NSAID, lek zmniejszający przekrwienie, lek tłumiący kaszel, lek przeciwbólowy, antagonista TNF, antagonista integryny, środek immunosupresyjny, antagonista IL-4, antagonista IL-13, podwójny antagonista IL-4/IL-13, DMARD, przeciwciało, które wiąże się z markerem powierzchniowym komórki B i antagonista BAFF; i/lub (iii) przeciwciało podaje się w połączonym schemacie leczenia, który obejmuje podawanie terapeutycznie skutecznej ilości przeciwciała anty-ige/m1' przed, jednocześnie lub po podaniu znanego sposobu leczenia zaburzeń alergicznych. 27. Przeciwciało do zastosowania w sposobie leczenia według zastrz. 26(iii), przy czym: (a) znane leczenie zaburzeń alergicznych obejmuje podawanie przeciwciała anty-ige leku przeciwhistaminowego, leku rozszerzającego oskrzela, glukokortykoidu, niesteroidowego leku przeciwzapalnego, leku immunosupresyjnego, antagonisty IL-4, antagonisty IL-13, podwójnego antagonisty IL-4/IL-13, leku zmniejszającego przekrwienie, leku tłumiącego kaszel lub leku przeciwbólowego; lub (b) znane leczenie zaburzeń alergicznych obejmuje schemat leczenia lub odczulanie alergenem. 28. Przeciwciało do zastosowania w sposobie leczenia według dowolnego z zastrz. 24 do 27, przy czym sposób jest do zapobiegania lub zmniejszania wytwarzania IgE wywołanej alergenem. 29. Zastosowanie przeciwciała określonego dowolnym z zastrzeżeń 1 do 11 do wytwarzania leku do leczenia zaburzenia mediowanego IgE. 30. Zastosowanie według zastrz. 29, przy czym: (i) zaburzenie mediowane IgE jest wybrane z grupy składającej się z: alergiczny nieżyt nosa, astma alergiczna, astma niealergiczna, atopowe zapalenie skóry, alergiczna gastroenteropatia, anafilaksja, pokrzywka, alergie pokarmowe, alergiczna aspergiloza oskrzelowopłucna, choroby pasożytnicze, śródmiąższowe zapalenie pęcherza moczowego, zespół hiper-ige, ataksja-teleangiektazja, zespół Wiskotta- Aldricha, atymiczna limfoplazja, szpiczak IgE, reakcja przeszczep przeciwko gospodarzowi i plamica alergiczna; i/lub (ii) przeciwciało podaje się w kombinacji z terapeutycznie skuteczną ilością co najmniej jednego leku wybranego z grupy składającej się z: przeciwciało anty-ige, lek przeciwhistaminowy, lek rozszerzający oskrzela, glikokortykoid, NSAID, lek zmniejszający przekrwienie, lek tłumiący kaszel, lek przeciwbólowy, antagonista
189 188- TNF, antagonista integryny, środek immunosupresyjny, antagonista IL-4, antagonista IL-13, podwójny antagonista IL-4/IL-13, DMARD, przeciwciało, które wiąże się z markerem powierzchniowym komórki B i antagonistą BAFF. 31. Mysia hybrydoma zdeponowana 21 marca 2007 w ATCC, wybrana z grupy składającej się z: PTA-8260, PTA-8261, PTA-8262, PTA-8263, PTA-8264, PTA-8265, PTA-8266, PTA- 8267, PTA-8268, PTA-8269, PTA Przeciwciało, które jest wydzielane przez hybrydomę określoną zastrzeżeniem Zwierzę transgeniczne, które eksprymuje odcinek ludzkiego M1' IgE, który zawiera reszty 317 do 351 z SEQ ID NO:1. Dorota Rzążewska Rzecznik patentowy
190 189-
191 190-
192 191-
193 192-
194 193-
195 194-
196 195-
197 196-
198 197-
199 198-
200 199-
201 200-
202 201-
203 202-
204 203-
205 204-
206 205-
207 206-
208 207-
209 208-
210 209-
211 210-
212 211-
213 212-
214 213-
215 214-
216 215-
217 216-
218 217-
219 218-
220 219-
221 220-
222 221-
223 222-
224 223-
225 224-
226 225-
227 226-
228 227-
229 228-
230 229-
231 230-
232 231-
233 232-
234 233-
235 234-
236 235-
237 236-
238 237-
239 238-
240 239-
241 240-
242 241-
243 242-
244 243-
245 244-
246 245-
247 246-
248 247-
249 248-
250 249-
251 250-
252 251-
253 252-
254 253-
255 254-
256 255-
257 256-
258 257-
259 258-
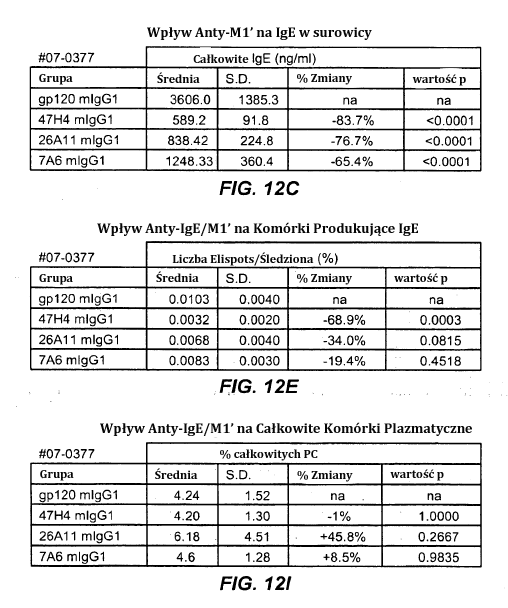
260 259-
261 260-
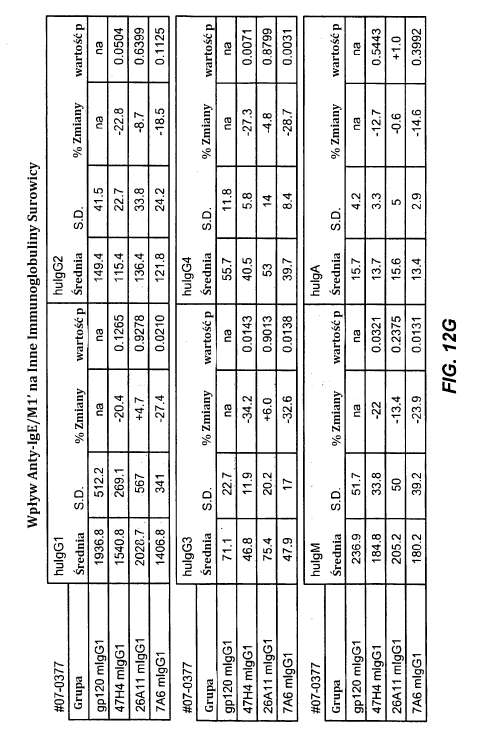
262 261-
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz. III): Aktywacja i funkcje efektorowe limfocytów B
: Aktywacja i funkcje efektorowe limfocytów B") PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz. III): Aktywacja i funkcje efektorowe limfocytów B Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt wykładu Rozpoznanie antygenu
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz. III): Aktywacja i funkcje efektorowe limfocytów B Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt wykładu Rozpoznanie antygenu
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej)
: wprowadzenie (komórki, receptory, rozwój odporności nabytej)") PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej) Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt do wykładu
