Porównywanie i dopasowywanie sekwencji
|
|
|
- Andrzej Witkowski
- 8 lat temu
- Przeglądów:
Transkrypt
1 Porównywanie i dopasowywanie sekwencji
2 Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna za przedmiot swoich badań uznaje cząsteczki DNA i białek a jej celem jest scharakteryzowanie mechanizmów mutacji i selekcji zachodzących na poziomie sekwencji. W ewolucji molekularnej nacisk kładzie się na porównywanie cząsteczek między różnymi gatunkami, co stanowi przewagę w stosunku do np. genetyki populacyjnej, która zajmuje się badaniem zróżnicowania genetycznego u osobników tego samego gatunku.
3 Filogenetyka to nauka wykorzystującą metody bioinformatyczne do analizy relacji ewolucyjnych na poziomie molekularnym Celem analizy filogenetycznej jest wysuwanie wniosków na temat tych relacji ewolucyjnych lub ich szacowanie. Filogenetyka molekularna obejmuje zestaw metod pozwalających wykorzystać informację zawartą w sekwencjach aminokwasowych lub nukleotydowych w celu odtworzenia historii ewolucyjnej, uwzględniając kolejność specjacji. Historia ewolucyjna odtwarzana dzięki analizie filogenetycznej, jest zwykle przedstawiana w postaci rozgałęziających się diagramów przypominających drzewo i odzwierciedlających przypuszczalne zależności genealogiczne między cząsteczkami lub organizmami.
4 Nic w bioinformatyce nie ma sensu, jeśli rozpatrywane jest w oderwaniu od ewolucji Za umowny początek filogenetyki - badań nad ewolucją molekularną przyjmuje się rok 1965 kiedy to ukazał się artykuł Zuckerkandala i Paulinga demonstrujący tzw. drzewo filogenetyczne skonstruowane w oparciu o sekwencje białek. T. Dobzhansky powiedział kiedyś: Nic w biologii nie ma sensu, jeśli rozpatrywane jest w oderwaniu od ewolucji
5 HOMOLOGIA kluczowe pojęcie, w odniesieniu do ewolucji molekularnej homologia - obecność podobnych własności ze względu na pochodzenie od wspólnego przodka, a rozbieżności między nimi są wynikiem ich różnicowania się w toku ewolucji homologię określamy na podstawie obserwowanych podobieństw homologia jest własnością binarną - nie ma stanów pośrednich homologia jest nieobserwowalna bezpośrednio, gdyż nie jesteśmy w stanie zaobserwować organizmów lub cząsteczek przodka

6 homologia - obecność podobnych własności ze względu na pochodzenie od wspólnego przodka, tj. jeśli związki między nimi są wynikiem ich różnicowania się w toku ewolucji

7 Sekwencje homologiczne mogą być: ortologami jeśli ich ostatni wspólny przodek istniał w momencie specjacji tzn. ortologami nazwywamy np. homologiczne geny w różnych organizmach, które kodują białka pełniące tę samą funkcję i które ewoluują bezpośrednio z pokolenia na pokolenie
8 Występowanie ortologów w różnych organizmów jest wynikiem rozdzielenia się gatunków czyli specjacji
9 Sekwencje homologiczne mogą być też: paralogami jeśli ich ostatni wspólny przodek istniał w momencie duplikacji w tym samym organizmie tzn. paralogami są homologiczne geny w organizmie, które kodują białka o pokrewnych, ale nie identycznych funkcjach Paralogami nazywamy spokrewnione ewolucyjnie sekwencje z jednego organizmu, których linie ewolucyjne rozeszły się w wyniku duplikacji genu.
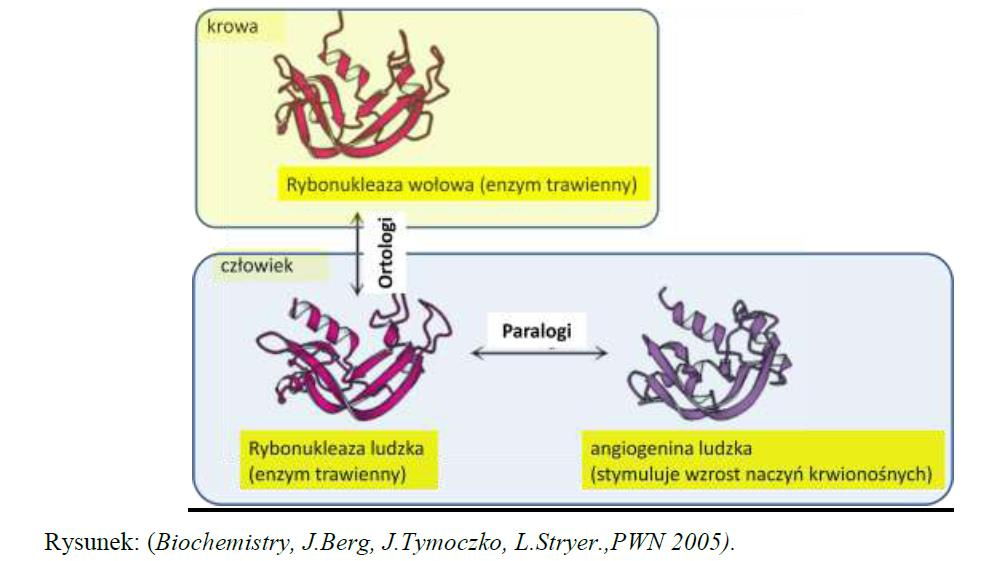
10
11 Jak możemy zobaczyć ewolucję molekularną: Podstawową i najczęściej stosowana procedurą w bioinformatyce jest porównywanie sekwencji poprzez wyznaczenie dopasowań par sekwencji lub wielu sekwencji jednocześnie Homologię obserwujemy na podstawie podobieństwa sekwencji Podobieństwo sekwencji ustalamy przez ich dopasowanie czyli alignment
12 Wyznaczanie homologii nie jest jedynym celem porównywania i dopasowywania sekwencji Po co porównywać sekwencje: w celu wyznaczenia homologii czyli określenia jak przebiegała ewolucja naszej cząsteczki z ciekawości - by dowiedzieć się czym jest nasza sekwencja i co zawiera w celu określenia jej przypuszczalnej funkcji poznać przybliżoną strukturę (białka) poznać położenie eksonów (DNA), lokalizacja domen funkcyjnych (białka)
13 Rodzaje dopasowań (alignmentów) Pokrycie sekwencji : globalny (wszystkie elementy sekwencji są dopasowane) lokalny (tylko fragmenty sekwencji są dopasowane) Liczba dopasowywanych sekwencji : dwie (Pairwise Sequence Alignment) więcej niż dwie (Multiple Sequence Alignment) Możemy dopasowywać oczywiście sekwencje nukleotydowe aminokwasowe
14 Żeby móc pokazać np. ewolucję molekularną lub określić funkcję nowo poznanego odcinka DNA musimy dopasować sekwencje w dość wysublimowany sposób dopasowanie (alignment) powinno być: liniowym przedstawieniem relacji pomiędzy sekwencjami z uwzględnieniem układu jeden-do-jednego pomiędzy resztami aminokwasowymi lub nukleotydami Specyfika dopasowania umieszczenie insercji i delecji (gaps) tak aby odzwierciedlały prawdziwe wydarzenia ewolucyjne największe wyzwanie algorytmów
15 Algorytmy używane do porównywania i dopasowywania sekwencji dot matrix metoda okienkowa programowanie dynamiczne BLAST FASTA metody hierarchiczne wykorzystywane przede wszystkim dla dopasowań wielokrotnych w filogenetyce
