Politechnika Wrocławska. Dopasowywanie sekwencji Sequence alignment
|
|
|
- Sabina Kubiak
- 6 lat temu
- Przeglądów:
Transkrypt
1 Dopasowywanie sekwencji Sequence alignment
2
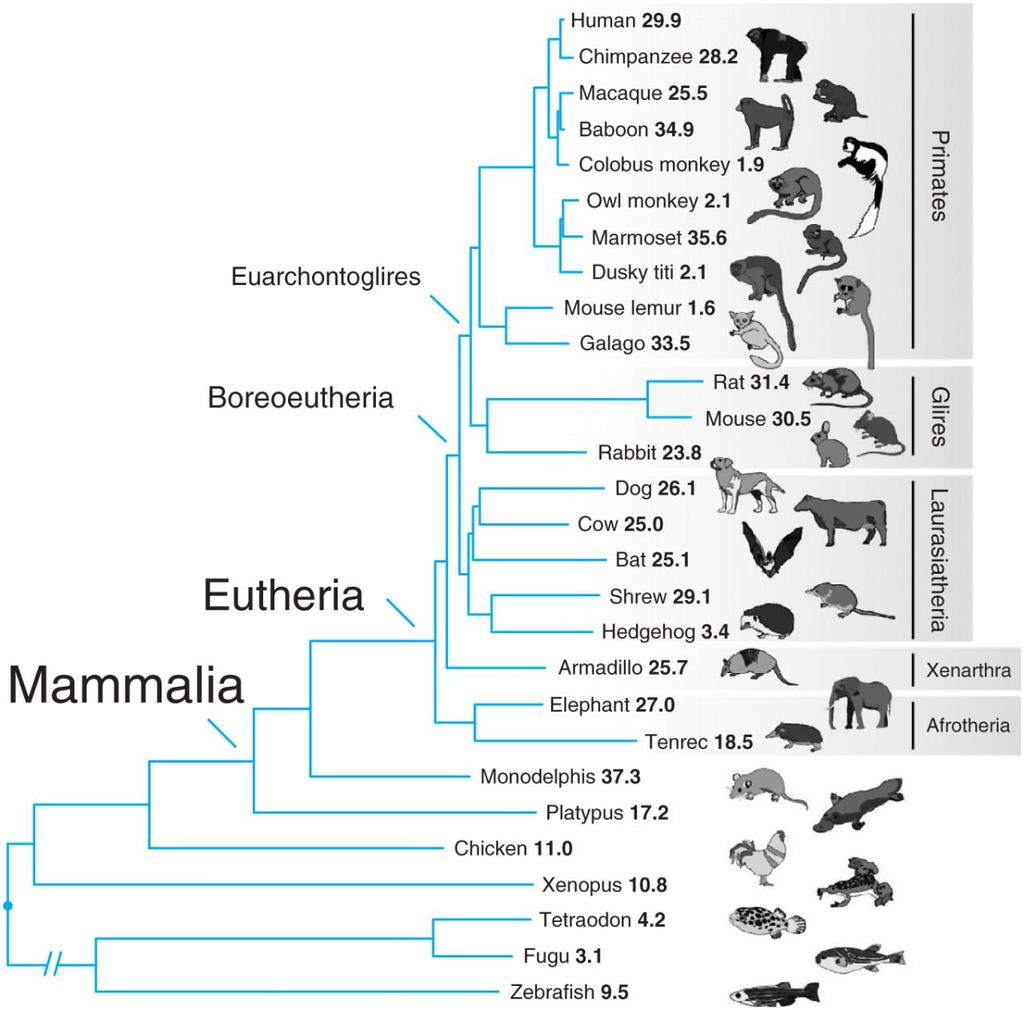
3 Drzewo filogenetyczne

4 Kserokopiarka zadanie: skopiować 300 stron. Co może pójść źle? 2x ta sama strona Opuszczona strona Nadmiarowa pusta strona Strona do góry nogami Przekrzywiona strona, część bez tekstu
5 Typowe mutacje w genach Substytucja Delecja Insercja Indel (przerwa) Przykładowy Efekt - przykłady Pojedynczy nukleotyd (Single Nucleotide Polymorphism, SNP) Mikrosatelity (Variable Number Tandem Repeats, VNTR) Minisatelity (Short Tandem Repeat Polymorphism, STRP) Pseudogeny