PODSTAWY IMMUNOLOGII Komórki i cząsteczki biorące udział w odporności nabytej (cz.i): wprowadzenie (komórki, receptory, rozwój odporności nabytej) Nadzieja Drela ndrela@biol.uw.edu.pl Konspekt do wykładu
Leczenie biologiczne co to znaczy?
 Leczenie biologiczne co to znaczy? lek med. Anna Bochenek Centrum Badawcze Współczesnej Terapii C B W T 26 Październik 2006 W oparciu o materiały źródłowe edukacyjnego Grantu, prezentowanego na DDW 2006
Leczenie biologiczne co to znaczy? lek med. Anna Bochenek Centrum Badawcze Współczesnej Terapii C B W T 26 Październik 2006 W oparciu o materiały źródłowe edukacyjnego Grantu, prezentowanego na DDW 2006
Lp. tydzień wykłady seminaria ćwiczenia
 Lp. tydzień wykłady seminaria ćwiczenia 21.02. Wprowadzeniedozag adnieńzwiązanychzi mmunologią, krótka historiaimmunologii, rozwójukładuimmun ologicznego. 19.02. 20.02. Wprowadzenie do zagadnień z immunologii.
Lp. tydzień wykłady seminaria ćwiczenia 21.02. Wprowadzeniedozag adnieńzwiązanychzi mmunologią, krótka historiaimmunologii, rozwójukładuimmun ologicznego. 19.02. 20.02. Wprowadzenie do zagadnień z immunologii.
starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg
 STRESZCZENIE Przewlekła białaczka limfocytowa (PBL) jest najczęstszą białaczką ludzi starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg kliniczny, zróżnicowane rokowanie. Etiologia
STRESZCZENIE Przewlekła białaczka limfocytowa (PBL) jest najczęstszą białaczką ludzi starszych na półkuli zachodniej. Typową cechą choroby jest heterogenny przebieg kliniczny, zróżnicowane rokowanie. Etiologia
Odporność nabyta: Nadzieja Drela Wydział Biologii UW, Zakład Immunologii
 Odporność nabyta: Komórki odporności nabytej: fenotyp, funkcje, powstawanie, krążenie w organizmie Cechy odporności nabytej Rozpoznawanie patogenów przez komórki odporności nabytej: receptory dla antygenu
Odporność nabyta: Komórki odporności nabytej: fenotyp, funkcje, powstawanie, krążenie w organizmie Cechy odporności nabytej Rozpoznawanie patogenów przez komórki odporności nabytej: receptory dla antygenu
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2300459. (96) Data i numer zgłoszenia patentu europejskiego: 22.06.2009 09772333.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2049 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.06.09 09772333.2 (97)
Tolerancja immunologiczna
 Tolerancja immunologiczna autotolerancja, tolerancja na alloantygeny i alergeny dr Katarzyna Bocian Zakład Immunologii kbocian@biol.uw.edu.pl Funkcje układu odpornościowego obrona bakterie alergie wirusy
Tolerancja immunologiczna autotolerancja, tolerancja na alloantygeny i alergeny dr Katarzyna Bocian Zakład Immunologii kbocian@biol.uw.edu.pl Funkcje układu odpornościowego obrona bakterie alergie wirusy
oporność odporność oporność odporność odporność oporność
 oporność odporność odporność nieswoista bierna - niskie ph na powierzchni skóry (mydła!) - enzymy - lizozym, pepsyna, kwas solny żołądka, peptydy o działaniu antybakteryjnym - laktoferyna- przeciwciała
oporność odporność odporność nieswoista bierna - niskie ph na powierzchni skóry (mydła!) - enzymy - lizozym, pepsyna, kwas solny żołądka, peptydy o działaniu antybakteryjnym - laktoferyna- przeciwciała
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE. Ewa Waszkowska ekspert UPRP
 WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
WYNALAZKI BIOTECHNOLOGICZNE W POLSCE Ewa Waszkowska ekspert UPRP Źródła informacji w biotechnologii projekt SLING Warszawa, 9-10.12.2010 PLAN WYSTĄPIENIA Umocowania prawne Wynalazki biotechnologiczne Statystyka
Wyklady IIIL 2016/ :00-16:30 środa Wprowadzenie do immunologii Prof. dr hab. med. ML Kowalski
 III rok Wydział Lekarski Immunologia ogólna z podstawami immunologii klinicznej i alergologii rok akademicki 2016/17 PROGRAM WYKŁADÓW Nr data godzina dzień tygodnia Wyklady IIIL 2016/2017 tytuł Wykladowca
III rok Wydział Lekarski Immunologia ogólna z podstawami immunologii klinicznej i alergologii rok akademicki 2016/17 PROGRAM WYKŁADÓW Nr data godzina dzień tygodnia Wyklady IIIL 2016/2017 tytuł Wykladowca
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1674111 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.07.2005 05760156.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2135881 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 14.06.2006 09010789.7
Część praktyczna: Metody pozyskiwania komórek do badań laboratoryjnych cz. I
 Ćwiczenie 1 Część teoretyczna: Budowa i funkcje układu odpornościowego 1. Układ odpornościowy - główne funkcje, typy odpowiedzi immunologicznej, etapy odpowiedzi odpornościowej. 2. Komórki układu immunologicznego.