16 Zasada algorytmów dynamicznych, które stały się podstawą narzędzi do poszukiwania podobieństwa w bazach danych polega na przesuwaniu jednej sekwencji nad drugą i obliczaniu najlepszego wyniku dopasowania Co jest składową wyniku podobieństwa? Do ogólnego wyniku (współczynnika podobieństwa sekwencji) dodawane są 1. współczynniki podobieństwa poszczególnych punktów porównania sekwencji (czyli np. pary nukleotydów lub aminokwasów) odczytywane z tzw. tablic podobieństwa 2. punktów (zwykle ujemne) wynikających z wstawienia przerwy i jej wydłużania (w jednej lub drugiej sekwencji), które obniżają końcowy wynik
17 Tablice podobieństwa Punktacja podobieństwa na przykładzie porównywania i dopasowywania sekwencji białkowych Wszystkie algorytmy porównujące sekwencje białkowe opierają się na współczynnikach podobieństwa dla wszystkich 210 możliwych par aminokwasów (190 par różnych aminokwasów i 20 identycznych). Pierwsze tablice opierały się właśnie na takim podobieństwie fizykochemicznych właściwości danych aminokwasów. tablice podobieństwa uwzględniają podobny charakter aminokwasów (np. I - L) i te pary mają wyższy współczynnik niż aminokwasy o różnym charakterze.
18 Nowsze generacje tablic podobieństwa (obecnie w powszechnym użyciu) opierają się na obserwowanej częstości substytucji (zmiany jednego aminokwasu na drugi), w zależności od stopnia pokrewieństwa. Oczywiście częstość obserwowanych substytucji odzwierciedla najczęściej podobieństwo właściwości fizyko-chemicznych, ale także bierze pod uwagę mechanizmy ewolucji molekularnej. Najpopularniejszym schematem punktowania jest tablica opracowana przez Margaret O. Dayhoff i współpracowników (1978)
19 Tablice podobieństwa Dayhoff Oparta jest ona na modelu ewolucji białek blisko spokrewnionych - substytucje aminokwasów są wynikiem kolejnych mutacji w obrębie odpowiednich kodonów. Częstość zmian jednego aminokwasu na drugi, obserwowanych w obrębie porównania wielu sekwencji, była dla każdego aminokwasu standaryzowana (przeliczana tak aby była uwzględniona częstość występowania tego aminokwasu we wszystkich sekwencjach bazy danych). Tak otrzymane dane zostały jeszcze skorygowane o prawdopodobieństwo wystąpienia akceptowalnej mutacji w określonym czasie ewolucji - PAM (Percentage of Acceptable point Mutations per 10 8 years) czyli są specyficzne dla określonego dystansu filogenetycznego Co to znaczy? Przy dystansie filogenetycznym określanym jako 256 PAM, podobieństwo sekwencji leży blisko granicy jego wykrycia (ok. 80% aminokwasów zmienionych) Jones i inni (1992), opierając się o dużo nowsze dane uaktualnili tablice Dayhoff i stworzyli tablice PET91. Jako że tablice PET91 obliczane są w podobny sposób jak tablice Dayhoff to analogicznie można je stosować przy różnych dystansach filogenetycznych PAM
20 Zasady poszukiwania podobieństwa sekwencji w bazach danych za pomocą algorytmów dynamicznych Pierwsza podstawowa zasada: Przy przeszukiwaniu baz danych stworzenie precyzyjnego dopasowania dwu sekwencji i wyznaczenie homologii nie jest celem samym w sobie (choć efekt przeszukiwania obserwujemy w postaci serii dopasowań dwu sekwencji pokazanych jako rankigowa lista trafień) - celem jest znalezienie sekwencji podobnych, które mogą (ale nie muszą) być homologiczne, spokrewnione z naszą - badaną,
21 Zasady poszukiwania podobieństwa sekwencji w bazach danych c.d. Drugą podstawową zasadą przeszukiwania bazy danych jest porównanie sekwencji sprawdzanej (kwerendy, zapytania - query sequence) do każdej sekwencji w bazie danych (których jest baaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaardzo duuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuuużo Trzecie założenie - większość z sekwencji w bazie jest niepodobnych do sekwencji porównywanej należy więc znaleźć sposób na odrzucenie tych sekwencji Czwarte założenie - przy dopasowywaniu sekwencji nie muszą one być podobnej długości (to jest zaleta programowania dynamicznego)
22 Rodzaje przeszukiwań baz danych w celu znalezienia podobnych sekwencji nukleotydy do nukleotydów- niska czułość ze względu na 4-literowy alfabet i zazwyczaj jest używana jedynie matryca identyczności dobra przy porównywaniu dużych sekwencji genomowych oraz porównaniach wstępnych białko do białek- najlepiej dopracowane duża czułość nukleotydy do białek- zamienia nukleotydy na białko we wszystkich sześciu ramkach odczytu, służy do znajdowania homologicznych białek, dobrze spisuje się w przypadku genomów eukariotycznych Terminologia przy poszukiwaniu podobieństwa w bazach danych query (kwerenda, zapytanie)- sekwencja dla której chcemy znaleźć podobne sekwencje w bazie danych subject - sekwencja znaleziona w bazie (dopasowana do kwerendy, zwykle jest ich wiele)
23 Algorytmy do przeszukiwania baz danych: Jednym z pierwszych był algorytm Smith a i Waterman a (1981) w programie BLITZ, który dołączał do wyniku statystyczną istotność porównania. Dziś w powszechnym użyciu są dwa podstawowe narzędzia (algorytmy): FASTA BLAST Ich wspólnym mianownikiem jest rozpoczęcie wyszukiwania podobieństwa od krótkich odcinków zadanej sekwencji nt lub aa
24 Przybliżenia - algorytm FASTA Pierwszym powszechnie stosowanym programem do przeszukiwania baz stało się narzędzie oparte na algorytmie FASTA, stworzonym przez Pearsona i Lipmana (1985) a jego zasada polega na wyszukiwaniu najistotniejszych przekątnych analizy dot-plot.
