Rekonstrukcja Filoinformatyka
|
|
|
- Filip Cichoń
- 7 lat temu
- Przeglądów:
Transkrypt

1 Rekonstrukcja Filoinformatyka filogenii TTTTTTTTAAAAATTTTTTTCTTTTAAA Jacek Dabert Zakład Morfologii Zwierząt UAM
2 Do czego służy analiza filogenetyczna? Do rekonstrukcji historycznych związków filogenetycznych pomiędzy taksonami. W aplikacjach biomedycznych np. epidemiologii i medycynie sądowej W badaniach molekularnych nad organizacją genomu i strukturą genów. W badaniach nad powstawaniem nowych alleli i szczepów laboratoryjnych W studiach porównawczych w ekologii i etologii W badaniu procesów fizjologicznych Generalnie na wszystkich polach, w których dokonuje się porównań między obiektami lub procesami.
3 Tematyka wykładu 1. Podstawowe terminy stosowane w rekonstrukcji filogenii. 2. Przygotowanie matrycy danych. 3. Algorytmy do konstrukcji drzew filogenetycznych. 4. Poszczególne etapy komputerowej analizy filogenetycznej danych molekularnych. 5. Analiza statystyczna uzyskanych wyników. 6. Procedura total evidence. 7. Współbieżne zdarzenia ewolucyjne.
4 Taksony naturalne i sztuczne a. grupa monofiletyczna, b. grupa parafiletyczna, c. grupa polifiletyczna
5 Podstawowe terminy dotyczące drzew filogenetycznych


6 Przykładowe rodzaje cech stosowanych w rekonstrukcji filogenii

7 Typy matryc danych matryca dystansów matryca cech

8 Nexus Data Editor
9 Czy cechy morfologiczne są nadal istotne? Wady bardziej homoplastyczne niż molekularne (?) ukierunkowane konwergencje tanie Zalety możliwość korzystania ze źródeł muzealnych ograniczona liczba cech trudności w znalezieniu cech homologicznych między odległymi taksonami taksony wymarłe mogą być analizowane głównie na podstawie danych morfologicznych dane morfologiczne mogą być testowalne na wszystkich etapach analizy filogenetycznej
10 Metody konstruowania drzew filogenetycznych Metoda obliczeniowa optymalizacja Parsymonia Maximum Likelihood wnioskowanie Bayesowskie Minimum Evolution Least Squares analiza klastrów UPGMA Neighbor-Joining Cechy Dystanse

11 Zegar molekularny Koncepcja zegara molekularnego (Zuckerlandl i Pauling, 1965) postuluje równe tempo substytucji we wszystkich liniach ewolucyjnych. Dzięki danym fosylnym możliwe jest kalibrowanie zegara i określanie bezwzględnego czasu dywergencji. x mln lat
12 UPGMA UPGMA (unweighted pair group method with arithmetic mean) to najprostsza metoda grupująca taksony według ogólnego podobieństwa lub odległości. Pracuje wyłącznie na matrycach dystansowych np. hybrydyzacja DNA-DNA lub konstruowanych z danych sekwencyjnych na podstawie ilości substytucji. UPGMA umożliwia określenie długości gałęzi (odlegości ewolucyjnej) jak i uporządkowania gałęzi. Zakłada stały zegar molekularny możliwe jest teoretycznie oszacowanie czasu dywergencji na podstawie różnic w sekwencjach.

13 Jak działa UPGMA A C B D OTU A-C A BB C D A-C A - 8 8,5 7 11,5 12 B B - 14 C - 11 D -- Matryca dystansowa zredukowana substytucje/100 nukleotydów 1. Znajdź najbliższą parę gatunków. 2. Połącz oba te gatunki w klaster. 3. Policz na nowo pozostałe dystanse jako średnią od A-C. 4. Idź do kroku 1 i powtórz procedurę.
14 Warunek trzech punktów Aby analiza UPGMA mogła być przeprowadzona z sukcesem dane muszą być zultrametryzowane. Oznacza to, że dla dowolnych trzech taksonów (x, y, z) dystanse (d) pomiędzy nimi muszą spełniać następujące wyrażenie: d(x,z) max (d(x,y), d(y,z)) Powyższą formułę nazywa się także warunkiem trzech punktów.
15 Neighbor-Joining (NJ) Metoda koncepcyjnie zbliżona do analizy klastrów, jednak dopuszcza niejednakowe tempo zmian molekularnych wśród gałęzi. Zasada analizy NJ: Wyszukiwanie par taksonów (sąsiadów=neighbors), które minimalizują totalną długość gałęzi na każdym etapie grupowania taksonów początkowo zgrupowanych w całkowicie politomicznym drzewie ( gwiazda ).
16 Jak działa NJ 1. Inicjalne drzewo ma postać w pełni politomicznej gwiazdy. B B C (D,(C,E)) D (((A,B),H),(G,F)) ((((A,B),H),(G,F)),(D,(C,E))) ((A,B),H) (A,B) A (C,E) E A H H A B G F D F 2. Losowo wybierana jest para sekwencji i łączona gałęzią z centrum gwiazdy. Liczona jest całkowita długość gałęzi drzewa. Para jest zwracana do gwiazdy. 3. Powtarzane jest to ze wszystkimi możliwymi kombinacjami par, aż do znalezienia drzewa o najmniejszej całkowitej długości gałęzi. Para sekwencji z tego drzewa sąsiaduje ze sobą w finalnym drzewie. C 4. Para ta jest tymczasowo kombinowana (G,F) G w jednostkę, włączana do gwiazdy E krótszej o jedną gałąź i matryca dystansów liczona jest na nowo. 5. Procedura jest powtarzana tak długo, aż wszyscy sąsiedzi zostaną znalezieni i otrzymamy gotowe drzewo.
17 Maksymalna wiarygodność (maximum likelihood,, ML) Metoda stosowana niemal wyłącznie do danych sekwencyjnych Stosunkowo skomplikowana podstawa teoretyczna i znaczne wymagania co do mocy sprzętu obliczeniowego. ML zakłada określony, niekiedy złożony model ewolucji sekwencji. Celem analizy ML jest odpowiedź na pytanie: Jakie jest prawdopodobieństwo P powstania obserwowanych danych D (w tym wypadku alignmentu wielu sekwencji) dla danej topologii drzewa filogenetycznego T przy określonym modelu ewolucji?
18 Wnioskowanie bayesowskie (BI, Bayesian inference) Metoda zbliżona koncepcyjnie do ML Celem analizy BI jest odpowiedź na pytanie: Jakie jest prawdopodobieństwo P, że dana topologia drzewa T przy określonym modelu ewolucji jest prawdziwa dla obserwowanych danych D (w tym wypadku alignmentu wielu sekwencji)? ML: P(D/T) BI: P (T/D) Formuła Bayesa: P(T/D) P(T) P(D/T) = P(D)
19 Modele ewolucji sekwencji Modele mogą dotyczyć różnych aspektów ewolucji sekwencji: Różnorodnego stosunku transwersji do tranzycji. Odmiennej frekwencji nukleotydów. Różnorodnego tempa ewolucji w poszczególnych miejscach sekwencji. Różnorodnego tempa ewolucji (=substytucji) w ramach linii (poszczególnych taksonów) czy całych partii drzewa. Bogactwo parametrów modeli ma pozytywne i negatywne strony: im więcej parametrów do testowania tym lepiej można dopasować model do konkretnych danych. im więcej parametrów do testowania tym wyższa wariancja oszacowania.
20 Long-branch attraction (LBA) albo Strefa Felsensteina W przypadku taksonów wykazujących bardzo wysokie tempo ewolucji (=substytucji) liczba synapomorfii może być wyższa dla bardziej odległych taksonów. W ten sposób taksony o słabszym tempie substytucji mogą błędnie grupować się ze sobą, a o długich gałęziach ze sobą lub outgrupą. Procedury ML oraz wnioskowanie bayesowskie są najbardziej odporne na ten problem, jeśli zostanie zastosowany odpowiedni model substytucji prawidłowe drzewo błędne drzewo
21 Maksymalna Parsymonia (MP) Założeniem jest, że ewolucja przebiega najkrótszą z możliwych dróg (zasada parsymonii). Jest metodą bardziej ewolucyjnie rygorystyczną niż metody dystansowe. Zasada analizy MP: Porównywane są wszystkie możliwe topologie drzew. To drzewo, które wymaga w sumie najmniejszej liczby zmian poszczególnych cech (substytucji i delecji/insercji) jest najlepszym drzewem.
22 Odmiany analizy MP 1. parsymonia Wagnera zezwala na rewersje, wszystkie cechy uporządkowane. 2. parsymonia Dollo cecha pojawia się tylko raz, możliwa jest rewersja do cechy ancestralnej. 3. parsymonia Camina-Sokala najostrzejsza, zmiany ewolucyjne są nieodwracalne (=Dollo + brak rewersji). 4. parsymonia ogólna dopuszczane są wszystkie możliwe sytuacje z trzech poprzednich, stosowane indywidualnie do określonych cech lub ich grup.

23 Cechy Apomorfie i plezjomorfie B A 1 A 2 A 0 1 A A C konwergencja T G C lub paralelizm G G G C A C G T C A T A A T T C A apomorfie i plezjomorfie to homologie konwergencje, paralelizmy i rewersje to homoplazje synapomorfia symplezjomorfia Wspólny przodek A 1 i A 2 A C apomorfia plezjomorfia rewersja Wspólny przodek B i A 0

24 Cechy informatywne i nieinformatywne Sekwencja Pozycja 1: nieinformatywna Pozycja 1 G G G G 2 A T T T 3 C T G G 4 A C T G 5 A A G G Pozycja 2: nieinformatywna Pozycja 3: nieinformatywna Pozycja 4: nieinformatywna Pozycja 5: informatywna
25 Argumentacja cech Cechy binarne niespolaryzowane Cechy binarne spolaryzowane Nieuporządkowana i niespolaryzowana seria przekształceń Seria uporządkowana i niespolaryzowana Seria uporządkowana i spolaryzowana
26 Ważenie tranzycje/transwersje transwersje za pomocą step matrices Tranzycje substytucje między purynami (A G) lub pirymidynami (C T). Transwersje substytucje między purynami a pirymidynami (A T, C G, A C, T G).