Ćwiczenie 1 Część teoretyczna: Budowa i funkcje układu odpornościowego 1. Układ odpornościowy - główne funkcje, typy odpowiedzi immunologicznej, etapy odpowiedzi odpornościowej. 2. Komórki układu immunologicznego.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2224595 (96) Data i numer zgłoszenia patentu europejskiego: 10.02.2010 10001353.1 (13) (51) T3 Int.Cl. H03K 17/96 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2224595 (96) Data i numer zgłoszenia patentu europejskiego: 10.02.2010 10001353.1 (13) (51) T3 Int.Cl. H03K 17/96 (2006.01)
pteronyssinus i Dermatophagoides farinae (dodatnie testy płatkowe stwierdzono odpowiednio u 59,8% i 57,8% pacjentów) oraz żółtko (52,2%) i białko
 oraz żółtko (52,2%) i białko") 8. Streszczenie Choroby alergiczne są na początku XXI wieku są globalnym problemem zdrowotnym. Atopowe zapalenie skóry (AZS) występuje u 20% dzieci i u ok. 1-3% dorosłych, alergiczny nieżyt nosa dotyczy
8. Streszczenie Choroby alergiczne są na początku XXI wieku są globalnym problemem zdrowotnym. Atopowe zapalenie skóry (AZS) występuje u 20% dzieci i u ok. 1-3% dorosłych, alergiczny nieżyt nosa dotyczy
Rok akademicki:2017/2018
 Rok akademicki:2017/2018 Studia magisterskie Kierunek: Analityka medyczna Przedmiot: IMMUNOLOGIA Z IMMUNOPATOLOGIĄ Rok III Semestr V Wykłady 45 godzin Ćwiczenia 30 godzin Seminaria 15 godzin Forma zaliczenia:
Rok akademicki:2017/2018 Studia magisterskie Kierunek: Analityka medyczna Przedmiot: IMMUNOLOGIA Z IMMUNOPATOLOGIĄ Rok III Semestr V Wykłady 45 godzin Ćwiczenia 30 godzin Seminaria 15 godzin Forma zaliczenia:
Immunologia komórkowa
 Immunologia komórkowa ocena immunofenotypu komórek Mariusz Kaczmarek Immunofenotyp Definicja I Charakterystyczny zbiór antygenów stanowiących elementy różnych struktur komórki, związany z jej różnicowaniem,
Immunologia komórkowa ocena immunofenotypu komórek Mariusz Kaczmarek Immunofenotyp Definicja I Charakterystyczny zbiór antygenów stanowiących elementy różnych struktur komórki, związany z jej różnicowaniem,
Przedmiot: IMMUNOLOGIA Z IMMUNOPATOLOGIĄ Rok III Semestr V Wykłady 45 godzin Ćwiczenia 45 godzin Forma zaliczenia: Egzamin praktyczny i teoretyczny
 Rok akademicki 2016/2017 Studia magisterskie Kierunek: Analityka medyczna Przedmiot: IMMUNOLOGIA Z IMMUNOPATOLOGIĄ Rok III Semestr V Wykłady 45 godzin Ćwiczenia 45 godzin Forma zaliczenia: Egzamin praktyczny
Rok akademicki 2016/2017 Studia magisterskie Kierunek: Analityka medyczna Przedmiot: IMMUNOLOGIA Z IMMUNOPATOLOGIĄ Rok III Semestr V Wykłady 45 godzin Ćwiczenia 45 godzin Forma zaliczenia: Egzamin praktyczny
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
Jak żywiciel broni się przed pasożytem?
 https://www. Jak żywiciel broni się przed pasożytem? Autor: Anna Bartosik Data: 12 kwietnia 2019 W poprzedniej części naszego kompendium wiedzy o pasożytach świń omówiliśmy, w jaki sposób pasożyt dostaje
https://www. Jak żywiciel broni się przed pasożytem? Autor: Anna Bartosik Data: 12 kwietnia 2019 W poprzedniej części naszego kompendium wiedzy o pasożytach świń omówiliśmy, w jaki sposób pasożyt dostaje
Rodzaje autoprzeciwciał, sposoby ich wykrywania, znaczenie w ustaleniu diagnozy i monitorowaniu. Objawy związane z mechanizmami uszkodzenia.