6
7 Homologia vs. podobieństwo sekwencji Czym to się różni?
8 Kiedy możliwa homologia? Z 1 mln przeanalizowanych białek na poziomie struktury 3D i funkcji wynikło, że 90% par sekwencji białkowych o podobieństwie >30% (na poziomie sekwencji, cała długość) wykazuje podobieństwo strukturalne (B. Rost) 30% - granica redundancji (nadmiarowości) 20-30% - szara strefa (10% homologów) 20% - przy takim nałożeniu nie można zakładać homologii (przy braku wyraźnych innych przesłanek)
9 Dopasowanie sekwencji Co można osiągnąć? Pokrewieństwo (homologia) i drzewo filogenetyczna Geny (duże podobieństwo u różnych gatunków) Obszary bardzo konserwatywne (ważne)
10
11 Podobieństwo sekwencji jak to ocenić?
12 Porównanie różnych sekwencji T O J E S T T A S E K W E N C J A T A M T A J E S T T E Z S E K W E N C J A T O J E S T T A S E K W E N C J A T A M T A J E S T T E Z S E K W E N C J A
13
14 Podobieństwo sekwencji powtórzenia liter
15 Podobieństwo sekwencji powtórzenia liter
16 Przykłady DotPlot różnych sekwencji identyczna substytucja Insercje-delecje
17
18
19
20
21 Geny zawierające powtórzenia Geny BRCA1-chromosom 17 i Gen BRCA2-chromosom 13 (wrażliwość na raka piersi), który zawiera wielokrotne powtórzenie krótkich odcinków (BRC- 39 aminkokwasów)

22 DotPlot przy częstych powtórzeniach Dopasowanie genu BRCA2
23 Dotplot tylko wizualizacja Spośród różnych ścieżek przekątniowych chcemy wybrać tylko jedną. Czy to jest jednoznaczne?
24

25 Szukanie optymalnej ścieżki 37 Programowanie dynamiczne Znajduje optymalną wartość funkcji celu dla całego zagadnienia rozwiązując podproblemy od najmniejszego do największego i zapisując optymalne wartości w tablicy. Pozwala to zastąpić wywołania rekurencyjne odwołaniami do odpowiednich komórek wspomnianej tablicy i gwarantuje, że każdy podproblem jest rozwiązywany tylko raz. wynik: km
26 Metoda optymalnej ścieżki Algorytm globalnego dopasowania sekwencji Needlemana-Wunscha (1970) Przykład programowania dynamicznego. Pierwszy w bioinformatyce
27 Programowanie dynamiczne Pełna przestrzeń możliwych dopasowań pomiędzy 2 białkami o długości 1000 aa to 10 (3*20) możliwych dopasowań Algorytm podzielony na podproblemy Efektywny, nie przeszukujemy bezładnie przestrzeni możliwych rozwiązań
28 Dopasowanie par (sekwencji) C A T W A L K C O W A R D
29 Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X