27 Ograniczenia metody exhaustive exhaustive dla MP Analizowane są wszystkie możliwe drzewa, dzięki czemu gwarantowane jest znalezienie najkrótszego drzewa. Bardzo czasochłonna i możliwa do zastosowania jedynie dla małych matryc danych (do 12 taksonów).

28 Procedura branch-and and-bound i heurystyczna B&B znajduje optymalne drzewo (-a), ale tylko dla matryc ~ 20 taksonów. LA>L A L=LCL H nie gwarantuje znalezienia optymalnego drzewa, ale matryce mogą być znacznie większe. LB=L B LC<L C D LD>L
29 Wady i zalety poszczególnych algorytmów Metoda UPGMA NJ ML Zalety bardzo prosta i bardzo szybka bardzo szybka (długie sekwencje, bootstrap) akceptuje linie wykazujące różne tempo ewolucji odporna na LBA dobre statystyczne podstawy sprawdza różne topologie używa całą informację z sekwencji dedykowana do danych sekwencyjnych Wady bardzo czuła na różne tempo ewolucji grupowanie możliwe jest jedynie, jeśli dane są ultrametryczne tzn. spełniają warunek trzech punktów informacja z sekwencji jest zredukowana (dystanse) daje tylko jedno możliwe drzewo silnie zależy od rodzaju zastosowanego modelu ewolucji bardzo wolna i wymaga dużej mocy komputera (BI szybsza) rezultaty zależą od zastosowanego modelu ewolucji MP jedyna w pełni kladystyczna metoda można identyfikować obszary problematyczne nie redukuje informacji z sekwencji sprawdza różnorodne drzewa (hipotezy) bardzo powolna dla nawet niedużych matryc, nawet dla strategii branch-and-bound nie wykorzystuje pełnej informacji z sekwencji (tylko informatywne) nie zakłada modelu ewolucji nie daje informacji o długości gałęzi
30 Wady i zalety poszczególnych algorytmów Generalnie wszystkie metody dystansowe są fenetyczne - konstruują drzewa poprzez grupowanie OTU na podstawie ogólnego podobieństwa (morfologicznego, sekwencji itp.). A ogólne podobieństwo nie koniecznie musi odzwierciedlać prawdziwe pokrewieństwo filogenetyczne. Natomiast metody ML, BI i MP, choć koncepcyjnie lepiej zakotwiczone w procesach ewolucyjnych, są ekstremalnie wymagające w stosunku do mocy obliczeniowej komputerów, a mnogość parametrów opcjonalnych może w efekcie wpłynąć na rekonstrukcję w trudny do oszacowania sposób (subiektywizm badacza).
31 Który algorytm jest najlepszy dla danych sekwencyjnych? ML i BI lepsze od pozostałych jeśli nierówne tempo substytucji pomiędzy liniami ewolucyjnymi
32 Podstawowe etapy analizy filogenetycznej na przykładzie procedury MP 1. Wybór markera do rozwiązania problemu na określonym poziomie taksonomicznym. 2. Alignment. 3. Skonstruowanie matrycy danych. 4. Zadanie odpowiednich parametrów wejściowych. 5. Przeprowadzenie analizy komputerowej. 6. Analiza statystyczna uzyskanych wyników. 6. Wizualizacja i interpretacja wyników.
33 Metoda a poziom taksonomiczny

34 Marker a poziom taksonomiczny 16S rrna
35 Test skośności na obecność sygnału filogenetycznego g 1 = n i= 1 ns 3 T T i 3 Liczba drzew silny sygnał słaby sygnał T - długość drzewa n liczba drzew o długości T s - odchylenie standardowe Długość drzewa
36 Podstawowe parametry statystyczne drzew MP - Ilość kroków (L) suma zmian cech na wszystkich gałęziach. Dla danego zestawu cech najlepszym jest drzewo o najmniejszej liczbie kroków. - Współczynnik konsystencji cechy m minimalna możliwa liczba kroków s faktyczna liczba kroków - Współczynnik retencji cechy g minimalna liczba kroków dla najgorszej możliwości tzn. całkowitej politomii c = r = m s g s g m - Współczynniki złożone M = m1+m2...+mn S = s1+s2...+sn G = g1+g2...+gn CI = M S R = G S G M

37 Wartość krytyczna CI
38 Metody próbkowania (resampling( resampling) Są to metody statystyczne służące do określenia stabilności kladów. Pobierane są wielokrotnie losowe próbki (pseudoreplikacje) z danych. Konstruowane są drzewka z wszystkich pseudoreplikacji i procedura powtarzana jest wielokrotnie ( np razy) Następnie liczony jest 50% majority rule consensus. Częstotliwość pojawiania się poszczególnych kladów w drzewie konsensusowym stanowi miarę stabilności testowanej topologii drzewa filogenetycznego. Stosowane do wszelkich danych dyskretnych, także dystansowych.
39 Jackknife Losowo pobierane do próbki cechy (dystanse) bez zwracania danych do oryginalnej matrycy. Symulacje wykazały, że najodpowiedniejszą wielkością próbki jest 1/e (ok. 36,8%) danych. oryginalna matryca N=42 próbka N=15 Av.calidridis1 tatgaatgaattttctgagaactgttttttctg--ttttt-c Av.calidridis2 tatgaatgaattttctgagaactgttttttctg--ttttt-c Av.calidridis3 tatgaatgaattttctgagaactgttttttctg--ttttt-c Av.calidridisalp tatgaatgaattttctaagggttggtttttttg--ttttt-c Av.phalaropi tatgaatgaatttactatgaattttttct---gaattttttc Av.philomachi tatgaatgaattttctgaaaattttttttt--a--tttattc Av.totanigla1 tatgaatgaatgttctaaaaattttttttt--g--ttttttc Av.totanigla2 tatgaatgaatgttctaaaaattttttttt--g--ttttttc Av.totanitot1 tatgaatgaattttctaaaaattttttttt--g--ttttttc Av.totanitot2 tatgaatgaattttctaaaaattttttttt--g--ttttttc Av.tretekiae tatgaatgaattttctaataattttttttt--g--ttttttc Av.tringae tatgaatgaattttctaataatttttattattg--ttttt-- Procedura ta jest powtarzana wielokrotnie (np razy) i za każdym razem z próbki budowane jest drzewo (-a) filogenetyczne. Następnie konstruowany jest konsensus.
40 Bootstrap Losowo pobierane są pseudoreplikacje i, w odróżnieniu do jackknife, dane zwracane są do oryginalnej matrycy. Wielkość próbki jest taka sama jak matrycy oryginalnej. Oznacza to, że pewne pseudoreplikacje są pobierane więcej niż jeden raz. oryginalna matryca N=42 próbka N=42 Av.calidridis1 Av.calidridis2 Av.calidridis3 Av.calidridisalp Av.phalaropi Av.philomachi Av.totanigla1 Av.totanigla2 Av.totanitot1 Av.totanitot2 Av.tretekiae Av.tringae tatgaatgaattttctgagaactgttttttctg--ttttt-c tatgaatgaattttctgagaactgttttttctg--ttttt-c tatgaatgaattttctgagaactgttttttctg--ttttt-c tatgaatgaattttctaagggttggtttttttg--ttttt-c tatgaatgaatttactatgaattttttct---gaattttttc tatgaatgaattttctgaaaattttttttt--a--tttattc tatgaatgaatgttctaaaaattttttttt--g--ttttttc tatgaatgaatgttctaaaaattttttttt--g--ttttttc tatgaatgaattttctaaaaattttttttt--g--ttttttc tatgaatgaattttctaaaaattttttttt--g--ttttttc tatgaatgaattttctaataattttttttt--g--ttttttc tatgaatgaattttctaataatttttattattg--ttttt--
41 Bootstrap wartości krytyczne
42 Indeks Bremera (decay index) Indeks ten wskazuje jaka liczba dodatkowych kroków ewolucyjnych jest potrzebna by uzyskać drzewo, w którym dany klad jest zlikwidowany tzn. tworzy politomię. Stosowany wyłącznie w analizie parsymonii. Plutarchusia chelopus Sikyonemus diplectron Eurysyringobia spinigera 1 drzewo MP, L = drzewa MP, L Limosilichus limosae Phyllochaeta bouveti Phyllochaeta interifolia
43 Consensus tree Rodzaj drzewa Odpowiada na pytanie Wady i zalety Strict Adams Majority Jakie grupy są zawsze monofiletyczne? 1. Jakie jest drzewko o najwyższej rozdzielczości, które będzie rozpoznawało problematyczne taksony? 2. Czy drzewka są logicznie spójne? Jakie jest podsumowanie konkurujących drzewek gdzie przeważa dominujący wzór? 1. Możliwa ekstremalna utrata rozdzielczości 2. Użyteczne jako rekonstrukcja filogenii wyłącznie, jeśli identyczne z jednym z oryginalnych MP drzewek 1. Dziwne umiejscowienie taksonów, które nie występuje w żadnym oryginalnym drzewku 2. Użyteczne jako rekonstrukcja filogenii wyłącznie, jeśli identyczne z jednym z oryginalnych MP drzewek 1. Najużyteczniejszy, gdy jest bardzo mało sprzecznych danych 2. Użyteczne jako rekonstrukcja filogenii wyłącznie, jeśli identyczne z jednym z oryginalnych MP drzewek
44 Optymizacja drzew ACCTRAN, DELTRAN Optymizacja drzew dokonywana jest a posteriori w celu prześledzenia ewolucji poszczególnych cech na gałęziach najbardziej parsymonicznego drzewa. ACCTRAN (Accelerated Transformation) przy jednakowo parsymonicznych rozwiązaniach faworyzowane są rewersje. Nowy stan cechy jest uruchamiany tak szybko, jak jest to możliwe (bliżej nasady drzewa). DELTRAN (Delayed Transformation) przy jednakowo parsymonicznych rozwiązaniach faworyzowane są paralelizmy. Nowy stan cechy jest uruchamiany tak późno, jak jest to możliwe (bliżej góry drzewa).
45 Śledzenie ewolucji cechy na drzewie rewersja paralelizm T A T A A T A T A A ACCTRAN DELTRAN A W obu przypadkach 2 kroki ewolucyjne A
46 Total evidence czy metoda konsensusowa? Total evidence jest definiowana jako równoczesna rekonstrukcja filogenetyczna wszystkich niepodzielonych danych (jedna matryca) dostępnych dla analizowanych taksonów. Metoda konsensusowa ( zgodność taksonomiczna ) poszukuje uzgodnienia hipotez uzyskanych z analizy różnych zbiorów danych.
47 Współbieżne zdarzenia ewolucyjne Wyróżnia się 4 główne typy równoległych zdarzeń ewolucyjnych zachodzących na różnych poziomach organizacji życia: Biogeografia Koewolucja Drzewa genowe Kodywergencja Wikariancja Kospecjacja Kodywergencja genów Duplikacja Specjacja sympatryczna Duplikacja linii Duplikacja genów Sortowanie Wymieranie Sortowanie linii Sortowanie genów Przeskok Dyspersja Zmiana żywiciela Transfer horyzontalny