 Zakres zagadnień do poszczególnych tematów zajęć I Choroby układowe tkanki łącznej 1. Toczeń rumieniowaty układowy 2. Reumatoidalne zapalenie stawów 3. Twardzina układowa 4. Zapalenie wielomięśniowe/zapalenie
Zakres zagadnień do poszczególnych tematów zajęć I Choroby układowe tkanki łącznej 1. Toczeń rumieniowaty układowy 2. Reumatoidalne zapalenie stawów 3. Twardzina układowa 4. Zapalenie wielomięśniowe/zapalenie
FOCUS Plus - Silniejsza ryba radzi sobie lepiej w trudnych warunkach
 FOCUS Plus - Silniejsza ryba radzi sobie lepiej w trudnych warunkach FOCUS Plus to dodatek dostępny dla standardowych pasz tuczowych BioMaru, dostosowany specjalnie do potrzeb ryb narażonych na trudne
FOCUS Plus - Silniejsza ryba radzi sobie lepiej w trudnych warunkach FOCUS Plus to dodatek dostępny dla standardowych pasz tuczowych BioMaru, dostosowany specjalnie do potrzeb ryb narażonych na trudne
Wirus zapalenia wątroby typu B
 Wirus zapalenia wątroby typu B Kliniczne następstwa zakażenia odsetek procentowy wyzdrowienie przewlekłe zakażenie Noworodki: 10% 90% Dzieci 1 5 lat: 70% 30% Dzieci starsze oraz 90% 5% - 10% Dorośli Choroby
Wirus zapalenia wątroby typu B Kliniczne następstwa zakażenia odsetek procentowy wyzdrowienie przewlekłe zakażenie Noworodki: 10% 90% Dzieci 1 5 lat: 70% 30% Dzieci starsze oraz 90% 5% - 10% Dorośli Choroby
Promotor: prof. dr hab. Katarzyna Bogunia-Kubik Promotor pomocniczy: dr inż. Agnieszka Chrobak
 INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ IM. LUDWIKA HIRSZFELDA WE WROCŁAWIU POLSKA AKADEMIA NAUK mgr Milena Iwaszko Rola polimorfizmu receptorów z rodziny CD94/NKG2 oraz cząsteczki HLA-E w patogenezie
INSTYTUT IMMUNOLOGII I TERAPII DOŚWIADCZALNEJ IM. LUDWIKA HIRSZFELDA WE WROCŁAWIU POLSKA AKADEMIA NAUK mgr Milena Iwaszko Rola polimorfizmu receptorów z rodziny CD94/NKG2 oraz cząsteczki HLA-E w patogenezie
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
1. Układ odpornościowy. Odporność humoralna
 Zakres zagadnień do poszczególnych tematów zajęć Seminaria 1. Układ odpornościowy. Odporność humoralna Ludzki układ odpornościowy Składowe i mechanizmy odporności wrodzonej Składowe i mechanizmy odpowiedzi
Zakres zagadnień do poszczególnych tematów zajęć Seminaria 1. Układ odpornościowy. Odporność humoralna Ludzki układ odpornościowy Składowe i mechanizmy odporności wrodzonej Składowe i mechanizmy odpowiedzi
Wpływ opioidów na układ immunologiczny
 Wpływ opioidów na układ immunologiczny Iwona Filipczak-Bryniarska Klinika Leczenia Bólu i Opieki Paliatywnej Katedry Chorób Wewnętrznych i Gerontologii Collegium Medicum Uniwersytet Jagielloński Kraków
Wpływ opioidów na układ immunologiczny Iwona Filipczak-Bryniarska Klinika Leczenia Bólu i Opieki Paliatywnej Katedry Chorób Wewnętrznych i Gerontologii Collegium Medicum Uniwersytet Jagielloński Kraków
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
Astma oskrzelowa. Zapalenie powoduje nadreaktywność oskrzeli ( cecha nabyta ) na różne bodźce.