25 Poszczególne etapy porównywania sekwencji programem FASTA. Początkowy etap polega na identyfikacji wszystkich identycznych odcinków dwóch sekwencji o długości k (k-tuples) i poszukiwaniu tych, które leżą na tej samej przekątnej.
26 FASTA - podsumowanie Oszacowuje istotność statystyczną otrzymanego alignmentu poprzez porównanie dystrybucji punktacji z białkami prawdziwie niehomologicznymi Co może być mylące dla tego algorytmu? niezwykła kompozycja sekwencji sekwencje transbłonowe sekwencje powtarzalne FASTA jest uważany za nieco wolniejszy od drugiego powszechnie używanego programu jakim jest BLAST, ale za to bardziej czuły i bardziej specyficzny (w zależności od ustawionych parametrów).

27 Program sugeruje wybór najlepszej bazy danych oraz formę uzyskania wyniku w zależności od wybranego algorytmu

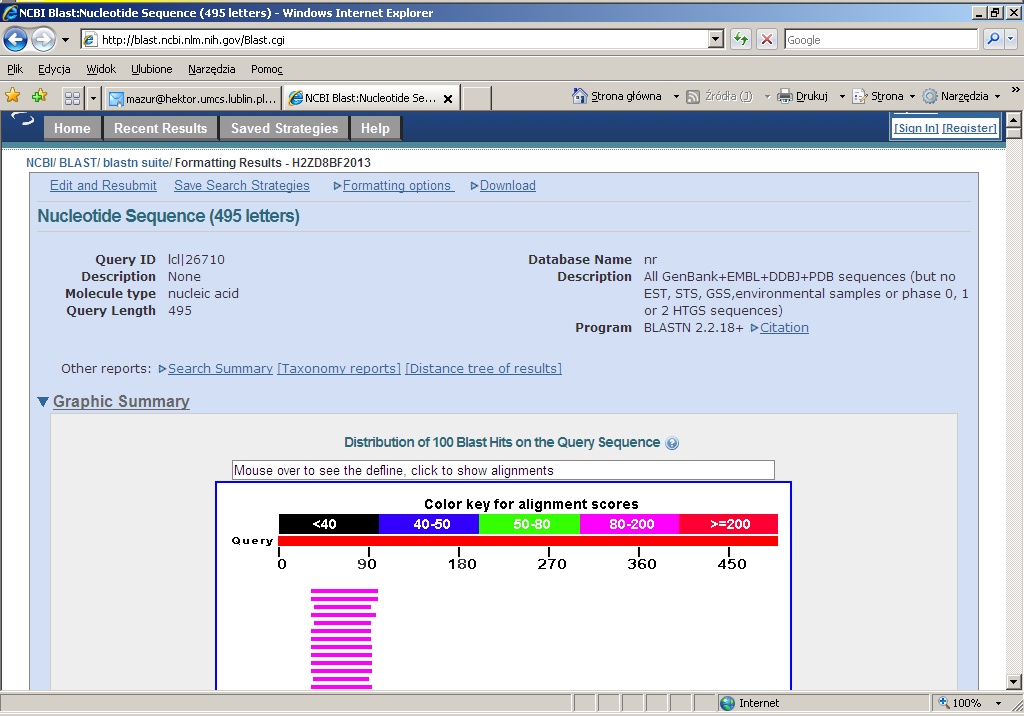
28 Trafienia uszeregowane w rankig wyników
29
30
31
32 Drugi szeroko używany program do wyszukiwania podobieństw - BLAST Basic Local Alignment Search Tool Znajduje najlepsze lokalne podobieństwa z sekwencją porównywaną
33 Algorytm BLAST BLAST zwiększył znacząco szybkość przeszukiwania baz danych nie zmniejszając jednocześnie w sposób istotny jego czułości poprzez podział query i sekwencji w bazie danych na fragmenty - słowa ("words"), i początkowym wyszukiwaniu podobieństw pomiędzy tymi fragmentami przy zastosowaniu określonej matrycy podstawień. Takie słowa są następnie wydłużane w obie strony i tworzony jest alignment jeśli wynik przekracza określoną wartość progową (threshold)
34 Poszczególne etapy poszukiwania podobieństwa bez przerw programem BLAST. 1. podzielenie query na nakładające się słowa 2. przeszukanie bazy w celu znalezienia słów pokrewnych 3. wydłużenie alignmentu zaczynając od słowa pokrewnego 4. wykonanie lokalnego programowania dynamicznego 5. obliczenie statystyczne wartości wyniku tzw. E-value
35 Wydłużenie zgodnego słowa "najdroższy" obliczeniowo etap BLASTa wydłużenie nie jest prowadzone do momentu gdy wyniki punktacji (score) spadnie do zera lecz do osiągnięcia pewnego minimalnego progu punktacji poniżej którego lepiej (dla BLASTA) jest przerwać dopasowanie (...i np. zacząć w innym miejscu)
36
37
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wykład 5 Dopasowywanie lokalne
 Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
 Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Dopasowywanie sekwencji Sequence alignment Drzewo filogenetyczne Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry
Generator testów Bioinformatyka wer / 0 Strona: 1
 Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
Generator testów 1.3.1 Bioinformatyka_zdalne wer. 1.0.13 / 0 Strona: 1
 Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW
 PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
Przyrównywanie sekwencji
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych???
 Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Wykład Bioinformatyka 2012-09-24. Bioinformatyka. Wykład 7. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM. Ewolucyjne podstawy Bioinformatyki
 Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Teoria ewolucji. Podstawowe pojęcia. Wspólne pochodzenie.
 Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego 5A pgolik@igib.uw.edu.pl Informacje, materiały: http://www.igib.uw.edu.pl/
Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego 5A pgolik@igib.uw.edu.pl Informacje, materiały: http://www.igib.uw.edu.pl/
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Dopasowanie sekwencji
 Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Analizy filogenetyczne
 BIOINFORMATYKA edycja 2016 / 2017 wykład 6 Analizy filogenetyczne dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Cele i zastosowania 2. Podstawy ewolucyjne
BIOINFORMATYKA edycja 2016 / 2017 wykład 6 Analizy filogenetyczne dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Cele i zastosowania 2. Podstawy ewolucyjne
Genomika Porównawcza. Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski
 Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Genomika Porównawcza Agnieszka Rakowska Instytut Informatyki i Matematyki Komputerowej Uniwersytet Jagiellooski 1 Plan prezentacji 1. Rodzaje i budowa drzew filogenetycznych 2. Metody ukorzeniania drzewa
Teoria ewolucji. Podstawowe pojęcia. Wspólne pochodzenie.
 Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Ewolucja Znaczenie ogólne: zmiany zachodzące stopniowo w czasie W biologii ewolucja biologiczna W astronomii i kosmologii ewolucja gwiazd i wszechświata
Teoria ewolucji Podstawowe pojęcia. Wspólne pochodzenie. Ewolucja Znaczenie ogólne: zmiany zachodzące stopniowo w czasie W biologii ewolucja biologiczna W astronomii i kosmologii ewolucja gwiazd i wszechświata
plezjomorfie: podobieństwa dziedziczone po dalszych przodkach (c. atawistyczna)
") Podobieństwa pomiędzy organizmami - cechy homologiczne: podobieństwa wynikające z dziedziczenia - apomorfie: podobieństwa dziedziczone po najbliższym przodku lub pojawiająca się de novo (c. ewolucyjnie
Podobieństwa pomiędzy organizmami - cechy homologiczne: podobieństwa wynikające z dziedziczenia - apomorfie: podobieństwa dziedziczone po najbliższym przodku lub pojawiająca się de novo (c. ewolucyjnie
dopasowanie sekwencji Porównywanie sekwencji Etapy dopasowywania sekwencji Homologia, podobieństwo i analogia
 Porównywanie sekwencji Homologia, podobieństwo i analogia dopasowanie sekwencji Dopasowanie/porównywanie Uliniowienie Alignment W bioinformatyce, dopasowanie sekwencji jest sposobem dopasowania struktur
Porównywanie sekwencji Homologia, podobieństwo i analogia dopasowanie sekwencji Dopasowanie/porównywanie Uliniowienie Alignment W bioinformatyce, dopasowanie sekwencji jest sposobem dopasowania struktur
Podstawy biologii. Informacja genetyczna. Co to jest ewolucja.
 Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Materiał genetyczny Materiałem genetycznym są kwasy nukleinowe Materiałem genetycznym organizmów komórkowych jest kwas deoksyrybonukleinowy
Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Materiał genetyczny Materiałem genetycznym są kwasy nukleinowe Materiałem genetycznym organizmów komórkowych jest kwas deoksyrybonukleinowy
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały i ewolucja molekularna
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych???
 Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych???
 Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Analizy DNA in silico - czyli czego można szukać i co można znaleźć w sekwencjach nukleotydowych??? Alfabet kwasów nukleinowych jest stosunkowo ubogi!!! Dla sekwencji DNA (RNA) stosuje się zasadniczo*
Samouczek: Konstruujemy drzewo
 ROZDZIAŁ 2 Samouczek: Konstruujemy drzewo Po co nam drzewa filogenetyczne? Drzewa filogenetyczne często pojawiają się dzisiaj w pracach z dziedziny biologii molekularnej, które nie mają związku z filogenetyką
ROZDZIAŁ 2 Samouczek: Konstruujemy drzewo Po co nam drzewa filogenetyczne? Drzewa filogenetyczne często pojawiają się dzisiaj w pracach z dziedziny biologii molekularnej, które nie mają związku z filogenetyką
Porównywanie sekwencji białkowych
 Bioinformatyka -9 Bioinformatyka Wykład 4. E. Banachowicz Zakład Biofizyki Molekularnej http://www.amu.edu.pl/~ewas Porównywanie sekwencji białkowych Wykład 4, Bioinformatyka -9 Porównywanie sekwencji
Bioinformatyka -9 Bioinformatyka Wykład 4. E. Banachowicz Zakład Biofizyki Molekularnej http://www.amu.edu.pl/~ewas Porównywanie sekwencji białkowych Wykład 4, Bioinformatyka -9 Porównywanie sekwencji
Ewolucja molekularna człowieka okiem bioinformatyka. Justyna Wojtczak Jarosław Jeleniewicz
 Ewolucja molekularna człowieka okiem bioinformatyka Justyna Wojtczak Jarosław Jeleniewicz Informatyka w biologii - bioinformatyka Jest to szeroka dziedzina zajmująca się tworzeniem zaawansowanych baz danych,
Ewolucja molekularna człowieka okiem bioinformatyka Justyna Wojtczak Jarosław Jeleniewicz Informatyka w biologii - bioinformatyka Jest to szeroka dziedzina zajmująca się tworzeniem zaawansowanych baz danych,
Dopasowanie par sekwencji
 BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
PODSTAWY BIOINFORMATYKI
 PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
PODSTAWY BIOINFORMATYKI Prowadzący: JOANNA SZYDA ADRIAN DROśDś WSTĘP 1. Katedra Genetyki badania bioinformatyczne 2. Tematyka przedmiotu 3. Charakterystyka wykładów 4. Charakterystyka ćwiczeń 5. Informacje
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Bioinformatyka Laboratorium, 30h. Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecnośd Literatura, materiały Bioinformatyka i ewolucja
3 Przeszukiwanie baz danych
 Spis treści 3 Przeszukiwanie baz danych 1 3.1 Heurystyczne algorytmy...................... 1 3.1.1 FASTA........................... 1 3.1.2 BLAST........................... 3 3.2 Macierze substytucyjne.......................