48 Drzewa gatunkowe a kladogramy areałowe Pangea Laurasia Eurazja Ameryka Północna Australia Amphilina foliacea Amphilina japonica Gigantolina elongata Gigantolina magna Gondwana Indo-Malaje Afryka Ameryka Południowa Schiz. paragonophora Schizochoerus janickii Schizochoerus africanus Schizochoerus liguloideus


49 Drzewa genowe a drzewa gatunkowe
 spóźnienie na łódkę wymarcie zjawisko X duplikacja zjawiska")
50 Zjawiska kofilogenetyczne kospecjacja inercja (brak specjacji) spóźnienie na łódkę wymarcie zjawisko X duplikacja zjawiska sortowania
51 Zjawiska kofilogenetyczne po zmianie żywiciela (transfer horyzontalny) specjacja po zmianie żywiciela specjacja i wymieranie po zmianie żywiciela brak specjacji po zmianie żywiciela brak specjacji i wymieranie po zmianie żywiciela
52 Zgodne topologie drzew gospodarzy i pasożytów Gospodarze czas tempo Pasożyty tempo ewolucji i czas specjacji różne - pseudokospecjacja tempo ewolucji różne, relatywny czas specjacji identyczny tempo ewolucji i czas specjacji identyczne
53 Niezgodne topologie drzew gospodarzy i pasożytów Transfer horyzontalny G1 G2 P1 P2 czas G3 P3 Duplikacja i sortowanie G1 P1 Po duplikacji wymieranie: 2 w linii P3 1 w linii P1-P2 G2 G3 P2 P3 P1 P2 P3
54 Niezgodne topologie c.d. Transfer horyzontalny G1 G2 P1 P2 P2 młodszy niż gospodarz czas G3 P3 Sortowanie (wymarcie) G1 P1 Oszacowanie czasu kladogenezy P1-P2 może pomóc w decyzji między możliwościami G2 G3 P2 P3 P2 starszy niż gospodarz
55 Założenia metodyczne analizy kospecjacji 1. Precyzyjnie opracowana systematyka, zarówno gospodarzy jak i pasożytów (symbiontów, komensali) 2. Solidnie udokumentowane rekonstrukcje filogenii obu partnerów 3. Intensywne (kompletne) zebranie pasożytów (symbiontów, komensali) z opracowywanych kladów (linii ewolucyjnych) 4. Molekularne filogenie oparte na homologicznych markerach dla obu partnerów 5. Porównanie rekonstrukcji filogenii obu partnerów za pomocą metod ilościowych w celu wykrycia zjawisk kospecjacji i statystyczne przetestowanie prawdopodobieństwa nieprzypadkowości obserwowanego paralelizmu ewolucyjnego
 TreeMap mapowanie topologii drzew - przecenia sortowania i duplikacje - niedocenia kospecjacje tak tak (TreeMap")
56 Parsymonia Brooksa kontra TreeMap założenia wady testowanie statystyczne oprogramowanie Parsymonia Brooksa pasożyty zamienione na kod binarny - przecenia przeskoki pomiędzy gospodarzami nie tak (wstępna wersja PACT) TreeMap mapowanie topologii drzew - przecenia sortowania i duplikacje - niedocenia kospecjacje tak tak (TreeMap )
57 Wszoły (Mallophaga( Mallophaga; Phthiraptera) ) i goffery (Rodentia; Geomyidae) wg Page, 2001
.")
58 Przykład filogenia Avenzoariinae Avenzoariinae to podrodzina roztoczy, które są komensalami występującymi na piórach ptaków siewkowych (Charadriiformes). 0,5 mm
59 MORPHOLOGY SEQUENCES Total evidence COMBINED Freyana anatina Bdellorhynchus Freyana polymorphus anatina Bonnetella Pteronyssoides fusca striatus 10 Zachvatkinia Scutulanyssus larica obscurus 3 Bdellorhynchus polymorphus Bychovskiata subcharadrii 16 Bonnetella fusca Bychovskiata squatarolae 4 13 Zachvatkinia larica 1 Bychovskiata intermedia 5 Bychovskiata subcharadrii 5 5 Bychovskiata charadrii Ovofreyana kurbanovae Bychovskiata semipalmati 8 3 Bychovskiata squatarolae Bychovskiata Bychovskiata hypoleuci intermedia Bychovskiata Bychovskiata dubia 14 charadrii Ovofreyana Bychovskiata kurbanovae semipalmati Pseudavenzoaria Bychovskiata indica hypoleuci 4 Bychovskiata dubia 4 Pseudavenzoaria ochropodis 3 20 Pomeranzevia ninnii Bregetovia mucronata Pseudavenzoaria 2indica 15 Bregetovia limosae Pseudavenzoaria 4 ochropodis Bregetovia obtusolobata 20 Bregetovia mucronata 2 5 Avenzoaria totani Bregetovia limosae Avenzoaria 27 Bregetovia totani 2-1 obtusolobata 4 Avenzoaria Avenzoaria tringae terekiae 3 Avenzoaria 1 Avenzoaria phalaropi calidridis Avenzoaria Avenzoaria calidridis 1-1 calidridis Avenzoaria philomachi Avenzoaria calidridis Avenzoaria phalaropi 3 1 Avenzoaria philomachi 1 Avenzoaria tringae Avenzoaria terekiae 3 Avenzoaria totani Pomeranzevia 4 ninnii Avenzoaria totani 2-1 Pteronyssoides striatus 20 Scutulanyssus obscurus 4 Filogenia Avenzoariinae (c.d.) Drzewo morfologiczne jest stabilne w bazalnych partiach i słabo potwierdzone na szczycie. Drzewo molekularne ma słabe potwierdzenie u nasady, lecz ma bardzo dobrą rozdzielczość w szczytowych partiach. Drzewo kombinowane ma zalety obu poprzednich.
60 Drzewa uzgodnione (reconciled( trees) Avenzoariinae i ptaków siewkowych (Charadriiformes) Liczba drzew kospecjacja duplikacja zmiana żywiciela sortowanie Liczba kos pe cjacji 2: Avenzoariinae ptaki siewkowe P - Numenius arquata M - Pomeranzevia ninnii P Limosa limosa M Bregetovia limosae P Phalaropus lobatus M Avenzoaria phalaropi P Xenus cinereus M Avenzoaria terekiae P Philomachus pugnax M Avenzoaria philomachi P Calidris alpina M - Avevzoaria calidridis 2 P - Calidris temminckii M - Avenzoaria calidridis 1-1 P Actitis hypoleucos M Bychovskiata charadrii P - Tringa glareola M - Avenzoaria totani 1-1 P - Tringa totanus M - Avenzoaria totani 2-1 P - Tringa nebularia M - Avenzoaria tringae M - Bregetovia obtusolobata P - Tringa erythropus M - Bregetovia mucronata P - Tringa solitaria M - Pseudavenzoaria indica P - Tringa ochropus M - Pseudavenzoaria ochropodis P - Himantopus himantopus M Bychovskiata subcharadrii P - Pluvialis squatarola M Bychovskiata squatarolae P - Vanellus leucurus M - Ovofreyana kurbanovae P - Charadrius leschenaulti M - Bychovskiata intermedia P - Charadrius dubius M - Bychovskiata dubia P - Charadrius hiaticula M - Bychovskiata charadrii P - Charadrius semipalmatus M - Bychovskiata semipalmati