 na różne bodźce.") Astma oskrzelowa Astma jest przewlekłym procesem zapalnym dróg oddechowych, w którym biorą udział liczne komórki, a przede wszystkim : mastocyty ( komórki tuczne ), eozynofile i limfocyty T. U osób podatnych
Astma oskrzelowa Astma jest przewlekłym procesem zapalnym dróg oddechowych, w którym biorą udział liczne komórki, a przede wszystkim : mastocyty ( komórki tuczne ), eozynofile i limfocyty T. U osób podatnych
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1658064 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.6 (51) Int. Cl. A61K31/37 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1658064 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 06.07.2004 04740699.6 (51) Int. Cl. A61K31/37 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890471 (96) Data i numer zgłoszenia patentu europejskiego: 19.10.2006 06791271.7 (13) (51) T3 Int.Cl. H04M 3/42 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI
 Załącznik nr 12 do zarządzenia Nr 59/2011/DGL Prezesa NFZ z dnia 10 października 2011 roku Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI ICD 10 D80 w tym D80.0, D80.1, D80.3, D80.4,
Załącznik nr 12 do zarządzenia Nr 59/2011/DGL Prezesa NFZ z dnia 10 października 2011 roku Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI ICD 10 D80 w tym D80.0, D80.1, D80.3, D80.4,
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii
 Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
Inwestycja w przyszłość czyli znaczenie ochrony własności przemysłowej dla współczesnej biotechnologii Zdolność patentowa wynalazków biotechnologicznych dr Małgorzata Kozłowska Ekspert w UPRP dr Ewa Waszkowska
Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI
 Załącznik nr 11 do Zarządzenia Nr 41/2009 Prezesa NFZ z dnia 15 września 2009 roku Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI ICD 10 D80 w tym D80.0, D80.1, D80.3, D80.4, D80.5,
Załącznik nr 11 do Zarządzenia Nr 41/2009 Prezesa NFZ z dnia 15 września 2009 roku Nazwa programu: LECZENIE PIERWOTNYCH NIEDOBORÓW ODPORNOŚCI U DZIECI ICD 10 D80 w tym D80.0, D80.1, D80.3, D80.4, D80.5,
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
PL 204536 B1. Szczepanik Marian,Kraków,PL Selmaj Krzysztof,Łódź,PL 29.12.2003 BUP 26/03 29.01.2010 WUP 01/10
 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 204536 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 354698 (22) Data zgłoszenia: 24.06.2002 (51) Int.Cl. A61K 38/38 (2006.01)
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 204536 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 354698 (22) Data zgłoszenia: 24.06.2002 (51) Int.Cl. A61K 38/38 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 223771 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.08 0886773.1 (13) (1) T3 Int.Cl. A47L 1/42 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 223771 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.08 0886773.1 (13) (1) T3 Int.Cl. A47L 1/42 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787644 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.11.2006 06123574.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787644 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.11.2006 06123574.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2326237 (96) Data i numer zgłoszenia patentu europejskiego: 07.07.2009 09780285.4 (13) (51) T3 Int.Cl. A47L 15/50 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2326237 (96) Data i numer zgłoszenia patentu europejskiego: 07.07.2009 09780285.4 (13) (51) T3 Int.Cl. A47L 15/50 (2006.01)
Spis tre 1. Podstawy immunologii 11 2. Mechanizmy immunopatologiczne 61
 Spis treści Przedmowa do wydania polskiego 6 Przedmowa do wydania pierwszego oryginalnego 6 Przedmowa do wydania drugiego oryginalnego 7 Przedmowa do wydania drugiego oryginalnego zmienionego i uaktualnionego
Spis treści Przedmowa do wydania polskiego 6 Przedmowa do wydania pierwszego oryginalnego 6 Przedmowa do wydania drugiego oryginalnego 7 Przedmowa do wydania drugiego oryginalnego zmienionego i uaktualnionego
Białka układu immunologicznego. Układ immunologiczny
 Białka układu immunologicznego Układ immunologiczny 1 Białka nadrodziny immunoglobulin Białka MHC 2 Białka MHC typu I Łańcuch ciężki (alfa) 45 kda Łańcuch lekki (beta 2 ) 12 kda Występują na powierzchni
Białka układu immunologicznego Układ immunologiczny 1 Białka nadrodziny immunoglobulin Białka MHC 2 Białka MHC typu I Łańcuch ciężki (alfa) 45 kda Łańcuch lekki (beta 2 ) 12 kda Występują na powierzchni
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1837599 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.03.2007 07004628.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1837599 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.03.2007 07004628.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2555663 (96) Data i numer zgłoszenia patentu europejskiego: 06.04.2011 11730434.5 (13) (51) T3 Int.Cl. A47L 15/42 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2555663 (96) Data i numer zgłoszenia patentu europejskiego: 06.04.2011 11730434.5 (13) (51) T3 Int.Cl. A47L 15/42 (2006.01)
Mam Haka na Raka. Chłoniak
 Mam Haka na Raka Chłoniak Nowotwór Pojęciem nowotwór określa się niekontrolowany rozrost nieprawidłowych komórek w organizmie człowieka. Nieprawidłowość komórek oznacza, że różnią się one od komórek otaczających
Mam Haka na Raka Chłoniak Nowotwór Pojęciem nowotwór określa się niekontrolowany rozrost nieprawidłowych komórek w organizmie człowieka. Nieprawidłowość komórek oznacza, że różnią się one od komórek otaczających
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1508941 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.08.2004 04018799.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1508941 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.08.2004 04018799.9
Dr. habil. Anna Salek International Bio-Consulting 1 Germany
 1 2 3 Drożdże są najprostszymi Eukariontami 4 Eucaryota Procaryota 5 6 Informacja genetyczna dla każdej komórki drożdży jest identyczna A zatem każda komórka koduje w DNA wszystkie swoje substancje 7 Przy
1 2 3 Drożdże są najprostszymi Eukariontami 4 Eucaryota Procaryota 5 6 Informacja genetyczna dla każdej komórki drożdży jest identyczna A zatem każda komórka koduje w DNA wszystkie swoje substancje 7 Przy
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
Model Marczuka przebiegu infekcji.