Spis treści 3 Przeszukiwanie baz danych 1 3.1 Heurystyczne algorytmy...................... 1 3.1.1 FASTA........................... 1 3.1.2 BLAST........................... 3 3.2 Macierze substytucyjne.......................
Dopasowanie sekwencji c.d. Sequence alignment. Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl
 krzysztof_pawlowski@sggw.pl") Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl dopasowanie - metody dopasowanie par sekwencji: Macierz punktów - dot matrix, dotplot Programowanie
Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl dopasowanie - metody dopasowanie par sekwencji: Macierz punktów - dot matrix, dotplot Programowanie
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii
 Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Technologia rekombinowanego DNA jest podstawą uzyskiwania genetycznie zmodyfikowanych organizmów 2. Medycyna i ochrona zdrowia
Algorytmika dla bioinformatyki
 Algorytmika dla bioinformatyki kurs 2018/2019 Prof. Danuta Makowiec Instytut Fizyki Teoretycznej i Astrofizyki pok. 353, danuta.makowiec@gmail.com Cele kursu 2 Treści wykładu będą skoncentrowane wokół
Algorytmika dla bioinformatyki kurs 2018/2019 Prof. Danuta Makowiec Instytut Fizyki Teoretycznej i Astrofizyki pok. 353, danuta.makowiec@gmail.com Cele kursu 2 Treści wykładu będą skoncentrowane wokół
Zmienność ewolucyjna. Ewolucja molekularna
 Zmienność ewolucyjna Ewolucja molekularna Mechanizmy ewolucji Generujące zmienność mutacje rearanżacje genomu horyzontalny transfer genów! Działające na warianty wytworzone przez zmienność dobór naturalny
Zmienność ewolucyjna Ewolucja molekularna Mechanizmy ewolucji Generujące zmienność mutacje rearanżacje genomu horyzontalny transfer genów! Działające na warianty wytworzone przez zmienność dobór naturalny
Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online
 Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
Biologia medyczna, materiały dla studentów
 Jaka tam ewolucja. Zanim trafię na jednego myślącego, muszę stoczyć bitwę zdziewięcioma orangutanami Carlos Ruis Zafon Wierzbownica drobnokwiatowa Fitosterole, garbniki, flawonoidy Właściwości przeciwzapalne,
Jaka tam ewolucja. Zanim trafię na jednego myślącego, muszę stoczyć bitwę zdziewięcioma orangutanami Carlos Ruis Zafon Wierzbownica drobnokwiatowa Fitosterole, garbniki, flawonoidy Właściwości przeciwzapalne,
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19
 WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
Ćwiczenie 5/6. Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST.
 Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Generator testów bioinformatyka wer / Strona: 1
 Przedmiot: wyklad monograficzny Nazwa testu: bioinformatyka wer. 1.0.6 Nr testu 10469906 Klasa: 5 IBOS Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Aminokwas jest to związek organiczny zawierający A) grupę
Przedmiot: wyklad monograficzny Nazwa testu: bioinformatyka wer. 1.0.6 Nr testu 10469906 Klasa: 5 IBOS Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Aminokwas jest to związek organiczny zawierający A) grupę
Algorytmy genetyczne w optymalizacji
 Algorytmy genetyczne w optymalizacji Literatura 1. David E. Goldberg, Algorytmy genetyczne i ich zastosowania, WNT, Warszawa 1998; 2. Zbigniew Michalewicz, Algorytmy genetyczne + struktury danych = programy
Algorytmy genetyczne w optymalizacji Literatura 1. David E. Goldberg, Algorytmy genetyczne i ich zastosowania, WNT, Warszawa 1998; 2. Zbigniew Michalewicz, Algorytmy genetyczne + struktury danych = programy
Urszula Poziomek, doradca metodyczny w zakresie biologii Materiał dydaktyczny przygotowany na konferencję z cyklu Na miarę Nobla, 14 stycznia 2010 r.
 Ćwiczenie 1 1 Wstęp Rozważając możliwe powiązania filogenetyczne gatunków, systematyka porównuje dane molekularne. Najskuteczniejszym sposobem badania i weryfikacji różnych hipotez filogenetycznych jest
Ćwiczenie 1 1 Wstęp Rozważając możliwe powiązania filogenetyczne gatunków, systematyka porównuje dane molekularne. Najskuteczniejszym sposobem badania i weryfikacji różnych hipotez filogenetycznych jest
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
PODSTAWY BIOINFORMATYKI 6 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa - przykłady 4. Etapy konstrukcji drzewa
Konspekt do zajęć z przedmiotu Genetyka dla kierunku Położnictwo dr Anna Skorczyk-Werner Katedra i Zakład Genetyki Medycznej
 Seminarium 1 część 1 Konspekt do zajęć z przedmiotu Genetyka dla kierunku Położnictwo dr Anna Skorczyk-Werner Katedra i Zakład Genetyki Medycznej Genom człowieka Genomem nazywamy całkowitą ilość DNA jaka
Seminarium 1 część 1 Konspekt do zajęć z przedmiotu Genetyka dla kierunku Położnictwo dr Anna Skorczyk-Werner Katedra i Zakład Genetyki Medycznej Genom człowieka Genomem nazywamy całkowitą ilość DNA jaka
Mechanizmy zmienności ewolucyjnej. Podstawy ewolucji molekularnej.