61 Problematyczna filogenia mew co na to Thecarthra?

62 227 Oprogramowanie filogenetyczne
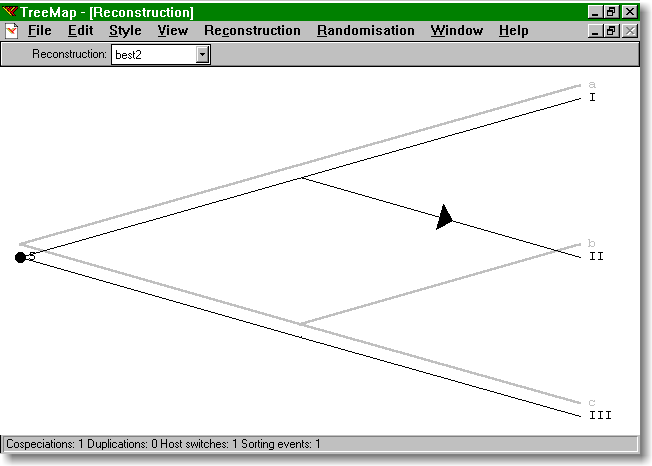

63 Niezbędnik Mały filogenetyk
64 PAUP Maksymalna Parsymonia Maximul Likelihood Neighbor-joining

65 Neighbor-joining MEGA

66 Maximum Likelihood PHYML


67 Wnioskowanie Bayesowskie MrBayes
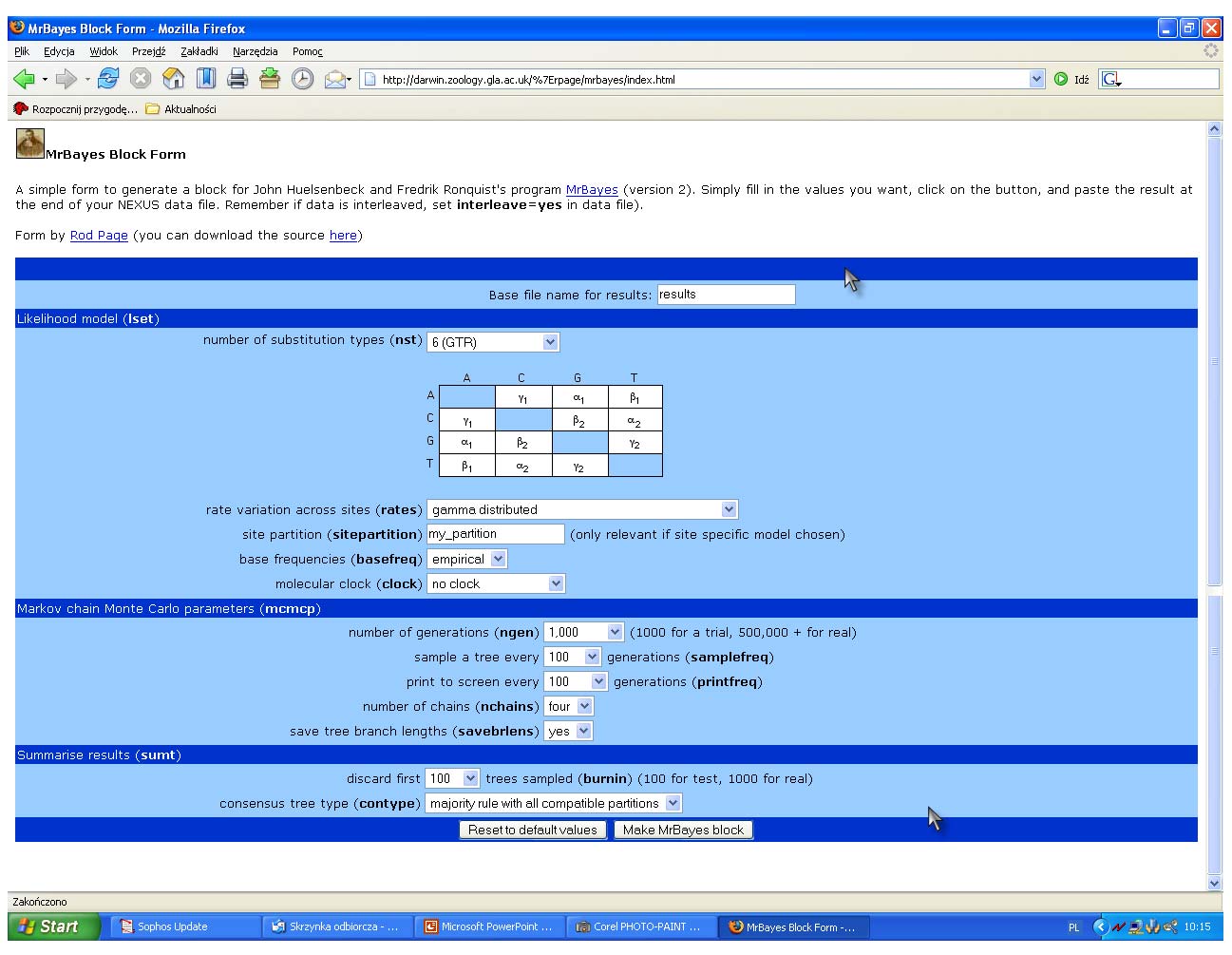
68 MrBayes

69 Testowanie zegara molekularnego + -

70 Kalkulacja modeli substytucji


71 Graficzna prezentacja wyników TreeView Macclade WinClada Mesquite
72 Programy filogenetyczne EXECUTOR (emulator Maca): PAUP: PHYLIP: MEGA: MrBAYES: PHYML: MODELTEST: MrMODELTEST: AUTODECAY: TIMER: r8s: TREEVIEW, NDE, TREEMAP, COMPONENT, GENETREE: MACCLADE: w/w i inne programy:
Genomika Porównawcza. Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski
 Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Parazytologia W6. Dabert Wstęp do parazytologii ewolucyjnej Teoria analizy ko filogenetycznej
 Dabert Wstęp do parazytologii ewolucyjnej Teoria analizy ko filogenetycznej Parazytologia W6 Rodzaje interakcji Z punktu widzenia osobników dowolnego gatunku większość innych gatunków, z którymi osobniki
Dabert Wstęp do parazytologii ewolucyjnej Teoria analizy ko filogenetycznej Parazytologia W6 Rodzaje interakcji Z punktu widzenia osobników dowolnego gatunku większość innych gatunków, z którymi osobniki
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
Filogenetyka molekularna. Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji
 Filogenetyka molekularna Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji Co to jest filogeneza? Filogeneza=drzewo filogenetyczne=drzewo rodowe=drzewo to rozgałęziający się diagram, który
Filogenetyka molekularna Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji Co to jest filogeneza? Filogeneza=drzewo filogenetyczne=drzewo rodowe=drzewo to rozgałęziający się diagram, który
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
klasyfikacja fenetyczna (numeryczna)
") Teorie klasyfikacji klasyfikacja fenetyczna (numeryczna) systematyka powinna być wolna od wszelkiej teorii (a zwłaszcza od teorii ewolucji) filogeneza jako ciąg zdarzeń jest niepoznawalna opiera się na
Teorie klasyfikacji klasyfikacja fenetyczna (numeryczna) systematyka powinna być wolna od wszelkiej teorii (a zwłaszcza od teorii ewolucji) filogeneza jako ciąg zdarzeń jest niepoznawalna opiera się na
Filogenetyka molekularna I. Krzysztof Spalik
 Filogenetyka molekularna I Krzysztof Spalik Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w badaniach ewolucyjnych, Kosmos 58(3-4): 485-498
Filogenetyka molekularna I Krzysztof Spalik Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w badaniach ewolucyjnych, Kosmos 58(3-4): 485-498
Analizy filogenetyczne
 BIOINFORMATYKA edycja 2016 / 2017 wykład 6 Analizy filogenetyczne dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Cele i zastosowania 2. Podstawy ewolucyjne
BIOINFORMATYKA edycja 2016 / 2017 wykład 6 Analizy filogenetyczne dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Cele i zastosowania 2. Podstawy ewolucyjne
Acknowledgement. Drzewa filogenetyczne
 Wykład 8 Drzewa Filogenetyczne Lokalizacja genów Some figures from: Acknowledgement M. Zvelebil, J.O. Baum, Introduction to Bioinformatics, Garland Science 2008 Tradycyjne drzewa pokrewieństwa Drzewa oparte
Wykład 8 Drzewa Filogenetyczne Lokalizacja genów Some figures from: Acknowledgement M. Zvelebil, J.O. Baum, Introduction to Bioinformatics, Garland Science 2008 Tradycyjne drzewa pokrewieństwa Drzewa oparte
Filogenetyka molekularna I. Krzysztof Spalik Zakład Filogenetyki Molekularnej i Ewolucji
 Filogenetyka molekularna I Krzysztof Spalik Zakład Filogenetyki Molekularnej i Ewolucji 3 Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w
Filogenetyka molekularna I Krzysztof Spalik Zakład Filogenetyki Molekularnej i Ewolucji 3 Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w
Filogenetyka molekularna I
 2 Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w badaniach ewolucyjnych, Kosmos 58(3-4): 485-498 Filogenetyka molekularna I John C. Avise
2 Literatura Krzysztof Spalik, Marcin Piwczyński (2009), Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w badaniach ewolucyjnych, Kosmos 58(3-4): 485-498 Filogenetyka molekularna I John C. Avise
plezjomorfie: podobieństwa dziedziczone po dalszych przodkach (c. atawistyczna)
") Podobieństwa pomiędzy organizmami - cechy homologiczne: podobieństwa wynikające z dziedziczenia - apomorfie: podobieństwa dziedziczone po najbliższym przodku lub pojawiająca się de novo (c. ewolucyjnie
Podobieństwa pomiędzy organizmami - cechy homologiczne: podobieństwa wynikające z dziedziczenia - apomorfie: podobieństwa dziedziczone po najbliższym przodku lub pojawiająca się de novo (c. ewolucyjnie
Recenzja rozprawy doktorskiej. mgr Marcina Jana Kamińskiego. pt. Grupa rodzajowa Ectateus (Coleoptera: Tenebrionidae) filogeneza i klasyfikacja.
 filogeneza i klasyfikacja.") Dr hab. Grzegorz Paśnik Instytut Systematyki i Ewolucji Zwierząt Polska Akademia Nauk ul. Sławkowska 17, 31-016 Kraków Recenzja rozprawy doktorskiej mgr Marcina Jana Kamińskiego pt. Grupa rodzajowa Ectateus
Dr hab. Grzegorz Paśnik Instytut Systematyki i Ewolucji Zwierząt Polska Akademia Nauk ul. Sławkowska 17, 31-016 Kraków Recenzja rozprawy doktorskiej mgr Marcina Jana Kamińskiego pt. Grupa rodzajowa Ectateus
Ograniczenia środowiskowe nie budzą wielu kontrowersji, co nie znaczy że rozumiemy do końca proces powstawania adaptacji fizjologicznych.
 1 Ograniczenia środowiskowe nie budzą wielu kontrowersji, co nie znaczy że rozumiemy do końca proces powstawania adaptacji fizjologicznych. Wiadomo, że ściśle powiązane z zagadnieniem interakcji kompetencje
1 Ograniczenia środowiskowe nie budzą wielu kontrowersji, co nie znaczy że rozumiemy do końca proces powstawania adaptacji fizjologicznych. Wiadomo, że ściśle powiązane z zagadnieniem interakcji kompetencje
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Metoda NJ (przyłączania sąsiadów) umożliwia tworzenie drzewa addytywnego: odległości ewolucyjne między sekwencjami
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Metoda NJ (przyłączania sąsiadów) umożliwia tworzenie drzewa addytywnego: odległości ewolucyjne między sekwencjami
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Bioinformatyka Laboratorium, 30h. Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Wykład Bioinformatyka 2012-09-24. Bioinformatyka. Wykład 7. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM. Ewolucyjne podstawy Bioinformatyki
 Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE
 D: PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE I. Strategia zachłanna II. Problem przetasowań w genomie III. Sortowanie przez odwrócenia IV. Algorytmy przybliżone V. Algorytm zachłanny
D: PROBLEM: SORTOWANIE PRZEZ ODWRÓCENIA METODA: ALGORYTMY ZACHŁANNE I. Strategia zachłanna II. Problem przetasowań w genomie III. Sortowanie przez odwrócenia IV. Algorytmy przybliżone V. Algorytm zachłanny
Wszystkie wyniki w postaci ułamków należy podawać z dokładnością do czterech miejsc po przecinku!
 Pracownia statystyczno-filogenetyczna Liczba punktów (wypełnia KGOB) / 30 PESEL Imię i nazwisko Grupa Nr Czas: 90 min. Łączna liczba punktów do zdobycia: 30 Czerwona Niebieska Zielona Żółta Zaznacz znakiem
Pracownia statystyczno-filogenetyczna Liczba punktów (wypełnia KGOB) / 30 PESEL Imię i nazwisko Grupa Nr Czas: 90 min. Łączna liczba punktów do zdobycia: 30 Czerwona Niebieska Zielona Żółta Zaznacz znakiem
Aproksymacja funkcji a regresja symboliczna
 Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(x), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(x), zwaną funkcją aproksymującą
Drzewa filogenetyczne jako matematyczny model relacji pokrewieństwa. dr inż. Damian Bogdanowicz
 Drzewa filogenetyczne jako matematyczny model relacji pokrewieństwa dr inż. Damian Bogdanowicz Sprawa R. Schmidt a z Lafayette Podczas rutynowych badań u pielęgniarki Janet Allen stwierdzono obecność wirusa
Drzewa filogenetyczne jako matematyczny model relacji pokrewieństwa dr inż. Damian Bogdanowicz Sprawa R. Schmidt a z Lafayette Podczas rutynowych badań u pielęgniarki Janet Allen stwierdzono obecność wirusa
Mikroekonometria 5. Mikołaj Czajkowski Wiktor Budziński
 Mikroekonometria 5 Mikołaj Czajkowski Wiktor Budziński Zadanie 1. Wykorzystując dane me.medexp3.dta przygotuj model regresji kwantylowej 1. Przygotuj model regresji kwantylowej w którym logarytm wydatków
Mikroekonometria 5 Mikołaj Czajkowski Wiktor Budziński Zadanie 1. Wykorzystując dane me.medexp3.dta przygotuj model regresji kwantylowej 1. Przygotuj model regresji kwantylowej w którym logarytm wydatków
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Ewolucjonizm NEODARWINIZM. Dr Jacek Francikowski Uniwersyteckie Towarzystwo Naukowe Uniwersytet Śląski w Katowicach
 Ewolucjonizm NEODARWINIZM Dr Jacek Francikowski Uniwersyteckie Towarzystwo Naukowe Uniwersytet Śląski w Katowicach Główne paradygmaty biologii Wspólne początki życia Komórka jako podstawowo jednostka funkcjonalna
Ewolucjonizm NEODARWINIZM Dr Jacek Francikowski Uniwersyteckie Towarzystwo Naukowe Uniwersytet Śląski w Katowicach Główne paradygmaty biologii Wspólne początki życia Komórka jako podstawowo jednostka funkcjonalna
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Konstrukcja drzew filogenetycznych podstawy teoretyczne.
 Dorota Rogalla Urszula Rogalla Konstrukcja drzew filogenetycznych podstawy teoretyczne. Streszczenie Wśród wyzwań stojących przed genetyką wymienić można nie tylko kojarzone klasycznie poznawanie struktury
Dorota Rogalla Urszula Rogalla Konstrukcja drzew filogenetycznych podstawy teoretyczne. Streszczenie Wśród wyzwań stojących przed genetyką wymienić można nie tylko kojarzone klasycznie poznawanie struktury
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Mikroekonometria 9. Mikołaj Czajkowski Wiktor Budziński
 Mikroekonometria 9 Mikołaj Czajkowski Wiktor Budziński Wielomianowy model logitowy Uogólnienie modelu binarnego Wybór pomiędzy 2 lub większą liczbą alternatyw Np. wybór środka transportu, głos w wyborach,
Mikroekonometria 9 Mikołaj Czajkowski Wiktor Budziński Wielomianowy model logitowy Uogólnienie modelu binarnego Wybór pomiędzy 2 lub większą liczbą alternatyw Np. wybór środka transportu, głos w wyborach,
Metoda dokładnej rekonstrukcji drzew filogenetycznych genów. współczynników substytucji dla genów i gatunków
 Metoda dokładnej rekonstrukcji drzew filogenetycznych genów z zastosowaniem współczynników substytucji dla genów i gatunków Na podstawie artykułu Accurate gene-tree reconstruction by learning gene- and
Metoda dokładnej rekonstrukcji drzew filogenetycznych genów z zastosowaniem współczynników substytucji dla genów i gatunków Na podstawie artykułu Accurate gene-tree reconstruction by learning gene- and
Teoria ewolucji. Podstawowe pojęcia. Wspólne pochodzenie.
 Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Ewolucja Znaczenie ogólne: zmiany zachodzące stopniowo w czasie W biologii ewolucja biologiczna W astronomii i kosmologii ewolucja gwiazd i wszechświata
Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Ewolucja Znaczenie ogólne: zmiany zachodzące stopniowo w czasie W biologii ewolucja biologiczna W astronomii i kosmologii ewolucja gwiazd i wszechświata
w analizie wyników badań eksperymentalnych, w problemach modelowania zjawisk fizycznych, w analizie obserwacji statystycznych.
 Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(), zwaną funkcją aproksymującą
Aproksymacja funkcji a regresja symboliczna Problem aproksymacji funkcji polega na tym, że funkcję F(), znaną lub określoną tablicą wartości, należy zastąpić inną funkcją, f(), zwaną funkcją aproksymującą
Urszula Poziomek, doradca metodyczny w zakresie biologii Materiał dydaktyczny przygotowany na konferencję z cyklu Na miarę Nobla, 14 stycznia 2010 r.
 Ćwiczenie 1 1 Wstęp Rozważając możliwe powiązania filogenetyczne gatunków, systematyka porównuje dane molekularne. Najskuteczniejszym sposobem badania i weryfikacji różnych hipotez filogenetycznych jest
Ćwiczenie 1 1 Wstęp Rozważając możliwe powiązania filogenetyczne gatunków, systematyka porównuje dane molekularne. Najskuteczniejszym sposobem badania i weryfikacji różnych hipotez filogenetycznych jest
Rycina 1. Zasięg i zagęszczenie łosi (liczba osobników/1000 ha) w Polsce w roku 2010 oraz rozmieszczenie 29 analizowanych populacji łosi.
 w Polsce w roku 2010 oraz rozmieszczenie 29 analizowanych populacji łosi.") Ryciny 193 Rycina 1. Zasięg i zagęszczenie łosi (liczba osobników/1000 ha) w Polsce w roku 2010 oraz rozmieszczenie 29 analizowanych populacji łosi. Na fioletowo zaznaczone zostały populacje (nr 1 14)
Ryciny 193 Rycina 1. Zasięg i zagęszczenie łosi (liczba osobników/1000 ha) w Polsce w roku 2010 oraz rozmieszczenie 29 analizowanych populacji łosi. Na fioletowo zaznaczone zostały populacje (nr 1 14)
Teoria ewolucji. Podstawowe pojęcia. Wspólne pochodzenie.
 Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego 5A pgolik@igib.uw.edu.pl Informacje, materiały: http://www.igib.uw.edu.pl/
Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego 5A pgolik@igib.uw.edu.pl Informacje, materiały: http://www.igib.uw.edu.pl/
46 Olimpiada Biologiczna
 46 Olimpiada Biologiczna Pracownia statystyczno-filogenetyczna Łukasz Banasiak i Jakub Baczyński 22 kwietnia 2017 r. Statystyka i filogenetyka / 30 Liczba punktów (wypełnia KGOB) PESEL Imię i nazwisko
46 Olimpiada Biologiczna Pracownia statystyczno-filogenetyczna Łukasz Banasiak i Jakub Baczyński 22 kwietnia 2017 r. Statystyka i filogenetyka / 30 Liczba punktów (wypełnia KGOB) PESEL Imię i nazwisko
STATYSTYKA MATEMATYCZNA
 STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
STATYSTYKA MATEMATYCZNA 1. Wykład wstępny. Teoria prawdopodobieństwa i elementy kombinatoryki 2. Zmienne losowe i ich rozkłady 3. Populacje i próby danych, estymacja parametrów 4. Testowanie hipotez 5.
< K (2) = ( Adams, John ), P (2) = adres bloku 2 > < K (1) = ( Aaron, Ed ), P (1) = adres bloku 1 >
 = ( Adams, John ), P (2) = adres bloku 2 > < K (1) = ( Aaron, Ed ), P (1) = adres bloku 1 >") Typy indeksów Indeks jest zakładany na atrybucie relacji atrybucie indeksowym (ang. indexing field). Indeks zawiera wartości atrybutu indeksowego wraz ze wskaźnikami do wszystkich bloków dyskowych zawierających
Typy indeksów Indeks jest zakładany na atrybucie relacji atrybucie indeksowym (ang. indexing field). Indeks zawiera wartości atrybutu indeksowego wraz ze wskaźnikami do wszystkich bloków dyskowych zawierających
STATYSTYKA I DOŚWIADCZALNICTWO Wykład 8
 STATYSTYKA I DOŚWIADCZALNICTWO Wykład 8 Regresja wielokrotna Regresja wielokrotna jest metodą statystyczną, w której oceniamy wpływ wielu zmiennych niezależnych (X 1, X 2, X 3,...) na zmienną zależną (Y).
STATYSTYKA I DOŚWIADCZALNICTWO Wykład 8 Regresja wielokrotna Regresja wielokrotna jest metodą statystyczną, w której oceniamy wpływ wielu zmiennych niezależnych (X 1, X 2, X 3,...) na zmienną zależną (Y).
Wyróżniamy dwa typy zadań projektowych.