 Model Marczuka przebiegu infekcji. Karolina Szymaniuk 27 maja 2013 Karolina Szymaniuk () Model Marczuka przebiegu infekcji. 27 maja 2013 1 / 17 Substrat Związek chemiczny, który ulega przemianie w wyniku
Model Marczuka przebiegu infekcji. Karolina Szymaniuk 27 maja 2013 Karolina Szymaniuk () Model Marczuka przebiegu infekcji. 27 maja 2013 1 / 17 Substrat Związek chemiczny, który ulega przemianie w wyniku
Terapeutyczne Programy Zdrowotne 2008 Leczenie stwardnienia rozsianego
 załącznik nr 16 do zarządzenia Nr 36/2008/DGL Prezesa NFZ z dnia 19 czerwca 2008 r. Nazwa programu: LECZENIE STWARDNIENIA ROZSIANEGO ICD-10 G.35 - stwardnienie rozsiane Dziedzina medycyny: neurologia I.
załącznik nr 16 do zarządzenia Nr 36/2008/DGL Prezesa NFZ z dnia 19 czerwca 2008 r. Nazwa programu: LECZENIE STWARDNIENIA ROZSIANEGO ICD-10 G.35 - stwardnienie rozsiane Dziedzina medycyny: neurologia I.
Diagnostyka zakażeń EBV
 Diagnostyka zakażeń EBV Jakie wyróżniamy główne konsekwencje kliniczne zakażenia EBV: 1) Mononukleoza zakaźna 2) Chłoniak Burkitta 3) Potransplantacyjny zespół limfoproliferacyjny Jakie są charakterystyczne
Diagnostyka zakażeń EBV Jakie wyróżniamy główne konsekwencje kliniczne zakażenia EBV: 1) Mononukleoza zakaźna 2) Chłoniak Burkitta 3) Potransplantacyjny zespół limfoproliferacyjny Jakie są charakterystyczne
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2321656 (96) Data i numer zgłoszenia patentu europejskiego:.08.09 09807498.2 (13) (51) T3 Int.Cl. G01R /18 (06.01) G01R 19/
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2321656 (96) Data i numer zgłoszenia patentu europejskiego:.08.09 09807498.2 (13) (51) T3 Int.Cl. G01R /18 (06.01) G01R 19/
Immunoregulacyjne aktywności wybranych pochodnych izoksazolu o potencjalnej przydatności terapeutycznej
 Instytut Immunologiii Terapii Doświadczalnej PAN we Wrocławiu Zakład Terapii Doświadczalnej Laboratorium Immunobiologii Kierownik: prof. Michał Zimecki Immunoregulacyjne aktywności wybranych pochodnych
Instytut Immunologiii Terapii Doświadczalnej PAN we Wrocławiu Zakład Terapii Doświadczalnej Laboratorium Immunobiologii Kierownik: prof. Michał Zimecki Immunoregulacyjne aktywności wybranych pochodnych
(96) Data i numer zgłoszenia patentu europejskiego:
 Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2913207 (96) Data i numer zgłoszenia patentu europejskiego: 08.05.2014 14167514.0 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2913207 (96) Data i numer zgłoszenia patentu europejskiego: 08.05.2014 14167514.0 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445186 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2011 11184611.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445186 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2011 11184611.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18761 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 31.03.06 06726163.6 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18761 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 31.03.06 06726163.6 (97)
Plan. Sztuczne systemy immunologiczne. Podstawowy słownik. Odporność swoista. Architektura systemu naturalnego. Naturalny system immunologiczny
 Sztuczne systemy immunologiczne Plan Naturalny system immunologiczny Systemy oparte na selekcji klonalnej Systemy oparte na modelu sieci idiotypowej 2 Podstawowy słownik Naturalny system immunologiczny
Sztuczne systemy immunologiczne Plan Naturalny system immunologiczny Systemy oparte na selekcji klonalnej Systemy oparte na modelu sieci idiotypowej 2 Podstawowy słownik Naturalny system immunologiczny
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1648484 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 01.06.04 047366.4 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1648484 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 01.06.04 047366.4 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1730054 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 22.03.2005 05731932.9 (51) Int. Cl. B65G17/06 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1591364 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2005 05103299.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1591364 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2005 05103299.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
UKŁAD LIMFATYCZNY UKŁAD ODPORNOŚCIOWY. Nadzieja Drela Instytut Zoologii, Zakład Immunologii
 UKŁAD LIMFATYCZNY UKŁAD ODPORNOŚCIOWY Nadzieja Drela Instytut Zoologii, Zakład Immunologii ndrela@biol.uw.edu.pl Układ limfatyczny Narządy, naczynia limfatyczne, krążące limfocyty Centralne narządy limfoidalne
UKŁAD LIMFATYCZNY UKŁAD ODPORNOŚCIOWY Nadzieja Drela Instytut Zoologii, Zakład Immunologii ndrela@biol.uw.edu.pl Układ limfatyczny Narządy, naczynia limfatyczne, krążące limfocyty Centralne narządy limfoidalne
Podstawy immunologii. Odporność. Odporność nabyta. nieswoista. swoista
 Podstawy immunologii dr n. med. Jolanta Meller Odporność nieswoista mechanizmy obronne skóry i błon śluzowych substancje biologicznie czynne: interferon, lizozym, dopełniacz leukocyty (fagocyty) swoista