 Mechanizmy zmienności ewolucyjnej Podstawy ewolucji molekularnej. Mechanizmy ewolucji } Generujące zmienność } mutacje } rearanżacje genomu } horyzontalny transfer genów } Działające na warianty wytworzone
Mechanizmy zmienności ewolucyjnej Podstawy ewolucji molekularnej. Mechanizmy ewolucji } Generujące zmienność } mutacje } rearanżacje genomu } horyzontalny transfer genów } Działające na warianty wytworzone
Wyszukiwanie podobnych sekwencji w bazach danych. Wyszukiwanie w sekwencji nukleotydów czy aminokwasów? Czułość i selektywność
 Wersja 1.05 Wprowadzenie do Informatyki Biomedycznej Wykład 3: Wyszukiwanie w bazach sekwencji Przewidywanie genów Wydział Informatyki PB Marek Krętowski pokój 206 e-mail: m.kretowski@pb.edu.pl http://aragorn.pb.bialystok.pl/~mkret
Wersja 1.05 Wprowadzenie do Informatyki Biomedycznej Wykład 3: Wyszukiwanie w bazach sekwencji Przewidywanie genów Wydział Informatyki PB Marek Krętowski pokój 206 e-mail: m.kretowski@pb.edu.pl http://aragorn.pb.bialystok.pl/~mkret
Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej
 Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej Specjacja } Pojawienie się bariery reprodukcyjnej między populacjami dające początek gatunkom } Specjacja allopatryczna
Teoria ewolucji. Losy gatunków: specjacja i wymieranie. Podstawy ewolucji molekularnej Specjacja } Pojawienie się bariery reprodukcyjnej między populacjami dające początek gatunkom } Specjacja allopatryczna
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
Acknowledgement. Drzewa filogenetyczne
 Wykład 8 Drzewa Filogenetyczne Lokalizacja genów Some figures from: Acknowledgement M. Zvelebil, J.O. Baum, Introduction to Bioinformatics, Garland Science 2008 Tradycyjne drzewa pokrewieństwa Drzewa oparte
Wykład 8 Drzewa Filogenetyczne Lokalizacja genów Some figures from: Acknowledgement M. Zvelebil, J.O. Baum, Introduction to Bioinformatics, Garland Science 2008 Tradycyjne drzewa pokrewieństwa Drzewa oparte
Podstawy ewolucji molekularnej. Ewolucja sekwencji DNA i białek
 Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Zmiany genetyczne w ewolucji } Mutacje } tworzą nowe allele genów } Inwersje } zmieniają układ genów na chromosomach } mogą uniemożliwić rekombinację
Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Zmiany genetyczne w ewolucji } Mutacje } tworzą nowe allele genów } Inwersje } zmieniają układ genów na chromosomach } mogą uniemożliwić rekombinację
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Teoria ewolucji. Podstawy wspólne pochodzenie.
 Teoria ewolucji. Podstawy wspólne pochodzenie. Ewolucja biologiczna } Znaczenie ogólne: } proces zmian informacji genetycznej (częstości i rodzaju alleli), } które to zmiany są przekazywane z pokolenia
Teoria ewolucji. Podstawy wspólne pochodzenie. Ewolucja biologiczna } Znaczenie ogólne: } proces zmian informacji genetycznej (częstości i rodzaju alleli), } które to zmiany są przekazywane z pokolenia
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Historia informacji genetycznej. Jak ewolucja tworzy nową informację (z ma ą dygresją).
.") Historia informacji genetycznej. Jak ewolucja tworzy nową informację (z ma ą dygresją). Czym jest życie? metabolizm + informacja (replikacja) 2 Cząsteczki organiczne mog y powstać w atmosferze pierwotnej
Historia informacji genetycznej. Jak ewolucja tworzy nową informację (z ma ą dygresją). Czym jest życie? metabolizm + informacja (replikacja) 2 Cząsteczki organiczne mog y powstać w atmosferze pierwotnej
Nuttall przeprowadził testy precypitacyjne białek surowicy, aby wykazać związek filogenetyczny między różnymi grupami zwierząt.
 1904 Nuttall przeprowadził testy precypitacyjne białek surowicy, aby wykazać związek filogenetyczny między różnymi grupami zwierząt. M. Prakash 2007.Encyclopaedia of Gene Evolution Vol. 2, Molecular Genetics,
1904 Nuttall przeprowadził testy precypitacyjne białek surowicy, aby wykazać związek filogenetyczny między różnymi grupami zwierząt. M. Prakash 2007.Encyclopaedia of Gene Evolution Vol. 2, Molecular Genetics,
Algorytmy genetyczne. Materiały do laboratorium PSI. Studia niestacjonarne
 Algorytmy genetyczne Materiały do laboratorium PSI Studia niestacjonarne Podstawowy algorytm genetyczny (PAG) Schemat blokowy algorytmu genetycznego Znaczenia, pochodzących z biologii i genetyki, pojęć
Algorytmy genetyczne Materiały do laboratorium PSI Studia niestacjonarne Podstawowy algorytm genetyczny (PAG) Schemat blokowy algorytmu genetycznego Znaczenia, pochodzących z biologii i genetyki, pojęć
Bioinformatyczne bazy danych
 Bioinformatyczne bazy danych Czym jest bioinformatyka? Bioinformatyka jest nauką integrującą różne dziedziny wiedzy Gruca (2010) Czym jest bioinformatyka? Bioinformatyka obejmuje technologie wykorzystujące
Bioinformatyczne bazy danych Czym jest bioinformatyka? Bioinformatyka jest nauką integrującą różne dziedziny wiedzy Gruca (2010) Czym jest bioinformatyka? Bioinformatyka obejmuje technologie wykorzystujące
Geny i działania na nich
 Metody bioinformatyki Geny i działania na nich prof. dr hab. Jan Mulawka Trzy królestwa w biologii Prokaryota organizmy, których komórki nie zawierają jądra, np. bakterie Eukaryota - organizmy, których
Metody bioinformatyki Geny i działania na nich prof. dr hab. Jan Mulawka Trzy królestwa w biologii Prokaryota organizmy, których komórki nie zawierają jądra, np. bakterie Eukaryota - organizmy, których
Podstawy biologii. Informacja genetyczna. Co to jest ewolucja.
 Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Zarys biologii molekularnej genu Podstawowe procesy genetyczne Replikacja powielanie informacji Ekspresja wyrażanie (realizowanie funkcji)
Podstawy biologii Informacja genetyczna. Co to jest ewolucja. Zarys biologii molekularnej genu Podstawowe procesy genetyczne Replikacja powielanie informacji Ekspresja wyrażanie (realizowanie funkcji)
Motywy i podobieństwo
 Motywy i podobieństwo Całość funkcja Modularna budowa białek Elementy składowe czyli miejsca wiązania, domeny 1 Motywy Motyw jest opisem określonej części trójwymiarowej struktury zawierającym charakterystyczny
Motywy i podobieństwo Całość funkcja Modularna budowa białek Elementy składowe czyli miejsca wiązania, domeny 1 Motywy Motyw jest opisem określonej części trójwymiarowej struktury zawierającym charakterystyczny
Wzorcowe efekty kształcenia dla kierunku studiów biotechnologia studia pierwszego stopnia profil ogólnoakademicki
 Załącznik nr 2 do Uchwały Rady Wydziału Biochemii, Biofizyki i Biotechnologii UJ z dnia 19 czerwca 2018 r. w sprawie programu i planu studiów na kierunku BIOTECHNOLOGIA na poziomie studiów pierwszego stopnia
Załącznik nr 2 do Uchwały Rady Wydziału Biochemii, Biofizyki i Biotechnologii UJ z dnia 19 czerwca 2018 r. w sprawie programu i planu studiów na kierunku BIOTECHNOLOGIA na poziomie studiów pierwszego stopnia
MACIERZE MUTACYJNE W ANALIZIE GENOMÓW czy możliwa jest rekonstrukcja filogenetyczna? Aleksandra Nowicka
 MAIERZE MUTAYJNE W ANALIZIE GENOMÓW czy możliwa jest rekonstrukcja filogenetyczna? Aleksandra Nowicka Zadaniem FILOGENETYKI jest : zrekonstruowanie ewolucyjnej historii wszystkich organizmów odkrycie przodka
MAIERZE MUTAYJNE W ANALIZIE GENOMÓW czy możliwa jest rekonstrukcja filogenetyczna? Aleksandra Nowicka Zadaniem FILOGENETYKI jest : zrekonstruowanie ewolucyjnej historii wszystkich organizmów odkrycie przodka
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu w porównaniu z analizą trankryptomu:
Badanie doboru naturalnego na poziomie molekularnym
 Badanie doboru naturalnego na poziomie molekularnym Podstawy ewolucji molekulanej Jak ewoluują sekwencje Zmiany genetyczne w ewolucji Mutacje tworzą nowe allele genów Inwersje zmieniają układ genów na
Badanie doboru naturalnego na poziomie molekularnym Podstawy ewolucji molekulanej Jak ewoluują sekwencje Zmiany genetyczne w ewolucji Mutacje tworzą nowe allele genów Inwersje zmieniają układ genów na
Wybrane techniki badania białek -proteomika funkcjonalna
 Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Wybrane techniki badania białek -proteomika funkcjonalna Proteomika: umożliwia badanie zestawu wszystkich (lub prawie wszystkich) białek komórkowych Zalety analizy proteomu np. w porównaniu z analizą trankryptomu:
Podstawy ewolucji molekularnej. Ewolucja sekwencji DNA i białek
 Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Zmiany genetyczne w ewolucji Mutacje tworzą nowe allele genów Inwersje zmieniają układ genów na chromosomach mogą uniemożliwić rekombinację
Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Zmiany genetyczne w ewolucji Mutacje tworzą nowe allele genów Inwersje zmieniają układ genów na chromosomach mogą uniemożliwić rekombinację
Bioinformatyka. Porównywanie sekwencji
 Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
Algorytmy genetyczne. Materiały do laboratorium PSI. Studia stacjonarne i niestacjonarne
 Algorytmy genetyczne Materiały do laboratorium PSI Studia stacjonarne i niestacjonarne Podstawowy algorytm genetyczny (PAG) Schemat blokowy algorytmu genetycznego Znaczenia, pochodzących z biologii i genetyki,
Algorytmy genetyczne Materiały do laboratorium PSI Studia stacjonarne i niestacjonarne Podstawowy algorytm genetyczny (PAG) Schemat blokowy algorytmu genetycznego Znaczenia, pochodzących z biologii i genetyki,
Filogenetyka molekularna. Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji
 Filogenetyka molekularna Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji Co to jest filogeneza? Filogeneza=drzewo filogenetyczne=drzewo rodowe=drzewo to rozgałęziający się diagram, który
Filogenetyka molekularna Dr Anna Karnkowska Zakład Filogenetyki Molekularnej i Ewolucji Co to jest filogeneza? Filogeneza=drzewo filogenetyczne=drzewo rodowe=drzewo to rozgałęziający się diagram, który
Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka
 Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka Słowo wstępne XIII Przedmowa XV 1. Bioinformatyka i Internet Andreas D. Baxevanis 1 1.1. Podstawy Internetu 2 1.2. Połączenie z Internetem
Księgarnia PWN: A.D. Baxevanis, B.F.F. Ouellette Bioinformatyka Słowo wstępne XIII Przedmowa XV 1. Bioinformatyka i Internet Andreas D. Baxevanis 1 1.1. Podstawy Internetu 2 1.2. Połączenie z Internetem
Dopasowanie sekwencji c.d. Sequence alignment. Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl
 krzysztof_pawlowski@sggw.pl") Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl Dopasowanie sekwencji - znaczenie Podobieństwo porównywanych sekwencji (similarity) może świadczyć
Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl Dopasowanie sekwencji - znaczenie Podobieństwo porównywanych sekwencji (similarity) może świadczyć
MSA i analizy filogenetyczne
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
października 2013: Elementarz biologii molekularnej. Wykład nr 2 BIOINFORMATYKA rok II
 10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
Bioinformatyczne bazy danych
 Bioinformatyczne bazy danych Czym jest bioinformatyka? Bioinformatyka jest nauką integrującą różne dziedziny wiedzy Gruca (2010) http://bioinformaticsonline.com/file/view/4482/bioinformatics-definitions-and-applications
Bioinformatyczne bazy danych Czym jest bioinformatyka? Bioinformatyka jest nauką integrującą różne dziedziny wiedzy Gruca (2010) http://bioinformaticsonline.com/file/view/4482/bioinformatics-definitions-and-applications
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Filogenetyka molekularna wykorzystuje informację zawartą w sekwencjach aminokwasów lub nukleotydów do kontrukcji drzew
Księgarnia PWN: Paul G. Higgs, Teresa K. Attwood - Bioinformatyka i ewolucja molekularna
 Księgarnia PWN: Paul G. Higgs, Teresa K. Attwood - Bioinformatyka i ewolucja molekularna Przedmowa...................................................... 1 1. Rewolucja informatyczna w naukach biomedycznych...........................
Księgarnia PWN: Paul G. Higgs, Teresa K. Attwood - Bioinformatyka i ewolucja molekularna Przedmowa...................................................... 1 1. Rewolucja informatyczna w naukach biomedycznych...........................
Podstawy biologiczne - komórki. Podstawy biologiczne - cząsteczki. Model komórki eukariotycznej. Wprowadzenie do Informatyki Biomedycznej
 Wprowadzenie do Informatyki Biomedycznej Wykład 1: Podstawy bioinformatyki Wydział Informatyki PB Podstawy biologiczne - komórki Wszystkie organizmy zbudowane są z komórek komórka jest skomplikowanym systemem
Wprowadzenie do Informatyki Biomedycznej Wykład 1: Podstawy bioinformatyki Wydział Informatyki PB Podstawy biologiczne - komórki Wszystkie organizmy zbudowane są z komórek komórka jest skomplikowanym systemem
Bioinformatyka II Modelowanie struktury białek
 Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Bioinformatyka II Modelowanie struktury białek 1. Który spośród wymienionych szablonów wybierzesz do modelowania dla każdego z podanych przypadków? Dlaczego? Struktura krystaliczną czy NMR (to samo białko,
Ewolucja informacji genetycznej
 1 Ewolucja informacji genetycznej Czym jest życie? metabolizm + informacja (replikacja) Cząsteczki organiczne mog y powstać w atmosferze pierwotnej Ziemi Oparin, Haldane Miller, 1953 Co by o najpierw?
1 Ewolucja informacji genetycznej Czym jest życie? metabolizm + informacja (replikacja) Cząsteczki organiczne mog y powstać w atmosferze pierwotnej Ziemi Oparin, Haldane Miller, 1953 Co by o najpierw?
Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO
 Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO Bogumil Konopka 1, Jean-Christophe Nebel 2, Malgorzata Kotulska 1 * 1 Politechnika
Ocena jakości modeli strukturalnych białek w oparciu o podobieństwo strukturalne i semantyczny opis funkcji w ontologii GO Bogumil Konopka 1, Jean-Christophe Nebel 2, Malgorzata Kotulska 1 * 1 Politechnika
Podstawy ewolucji molekularnej. Ewolucja sekwencji DNA i białek
 Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Egzamin: 29.01.2018 16:00, sala 9B Pierwsza synteza Ewolucja jako zmiany częstości alleli w populacji Mutacje jako źródło nowych alleli Dobór
Podstawy ewolucji molekularnej Ewolucja sekwencji DNA i białek Egzamin: 29.01.2018 16:00, sala 9B Pierwsza synteza Ewolucja jako zmiany częstości alleli w populacji Mutacje jako źródło nowych alleli Dobór
MultiSETTER: web server for multiple RNA structure comparison. Sandra Sobierajska Uniwersytet Jagielloński
 MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
MultiSETTER: web server for multiple RNA structure comparison Sandra Sobierajska Uniwersytet Jagielloński Wprowadzenie Budowa RNA: - struktura pierwszorzędowa sekwencja nukleotydów w łańcuchu: A, U, G,
Podstawy teorii ewolucji. Informacja i ewolucja
 Podstawy teorii ewolucji Informacja i ewolucja Podręczniki 2 Dla zainteresowanych http://wps.prenhall.com/esm_freeman_evol_4/ 3 Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego
Podstawy teorii ewolucji Informacja i ewolucja Podręczniki 2 Dla zainteresowanych http://wps.prenhall.com/esm_freeman_evol_4/ 3 Informacje Kontakt: Paweł Golik Instytut Genetyki i Biotechnologii, Pawińskiego
Porównywanie sekwencji białek i kwasów nukleinowych
 Porównywanie sekwencji białek i kwasów nukleinowych Krzysztof Lewiński 1. Podobieństwo i jego miara Wprawdzie podobieństwo jest pojęciem często używanym w życiu codziennym ale nie oznacza to, że możemy
Porównywanie sekwencji białek i kwasów nukleinowych Krzysztof Lewiński 1. Podobieństwo i jego miara Wprawdzie podobieństwo jest pojęciem często używanym w życiu codziennym ale nie oznacza to, że możemy