 Obowiązkowymi do zaliczenia projektu jest realizacja 2-3 zadań programistycznych. Zadania realizowane są w grupach 2-3 osobowych (zależnie od stopnia trudności zadania i liczebności całej klasy laboratoryjnej).
Obowiązkowymi do zaliczenia projektu jest realizacja 2-3 zadań programistycznych. Zadania realizowane są w grupach 2-3 osobowych (zależnie od stopnia trudności zadania i liczebności całej klasy laboratoryjnej).
CLUSTERING. Metody grupowania danych
 CLUSTERING Metody grupowania danych Plan wykładu Wprowadzenie Dziedziny zastosowania Co to jest problem klastrowania? Problem wyszukiwania optymalnych klastrów Metody generowania: k centroidów (k - means
CLUSTERING Metody grupowania danych Plan wykładu Wprowadzenie Dziedziny zastosowania Co to jest problem klastrowania? Problem wyszukiwania optymalnych klastrów Metody generowania: k centroidów (k - means
Hierarchiczna analiza skupień
 Hierarchiczna analiza skupień Cel analizy Analiza skupień ma na celu wykrycie w zbiorze obserwacji klastrów, czyli rozłącznych podzbiorów obserwacji, wewnątrz których obserwacje są sobie w jakimś określonym
Hierarchiczna analiza skupień Cel analizy Analiza skupień ma na celu wykrycie w zbiorze obserwacji klastrów, czyli rozłącznych podzbiorów obserwacji, wewnątrz których obserwacje są sobie w jakimś określonym
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Transformacja wiedzy w budowie i eksploatacji maszyn
 Uniwersytet Technologiczno Przyrodniczy im. Jana i Jędrzeja Śniadeckich w Bydgoszczy Wydział Mechaniczny Transformacja wiedzy w budowie i eksploatacji maszyn Bogdan ŻÓŁTOWSKI W pracy przedstawiono proces
Uniwersytet Technologiczno Przyrodniczy im. Jana i Jędrzeja Śniadeckich w Bydgoszczy Wydział Mechaniczny Transformacja wiedzy w budowie i eksploatacji maszyn Bogdan ŻÓŁTOWSKI W pracy przedstawiono proces
Teoria ewolucji. Podstawy wspólne pochodzenie.
 Teoria ewolucji. Podstawy wspólne pochodzenie. Ewolucja biologiczna } Znaczenie ogólne: } proces zmian informacji genetycznej (częstości i rodzaju alleli), } które to zmiany są przekazywane z pokolenia
Teoria ewolucji. Podstawy wspólne pochodzenie. Ewolucja biologiczna } Znaczenie ogólne: } proces zmian informacji genetycznej (częstości i rodzaju alleli), } które to zmiany są przekazywane z pokolenia
Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej
 Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej Specjacja } Pojawienie się bariery reprodukcyjnej między populacjami dające początek gatunkom } Specjacja allopatryczna
Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej Specjacja } Pojawienie się bariery reprodukcyjnej między populacjami dające początek gatunkom } Specjacja allopatryczna
Kierunek i poziom studiów: Biologia, poziom drugi Sylabus modułu: Filogenetyka i taksonomia roślin i zwierząt dla EKOP
 Uniwersytet Śląski w Katowicach str. 1 Kierunek i poziom studiów: Biologia, poziom drugi Sylabus modułu: Filogenetyka i taksonomia roślin i zwierząt dla EKOP kod modułu: 2BL_12 1. Informacje ogólne koordynator
Uniwersytet Śląski w Katowicach str. 1 Kierunek i poziom studiów: Biologia, poziom drugi Sylabus modułu: Filogenetyka i taksonomia roślin i zwierząt dla EKOP kod modułu: 2BL_12 1. Informacje ogólne koordynator
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
Wykład 5: Statystyki opisowe (część 2)
") Wykład 5: Statystyki opisowe (część 2) Wprowadzenie Na poprzednim wykładzie wprowadzone zostały statystyki opisowe nazywane miarami położenia (średnia, mediana, kwartyle, minimum i maksimum, modalna oraz
Wykład 5: Statystyki opisowe (część 2) Wprowadzenie Na poprzednim wykładzie wprowadzone zostały statystyki opisowe nazywane miarami położenia (średnia, mediana, kwartyle, minimum i maksimum, modalna oraz
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Eksploracja Danych. wykład 4. Sebastian Zając. 10 maja 2017 WMP.SNŚ UKSW. Sebastian Zając (WMP.SNŚ UKSW) Eksploracja Danych 10 maja / 18
 Eksploracja Danych 10 maja / 18") Eksploracja Danych wykład 4 Sebastian Zając WMP.SNŚ UKSW 10 maja 2017 Sebastian Zając (WMP.SNŚ UKSW) Eksploracja Danych 10 maja 2017 1 / 18 Klasyfikacja danych Klasyfikacja Najczęściej stosowana (najstarsza)
Eksploracja Danych wykład 4 Sebastian Zając WMP.SNŚ UKSW 10 maja 2017 Sebastian Zając (WMP.SNŚ UKSW) Eksploracja Danych 10 maja 2017 1 / 18 Klasyfikacja danych Klasyfikacja Najczęściej stosowana (najstarsza)
Algorytmy genetyczne w interpolacji wielomianowej
 Algorytmy genetyczne w interpolacji wielomianowej (seminarium robocze) Seminarium Metod Inteligencji Obliczeniowej Warszawa 22 II 2006 mgr inż. Marcin Borkowski Plan: Przypomnienie algorytmu niszowego
Algorytmy genetyczne w interpolacji wielomianowej (seminarium robocze) Seminarium Metod Inteligencji Obliczeniowej Warszawa 22 II 2006 mgr inż. Marcin Borkowski Plan: Przypomnienie algorytmu niszowego
Analiza składowych głównych. Wprowadzenie
 Wprowadzenie jest techniką redukcji wymiaru. Składowe główne zostały po raz pierwszy zaproponowane przez Pearsona(1901), a następnie rozwinięte przez Hotellinga (1933). jest zaliczana do systemów uczących
Wprowadzenie jest techniką redukcji wymiaru. Składowe główne zostały po raz pierwszy zaproponowane przez Pearsona(1901), a następnie rozwinięte przez Hotellinga (1933). jest zaliczana do systemów uczących
Mikroekonometria 6. Mikołaj Czajkowski Wiktor Budziński
 Mikroekonometria 6 Mikołaj Czajkowski Wiktor Budziński Metody symulacyjne Monte Carlo Metoda Monte-Carlo Wykorzystanie mocy obliczeniowej komputerów, aby poznać charakterystyki zmiennych losowych poprzez
Mikroekonometria 6 Mikołaj Czajkowski Wiktor Budziński Metody symulacyjne Monte Carlo Metoda Monte-Carlo Wykorzystanie mocy obliczeniowej komputerów, aby poznać charakterystyki zmiennych losowych poprzez
Data Mining Wykład 9. Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster. Plan wykładu. Sformułowanie problemu
 Grupowanie hierarchiczne O-Cluster. Plan wykładu. Sformułowanie problemu") Data Mining Wykład 9 Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster Plan wykładu Wprowadzanie Definicja problemu Klasyfikacja metod grupowania Grupowanie hierarchiczne Sformułowanie problemu
Data Mining Wykład 9 Analiza skupień (grupowanie) Grupowanie hierarchiczne O-Cluster Plan wykładu Wprowadzanie Definicja problemu Klasyfikacja metod grupowania Grupowanie hierarchiczne Sformułowanie problemu
znalezienia elementu w zbiorze, gdy w nim jest; dołączenia nowego elementu w odpowiednie miejsce, aby zbiór pozostał nadal uporządkowany.
 Przedstawiamy algorytmy porządkowania dowolnej liczby elementów, którymi mogą być liczby, jak również elementy o bardziej złożonej postaci (takie jak słowa i daty). Porządkowanie, nazywane również często
Przedstawiamy algorytmy porządkowania dowolnej liczby elementów, którymi mogą być liczby, jak również elementy o bardziej złożonej postaci (takie jak słowa i daty). Porządkowanie, nazywane również często
Statystyczne sterowanie procesem
 Statystyczne sterowanie procesem SPC (ang. Statistical Process Control) Trzy filary SPC: 1. sporządzenie dokładnego diagramu procesu produkcji; 2. pobieranie losowych próbek (w regularnych odstępach czasu
Statystyczne sterowanie procesem SPC (ang. Statistical Process Control) Trzy filary SPC: 1. sporządzenie dokładnego diagramu procesu produkcji; 2. pobieranie losowych próbek (w regularnych odstępach czasu
Testowanie hipotez statystycznych
 9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
9 października 2008 ...czyli definicje na rozgrzewkę n-elementowa próba losowa - wektor n zmiennych losowych (X 1,..., X n ); intuicyjnie: wynik n eksperymentów realizacja próby (X 1,..., X n ) w ω Ω :