Podstawy immunologii dr n. med. Jolanta Meller Odporność nieswoista mechanizmy obronne skóry i błon śluzowych substancje biologicznie czynne: interferon, lizozym, dopełniacz leukocyty (fagocyty) swoista
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2003466 (96) Data i numer zgłoszenia patentu europejskiego: 12.06.2008 08460024.6 (13) (51) T3 Int.Cl. G01S 5/02 (2010.01)
Pytania Egzamin magisterski
 Pytania Egzamin magisterski Międzyuczelniany Wydział Biotechnologii UG i GUMed 1. Krótko omów jakie informacje powinny być zawarte w typowych rozdziałach publikacji naukowej: Wstęp, Materiały i Metody,
Pytania Egzamin magisterski Międzyuczelniany Wydział Biotechnologii UG i GUMed 1. Krótko omów jakie informacje powinny być zawarte w typowych rozdziałach publikacji naukowej: Wstęp, Materiały i Metody,
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
O PO P R O NOŚ O Ć Ś WR
 ODPORNOŚĆ WRODZONA Egzamin 3 czerwca 2015 godz. 17.30 sala 9B FUNKCJE UKŁADU ODPORNOŚCIOWEGO OBRONA NADZÓR OBCE BIAŁKA WIRUSY BAKTERIE GRZYBY PASOŻYTY NOWOTWORY KOMÓRKI USZKODZONE KOMÓRKI OBUNMIERAJĄCE
ODPORNOŚĆ WRODZONA Egzamin 3 czerwca 2015 godz. 17.30 sala 9B FUNKCJE UKŁADU ODPORNOŚCIOWEGO OBRONA NADZÓR OBCE BIAŁKA WIRUSY BAKTERIE GRZYBY PASOŻYTY NOWOTWORY KOMÓRKI USZKODZONE KOMÓRKI OBUNMIERAJĄCE
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
Spis treści. Wykaz używanych skrótów i symboli... 14. 1. Wprowadzenie... 18
 Spis treści Wykaz używanych skrótów i symboli... 14 1. Wprowadzenie... 18 1.1. Podstawowe zasady działania układu immunologicznego... 18 1.1.1. Formy odpowiedzi immunologicznej... 19 1.1.2. Rozpoznanie
Spis treści Wykaz używanych skrótów i symboli... 14 1. Wprowadzenie... 18 1.1. Podstawowe zasady działania układu immunologicznego... 18 1.1.1. Formy odpowiedzi immunologicznej... 19 1.1.2. Rozpoznanie
NON-HODGKIN S LYMPHOMA
 NON-HODGKIN S LYMPHOMA Klinika Hematologii, Nowotworów Krwi i Transplantacji Szpiku We Wrocławiu Aleksandra Bogucka-Fedorczuk DEFINICJA Chłoniaki Non-Hodgkin (NHL) to heterogeniczna grupa nowotworów charakteryzująca
NON-HODGKIN S LYMPHOMA Klinika Hematologii, Nowotworów Krwi i Transplantacji Szpiku We Wrocławiu Aleksandra Bogucka-Fedorczuk DEFINICJA Chłoniaki Non-Hodgkin (NHL) to heterogeniczna grupa nowotworów charakteryzująca
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 221611 (96) Data i numer zgłoszenia patentu europejskiego: 19.01. 000481.1 (13) (1) T3 Int.Cl. B28C /42 (06.01) B60P 3/16
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 221611 (96) Data i numer zgłoszenia patentu europejskiego: 19.01. 000481.1 (13) (1) T3 Int.Cl. B28C /42 (06.01) B60P 3/16
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1505553. (96) Data i numer zgłoszenia patentu europejskiego: 05.08.2004 04018511.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 3 (96) Data i numer zgłoszenia patentu europejskiego: 0.08.04 0401811.8 (13) (1) T3 Int.Cl. G08C 17/00 (06.01) Urząd Patentowy
Dobierając optymalny program szczepień, jesteśmy w stanie zapobiec chorobom, które mogą być zagrożeniem dla zdrowia Państwa pupila.
 SZCZEPIENIA KOTÓW Działamy według zasady: Lepiej zapobiegać niż leczyć Wychodząc naprzeciw Państwa oczekiwaniom oraz dbając o dobro Waszych pupili opisaliśmy program profilaktyczny chorób zakaźnych psów,
SZCZEPIENIA KOTÓW Działamy według zasady: Lepiej zapobiegać niż leczyć Wychodząc naprzeciw Państwa oczekiwaniom oraz dbając o dobro Waszych pupili opisaliśmy program profilaktyczny chorób zakaźnych psów,
Cytokiny jako nośniki informacji
 Wykład 2 15.10.2014 Cytokiny jako nośniki informacji Termin cytokiny (z greckiego: cyto = komórka i kinos = ruch) określa dużą grupę związków o różnym pochodzeniu i budowie, będących peptydami, białkami
Wykład 2 15.10.2014 Cytokiny jako nośniki informacji Termin cytokiny (z greckiego: cyto = komórka i kinos = ruch) określa dużą grupę związków o różnym pochodzeniu i budowie, będących peptydami, białkami
Przemiana materii i energii - Biologia.net.pl
 Ogół przemian biochemicznych, które zachodzą w komórce składają się na jej metabolizm. Wyróżnia się dwa antagonistyczne procesy metabolizmu: anabolizm i katabolizm. Szlak metaboliczny w komórce, to szereg
Ogół przemian biochemicznych, które zachodzą w komórce składają się na jej metabolizm. Wyróżnia się dwa antagonistyczne procesy metabolizmu: anabolizm i katabolizm. Szlak metaboliczny w komórce, to szereg
Spis treści. Komórki, tkanki i narządy układu odpornościowego 5. Swoista odpowiedź immunologiczna: mechanizmy 53. Odporność nieswoista 15
 Spis treści Komórki, tkanki i narządy układu odpornościowego 5 1. Wstęp: układ odpornościowy 7 2. Komórki układu odpornościowego 8 3. kanki i narządy układu odpornościowego 10 Odporność nieswoista 15 1.
Spis treści Komórki, tkanki i narządy układu odpornościowego 5 1. Wstęp: układ odpornościowy 7 2. Komórki układu odpornościowego 8 3. kanki i narządy układu odpornościowego 10 Odporność nieswoista 15 1.