Spis treści. Przedmowa... XI. Rozdział 1. Pomiar: jednostki miar... 1. Rozdział 2. Pomiar: liczby i obliczenia liczbowe... 16
 Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
Spis treści Przedmowa.......................... XI Rozdział 1. Pomiar: jednostki miar................. 1 1.1. Wielkości fizyczne i pozafizyczne.................. 1 1.2. Spójne układy miar. Układ SI i jego
Budowanie drzewa filogenetycznego
 Szkoła Festiwalu Nauki 134567 Wojciech Grajkowski Szkoła Festiwalu Nauki, ul. Ks. Trojdena 4, 02-109 Warszawa www.sfn.edu.pl sfn@iimcb.gov.pl Budowanie drzewa filogenetycznego Cel Ćwiczenie polega na budowaniu
Szkoła Festiwalu Nauki 134567 Wojciech Grajkowski Szkoła Festiwalu Nauki, ul. Ks. Trojdena 4, 02-109 Warszawa www.sfn.edu.pl sfn@iimcb.gov.pl Budowanie drzewa filogenetycznego Cel Ćwiczenie polega na budowaniu
Biologia medyczna, materiały dla studentów
 Jaka tam ewolucja. Zanim trafię na jednego myślącego, muszę stoczyć bitwę zdziewięcioma orangutanami Carlos Ruis Zafon Wierzbownica drobnokwiatowa Fitosterole, garbniki, flawonoidy Właściwości przeciwzapalne,
Jaka tam ewolucja. Zanim trafię na jednego myślącego, muszę stoczyć bitwę zdziewięcioma orangutanami Carlos Ruis Zafon Wierzbownica drobnokwiatowa Fitosterole, garbniki, flawonoidy Właściwości przeciwzapalne,
Klasyfikator. ˆp(k x) = 1 K. I(ρ(x,x i ) ρ(x,x (K) ))I(y i =k),k =1,...,L,
 = 1 K. I(ρ(x,x i ) ρ(x,x (K) ))I(y i =k),k =1,...,L,") Klasyfikator Jedną z najistotniejszych nieparametrycznych metod klasyfikacji jest metoda K-najbliższych sąsiadów, oznaczana przez K-NN. W metodzie tej zaliczamy rozpoznawany obiekt do tej klasy, do której
Klasyfikator Jedną z najistotniejszych nieparametrycznych metod klasyfikacji jest metoda K-najbliższych sąsiadów, oznaczana przez K-NN. W metodzie tej zaliczamy rozpoznawany obiekt do tej klasy, do której
1. Symulacje komputerowe Idea symulacji Przykład. 2. Metody próbkowania Jackknife Bootstrap. 3. Łańcuchy Markova. 4. Próbkowanie Gibbsa
 BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
BIOINFORMATYKA 1. Wykład wstępny 2. Bazy danych: projektowanie i struktura 3. Równowaga Hardyego-Weinberga, wsp. rekombinacji 4. Analiza asocjacyjna 5. Analiza asocjacyjna 6. Sekwencjonowanie nowej generacji
Charakterystyki liczbowe (estymatory i parametry), które pozwalają opisać właściwości rozkładu badanej cechy (zmiennej)
, które pozwalają opisać właściwości rozkładu badanej cechy (zmiennej)") Charakterystyki liczbowe (estymatory i parametry), które pozwalają opisać właściwości rozkładu badanej cechy (zmiennej) 1 Podział ze względu na zakres danych użytych do wyznaczenia miary Miary opisujące
Charakterystyki liczbowe (estymatory i parametry), które pozwalają opisać właściwości rozkładu badanej cechy (zmiennej) 1 Podział ze względu na zakres danych użytych do wyznaczenia miary Miary opisujące
ALGORYTMY KONSTRUOWANIA DENDROGRAMÓW STOSOWANYCH PRZY ANALIZIE FILOGENETYCZNEJ MIKROORGANIZMÓW
 ALGORYTMY KONSTRUOWANIA DENDROGRAMÓW STOSOWANYCH PRZY ANALIZIE FILOGENETYCZNEJ MIKROORGANIZMÓW Filip Zdziennicki, Anna Misiewicz Instytut Biotechnologii Przemysłu Rolno-Spożywczego im. prof. Wacława Dąbrowskiego
ALGORYTMY KONSTRUOWANIA DENDROGRAMÓW STOSOWANYCH PRZY ANALIZIE FILOGENETYCZNEJ MIKROORGANIZMÓW Filip Zdziennicki, Anna Misiewicz Instytut Biotechnologii Przemysłu Rolno-Spożywczego im. prof. Wacława Dąbrowskiego
Filogenetyka. Dr Marek D. Koter, dr hab. Marcin Filipecki. Katedra Genetyki, Hodowli i Biotechnologii Roślin, SGGW
 Filogenetyka Dr Marek D. Koter, dr hab. Marcin Filipecki Katedra Genetyki, Hodowli i Biotechnologii Roślin, SGGW 1 Twórcy teorii ewolucji Charles Darwin Jean Baptiste de Lamarck Podróż HMS Beagle 2 i zbrodniczy
Filogenetyka Dr Marek D. Koter, dr hab. Marcin Filipecki Katedra Genetyki, Hodowli i Biotechnologii Roślin, SGGW 1 Twórcy teorii ewolucji Charles Darwin Jean Baptiste de Lamarck Podróż HMS Beagle 2 i zbrodniczy
Klasyfikatory: k-nn oraz naiwny Bayesa. Agnieszka Nowak Brzezińska Wykład IV
 Klasyfikatory: k-nn oraz naiwny Bayesa Agnieszka Nowak Brzezińska Wykład IV Naiwny klasyfikator Bayesa Naiwny klasyfikator bayesowski jest prostym probabilistycznym klasyfikatorem. Zakłada się wzajemną
Klasyfikatory: k-nn oraz naiwny Bayesa Agnieszka Nowak Brzezińska Wykład IV Naiwny klasyfikator Bayesa Naiwny klasyfikator bayesowski jest prostym probabilistycznym klasyfikatorem. Zakłada się wzajemną
Algorytmy decyzyjne będące alternatywą dla sieci neuronowych
 Algorytmy decyzyjne będące alternatywą dla sieci neuronowych Piotr Dalka Przykładowe algorytmy decyzyjne Sztuczne sieci neuronowe Algorytm k najbliższych sąsiadów Kaskada klasyfikatorów AdaBoost Naiwny
Algorytmy decyzyjne będące alternatywą dla sieci neuronowych Piotr Dalka Przykładowe algorytmy decyzyjne Sztuczne sieci neuronowe Algorytm k najbliższych sąsiadów Kaskada klasyfikatorów AdaBoost Naiwny
FILOGENETYKA. Bioinformatyka, wykład. 8 c.d. 0)
") FILOGENETYKA Bioinformatyka, wykład 8 c.d. (7.XII.2010) 0) krzysztof_pawlowski@sggw.pl Filogenetyka Cel rekonstrukcja historii ewolucji wszystkich organizmów. Klasyczne podejście: historia ewolucji jest
FILOGENETYKA Bioinformatyka, wykład 8 c.d. (7.XII.2010) 0) krzysztof_pawlowski@sggw.pl Filogenetyka Cel rekonstrukcja historii ewolucji wszystkich organizmów. Klasyczne podejście: historia ewolucji jest
Algorytmy genetyczne
 Algorytmy genetyczne Motto: Zamiast pracowicie poszukiwać najlepszego rozwiązania problemu informatycznego lepiej pozwolić, żeby komputer sam sobie to rozwiązanie wyhodował! Algorytmy genetyczne służą
Algorytmy genetyczne Motto: Zamiast pracowicie poszukiwać najlepszego rozwiązania problemu informatycznego lepiej pozwolić, żeby komputer sam sobie to rozwiązanie wyhodował! Algorytmy genetyczne służą
Ekologia wyk. 1. wiedza z zakresu zarówno matematyki, biologii, fizyki, chemii, rozumienia modeli matematycznych
 Ekologia wyk. 1 wiedza z zakresu zarówno matematyki, biologii, fizyki, chemii, rozumienia modeli matematycznych Ochrona środowiska Ekologia jako dziedzina nauki jest nauką o zależnościach decydujących
Ekologia wyk. 1 wiedza z zakresu zarówno matematyki, biologii, fizyki, chemii, rozumienia modeli matematycznych Ochrona środowiska Ekologia jako dziedzina nauki jest nauką o zależnościach decydujących
BIOINFORMATYKA. Copyright 2011, Joanna Szyda
 BIOINFORMATYKA 1. Wykład wstępny 2. Struktury danych w badaniach bioinformatycznych 3. Bazy danych: projektowanie i struktura 4. Bazy danych: projektowanie i struktura 5. Powiązania pomiędzy genami: równ.
BIOINFORMATYKA 1. Wykład wstępny 2. Struktury danych w badaniach bioinformatycznych 3. Bazy danych: projektowanie i struktura 4. Bazy danych: projektowanie i struktura 5. Powiązania pomiędzy genami: równ.
Krzysztof Spalik 1, Marcin Piwczyński 2
 Tom 58 2009 Numer 3 4 (284 285) Strony 485 498 Krzysztof Spalik 1, Marcin Piwczyński 2 1 Zakład Systematyki i Geografii Roślin Instytut Botaniki Uniwersytet Warszawski Aleje Ujazdowskie 4, 00-478 Warszawa
Tom 58 2009 Numer 3 4 (284 285) Strony 485 498 Krzysztof Spalik 1, Marcin Piwczyński 2 1 Zakład Systematyki i Geografii Roślin Instytut Botaniki Uniwersytet Warszawski Aleje Ujazdowskie 4, 00-478 Warszawa
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
ALGORYTMICZNA I STATYSTYCZNA ANALIZA DANYCH
 1 ALGORYTMICZNA I STATYSTYCZNA ANALIZA DANYCH WFAiS UJ, Informatyka Stosowana II stopień studiów 2 Wnioskowanie statystyczne dla zmiennych numerycznych Porównywanie dwóch średnich Boot-strapping Analiza
1 ALGORYTMICZNA I STATYSTYCZNA ANALIZA DANYCH WFAiS UJ, Informatyka Stosowana II stopień studiów 2 Wnioskowanie statystyczne dla zmiennych numerycznych Porównywanie dwóch średnich Boot-strapping Analiza
MSA i analizy filogenetyczne
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
Algorytm. Krótka historia algorytmów
 Algorytm znaczenie cybernetyczne Jest to dokładny przepis wykonania w określonym porządku skończonej liczby operacji, pozwalający na rozwiązanie zbliżonych do siebie klas problemów. znaczenie matematyczne
Algorytm znaczenie cybernetyczne Jest to dokładny przepis wykonania w określonym porządku skończonej liczby operacji, pozwalający na rozwiązanie zbliżonych do siebie klas problemów. znaczenie matematyczne
Mitochondrialna Ewa;
 Mitochondrialna Ewa; jej sprzymierzeńcy i wrogowie Lien Dybczyńska Zakład genetyki, Uniwersytet Warszawski 01.05.2004 Milion lat temu Ale co dalej??? I wtedy wkracza biologia molekularna Analiza różnic
Mitochondrialna Ewa; jej sprzymierzeńcy i wrogowie Lien Dybczyńska Zakład genetyki, Uniwersytet Warszawski 01.05.2004 Milion lat temu Ale co dalej??? I wtedy wkracza biologia molekularna Analiza różnic
Algorytmy genetyczne
 9 listopada 2010 y ewolucyjne - zbiór metod optymalizacji inspirowanych analogiami biologicznymi (ewolucja naturalna). Pojęcia odwzorowujące naturalne zjawiska: Osobnik Populacja Genotyp Fenotyp Gen Chromosom
9 listopada 2010 y ewolucyjne - zbiór metod optymalizacji inspirowanych analogiami biologicznymi (ewolucja naturalna). Pojęcia odwzorowujące naturalne zjawiska: Osobnik Populacja Genotyp Fenotyp Gen Chromosom
STATYSTYKA MATEMATYCZNA WYKŁAD 4. Testowanie hipotez Estymacja parametrów
 STATYSTYKA MATEMATYCZNA WYKŁAD 4 Testowanie hipotez Estymacja parametrów WSTĘP 1. Testowanie hipotez Błędy związane z testowaniem hipotez Etapy testowana hipotez Testowanie wielokrotne 2. Estymacja parametrów
STATYSTYKA MATEMATYCZNA WYKŁAD 4 Testowanie hipotez Estymacja parametrów WSTĘP 1. Testowanie hipotez Błędy związane z testowaniem hipotez Etapy testowana hipotez Testowanie wielokrotne 2. Estymacja parametrów
Modelowanie jako sposób opisu rzeczywistości. Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka
 Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
Modelowanie jako sposób opisu rzeczywistości Katedra Mikroelektroniki i Technik Informatycznych Politechnika Łódzka 2015 Wprowadzenie: Modelowanie i symulacja PROBLEM: Podstawowy problem z opisem otaczającej
Podstawowe pojęcia. Własności próby. Cechy statystyczne dzielimy na
 Podstawowe pojęcia Zbiorowość statystyczna zbiór jednostek (obserwacji) nie identycznych, ale stanowiących logiczną całość Zbiorowość (populacja) generalna skończony lub nieskończony zbiór jednostek, które
Podstawowe pojęcia Zbiorowość statystyczna zbiór jednostek (obserwacji) nie identycznych, ale stanowiących logiczną całość Zbiorowość (populacja) generalna skończony lub nieskończony zbiór jednostek, które
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
Statystyka matematyczna i ekonometria
 Statystyka matematyczna i ekonometria prof. dr hab. inż. Jacek Mercik B4 pok. 55 jacek.mercik@pwr.wroc.pl (tylko z konta studenckiego z serwera PWr) Konsultacje, kontakt itp. Strona WWW Elementy wykładu.
Statystyka matematyczna i ekonometria prof. dr hab. inż. Jacek Mercik B4 pok. 55 jacek.mercik@pwr.wroc.pl (tylko z konta studenckiego z serwera PWr) Konsultacje, kontakt itp. Strona WWW Elementy wykładu.
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 7: drzewa filogenetyczne prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska rzewa filogenetyczne rzewa filogenetyczne odzwierciedlają
lgorytmy kombinatoryczne w bioinformatyce wykład 7: drzewa filogenetyczne prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska rzewa filogenetyczne rzewa filogenetyczne odzwierciedlają
Systemy uczące się Lab 4
 Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
Systemy uczące się Lab 4 dr Przemysław Juszczuk Katedra Inżynierii Wiedzy, Uniwersytet Ekonomiczny 26 X 2018 Projekt zaliczeniowy Podstawą zaliczenia ćwiczeń jest indywidualne wykonanie projektu uwzględniającego
Zmienność ewolucyjna. Ewolucja molekularna
 Zmienność ewolucyjna Ewolucja molekularna Mechanizmy ewolucji Generujące zmienność mutacje rearanżacje genomu horyzontalny transfer genów! Działające na warianty wytworzone przez zmienność dobór naturalny
Zmienność ewolucyjna Ewolucja molekularna Mechanizmy ewolucji Generujące zmienność mutacje rearanżacje genomu horyzontalny transfer genów! Działające na warianty wytworzone przez zmienność dobór naturalny
Metody probabilistyczne klasyfikatory bayesowskie
 Konwersatorium Matematyczne Metody Ekonomii narzędzia matematyczne w eksploracji danych First Prev Next Last Go Back Full Screen Close Quit Metody probabilistyczne klasyfikatory bayesowskie Wykład 8 Marcin
Konwersatorium Matematyczne Metody Ekonomii narzędzia matematyczne w eksploracji danych First Prev Next Last Go Back Full Screen Close Quit Metody probabilistyczne klasyfikatory bayesowskie Wykład 8 Marcin
Wprowadzenie do analizy korelacji i regresji
 Statystyka dla jakości produktów i usług Six sigma i inne strategie Wprowadzenie do analizy korelacji i regresji StatSoft Polska Wybrane zagadnienia analizy korelacji Przy analizie zjawisk i procesów stanowiących
Statystyka dla jakości produktów i usług Six sigma i inne strategie Wprowadzenie do analizy korelacji i regresji StatSoft Polska Wybrane zagadnienia analizy korelacji Przy analizie zjawisk i procesów stanowiących
FILOGENETYKA. Bioinformatyka, wykład 7 (24.XI.200..XI.2008)
") FILOGENETYKA Bioinformatyka, wykład 7 (24.XI.200.XI.2008) krzysztof_pawlowski@sggw.pl Filogenetyka Cel rekonstrukcja historii ewolucji wszystkich organizmów. Klasyczne podejście: historia ewolucji jest
FILOGENETYKA Bioinformatyka, wykład 7 (24.XI.200.XI.2008) krzysztof_pawlowski@sggw.pl Filogenetyka Cel rekonstrukcja historii ewolucji wszystkich organizmów. Klasyczne podejście: historia ewolucji jest
Wykład 3 Hipotezy statystyczne
 Wykład 3 Hipotezy statystyczne Hipotezą statystyczną nazywamy każde przypuszczenie dotyczące nieznanego rozkładu obserwowanej zmiennej losowej (cechy populacji generalnej) Hipoteza zerowa (H 0 ) jest hipoteza
Wykład 3 Hipotezy statystyczne Hipotezą statystyczną nazywamy każde przypuszczenie dotyczące nieznanego rozkładu obserwowanej zmiennej losowej (cechy populacji generalnej) Hipoteza zerowa (H 0 ) jest hipoteza
Przetarg nieograniczony na zakup specjalistycznej aparatury laboratoryjnej Znak sprawy: DZ-2501/6/17
 Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
Część nr 2: SEKWENATOR NASTĘPNEJ GENERACJI Z ZESTAWEM DEDYKOWANYCH ODCZYNNIKÓW Określenie przedmiotu zamówienia zgodnie ze Wspólnym Słownikiem Zamówień (CPV): 38500000-0 aparatura kontrolna i badawcza
METODY STATYSTYCZNE W BIOLOGII
 METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY STATYSTYCZNE W BIOLOGII 1. Wykład wstępny 2. Populacje i próby danych 3. Testowanie hipotez i estymacja parametrów 4. Planowanie eksperymentów biologicznych 5. Najczęściej wykorzystywane testy statystyczne
METODY BADAŃ NA ZWIERZĘTACH ze STATYSTYKĄ wykład 3-4. Parametry i wybrane rozkłady zmiennych losowych
 METODY BADAŃ NA ZWIERZĘTACH ze STATYSTYKĄ wykład - Parametry i wybrane rozkłady zmiennych losowych Parametry zmiennej losowej EX wartość oczekiwana D X wariancja DX odchylenie standardowe inne, np. kwantyle,
METODY BADAŃ NA ZWIERZĘTACH ze STATYSTYKĄ wykład - Parametry i wybrane rozkłady zmiennych losowych Parametry zmiennej losowej EX wartość oczekiwana D X wariancja DX odchylenie standardowe inne, np. kwantyle,