Odporność, stres, alergia
 Odporność, stres, alergia Odporność komórkowa Układ mikrofagocytarny Układ makrofagocytarny 1 Układ mikrofagocytarny Granulocyty obojętnochłonne Granulocyty kwasochłonne Granulocyty zasadochłonne Układ
Odporność, stres, alergia Odporność komórkowa Układ mikrofagocytarny Układ makrofagocytarny 1 Układ mikrofagocytarny Granulocyty obojętnochłonne Granulocyty kwasochłonne Granulocyty zasadochłonne Układ
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1734922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.03.2005 05728244.4
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1734922 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.03.2005 05728244.4
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2776315 (96) Data i numer zgłoszenia patentu europejskiego: 19.07.2013 13753588.6 (13) (51) T4 Int.Cl. B64C 29/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2776315 (96) Data i numer zgłoszenia patentu europejskiego: 19.07.2013 13753588.6 (13) (51) T4 Int.Cl. B64C 29/00 (2006.01)
KOŁO NAUKOWE IMMUNOLOGII. Mikrochimeryzm badania w hodowlach leukocytów in vitro
 KOŁO NAUKOWE IMMUNOLOGII Mikrochimeryzm badania w hodowlach leukocytów in vitro Koło Naukowe Immunolgii kolo_immunologii@biol.uw.edu.pl kolo_immunologii.kn@uw.edu.pl CEL I PRZEDMIOT PROJEKTU Celem doświadczenia
KOŁO NAUKOWE IMMUNOLOGII Mikrochimeryzm badania w hodowlach leukocytów in vitro Koło Naukowe Immunolgii kolo_immunologii@biol.uw.edu.pl kolo_immunologii.kn@uw.edu.pl CEL I PRZEDMIOT PROJEKTU Celem doświadczenia
ROLA WAPNIA W FIZJOLOGII KOMÓRKI
 ROLA WAPNIA W FIZJOLOGII KOMÓRKI Michał M. Dyzma PLAN REFERATU Historia badań nad wapniem Domeny białek wiążące wapń Homeostaza wapniowa w komórce Komórkowe rezerwuary wapnia Białka buforujące Pompy wapniowe
ROLA WAPNIA W FIZJOLOGII KOMÓRKI Michał M. Dyzma PLAN REFERATU Historia badań nad wapniem Domeny białek wiążące wapń Homeostaza wapniowa w komórce Komórkowe rezerwuary wapnia Białka buforujące Pompy wapniowe
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
ROZPORZĄDZENIE KOMISJI (UE) / z dnia r.
 / z dnia r.") KOMISJA EUROPEJSKA Bruksela, dnia 29.5.2018 C(2018) 3193 final ROZPORZĄDZENIE KOMISJI (UE) / z dnia 29.5.2018 r. zmieniające rozporządzenie (WE) nr 847/2000 w odniesieniu do definicji pojęcia podobnego
KOMISJA EUROPEJSKA Bruksela, dnia 29.5.2018 C(2018) 3193 final ROZPORZĄDZENIE KOMISJI (UE) / z dnia 29.5.2018 r. zmieniające rozporządzenie (WE) nr 847/2000 w odniesieniu do definicji pojęcia podobnego
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 21737 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2010 10790844.4 (13) (1) T3 Int.Cl. A47L 1/42 (2006.01) A47L
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 21737 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2010 10790844.4 (13) (1) T3 Int.Cl. A47L 1/42 (2006.01) A47L
Kod przedmiotu/modułu MK_39 Punkty ETCS: 6. Jednostka: Zakład Immunologii Katedry Immunologii Klinicznej, ul. Rokietnicka 5d, 60-806 Poznań
 Kod przedmiotu/modułu MK_39 Punkty ETCS: 6 Nazwa przedmiotu: Immunopatologia Jednostka: Zakład Immunologii Katedry Immunologii Klinicznej, ul. Rokietnicka 5d, 60-806 Poznań Osoba odpowiedzialna za przedmiot:
Kod przedmiotu/modułu MK_39 Punkty ETCS: 6 Nazwa przedmiotu: Immunopatologia Jednostka: Zakład Immunologii Katedry Immunologii Klinicznej, ul. Rokietnicka 5d, 60-806 Poznań Osoba odpowiedzialna za przedmiot:
Poradnia Immunologiczna
 Poradnia Immunologiczna Centrum Onkologii Ziemi Lubelskiej im. św. Jana z Dukli Lublin, 2011 Szanowni Państwo, Uprzejmie informujemy, że w Centrum Onkologii Ziemi Lubelskiej im. św. Jana z Dukli funkcjonuje
Poradnia Immunologiczna Centrum Onkologii Ziemi Lubelskiej im. św. Jana z Dukli Lublin, 2011 Szanowni Państwo, Uprzejmie informujemy, że w Centrum Onkologii Ziemi Lubelskiej im. św. Jana z Dukli funkcjonuje
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2949485 (96) Data i numer zgłoszenia patentu europejskiego: 06.10.2014 14187774.6 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2949485 (96) Data i numer zgłoszenia patentu europejskiego: 06.10.2014 14187774.6 (13) (51) T3 Int.Cl. B60C 23/04 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2057877 (96) Data i numer zgłoszenia patentu europejskiego: 03.11.2008 08019246.1 (13) (51) T3 Int.Cl. A01C 23/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2057877 (96) Data i numer zgłoszenia patentu europejskiego: 03.11.2008 08019246.1 (13) (51) T3 Int.Cl. A01C 23/00 (2006.01)