30 Schemat punktowania If przerwa (ruch nie po skosie) Else s= s-1 (liniowa kara) If a==b Else s=s+1 s=s+0 Wynik: C A T W A L K C Globalne dopasowanie O W A R D C 1 0 C A T W A L K O 0 W 1 A 2 R 2 D 2 2 (wynik sumowania:+2)
31 Dopasowanie globalne (Needleman & Wunsch) Znajdowanie optymalnej ścieżki 2 najlepsze dopasowania: C A T W A L K C O W A R D : +2 C A T W A L K C O - W A R D : +2 - C A T W A L K C O W A R D
32
33 Różne kary za przerwy (gap penalty) A) wysoka, B) niska
34 Dopasowanie lokalne sekwencji: Smith & Waterman Schemat score = 0; If (gap = = true) Else score=score - 1; If (letter1 = = letter2) score=score + 1; Else If (score<0) score=score - 0.5; score=0; Cofnij ścieżką od największej wartości aż do zera - C A T W A L K C 0 O 0 W 0 A 0 R 0 D 0
35 Smith & Waterman Global alignment: C A T W A L K C O W A R D or C A T W A L K C O - W A R D Local alignment: CATWALK COWARD - C A T W A L K C O W A R D
36 Globalne vs. lokalne
37
38 Dopasuj samodzielnie - C T T A G A - 0 G T A A 3 rozwiązania: CTTAGA G-TA-A CTTAGA GT-A-A CTTAGA -GTA-A
39 Needleman & Wunsch C A T W A L K C 1 0 O 0 W 1 A R D
40 Needleman & Wunsch C A T W A L K C 1 0 O 0 W 1 0 A 0 2 R D
41 Needleman & Wunsch C A T W A L K C 1 0 O 0 W A 0 2 R -1? D
42 Needleman & Wunsch C A T W A L K C 1 0 O 0 W A R D
43 Needleman & Wunsch C A T W A L K C 1 0 O 0 W A R D
44 Needleman & Wunsch Najlepsze dopasowanie do tyłu C A T W A L K Rozpocznij od najwyższego wyniku z prawa lub z dołu C 1 0 O 0 W A R D
45 Needleman & Wunsch Najlepsze dopasowanie do tyłu C A T W A L K Rozpocznij od najwyższego wyniku z prawa lub z dołu Cofaj się po strzałkach, do tyłu Może być kilka dróg! C 1 0 O 0 W A R D
46 Różne kary za przerwy (gap penalty) A) wysoka, B) niska
47 Dopasowanie lokalne sekwencji: Smith & Waterman Schemat score = 0; If (gap = = true) Else score=score - 1; If (letter1 = = letter2) score=score + 1; Else If (score<0) score=score - 0.5; score=0; Cofnij ścieżką od największej wartości aż do zera - C A T W A L K C 0 O 0 W 0 A 0 R 0 D 0
48 Smith & Waterman Global alignment: C A T W A L K C O W A R D or C A T W A L K C O - W A R D Local alignment: CATWALK COWARD - C A T W A L K C O W A R D

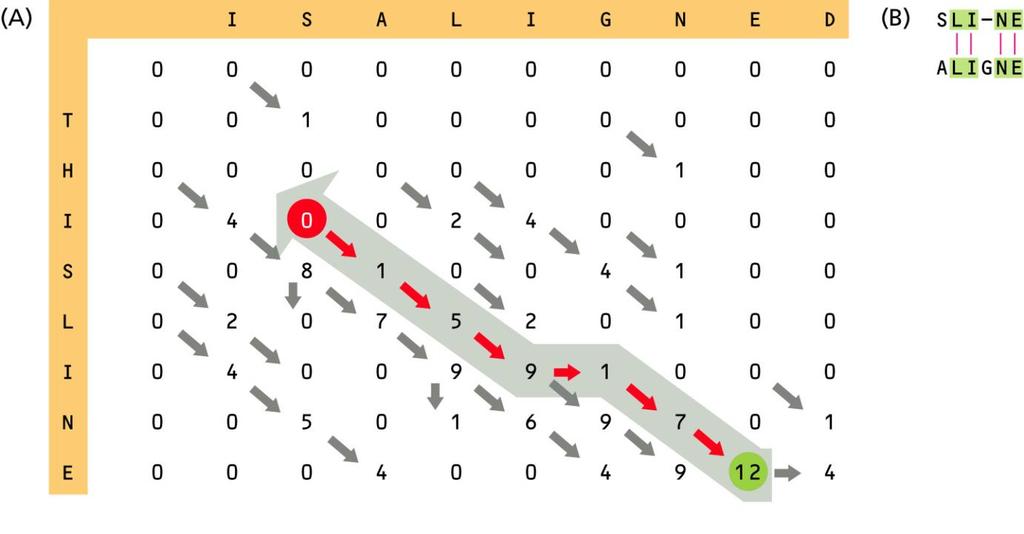
49 Globalne vs. lokalne
50 Dopasuj samodzielnie - R E D C E D K L - 0 A C E D E C A D E
Wykład 5 Dopasowywanie lokalne
 Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Wykład 5 Dopasowywanie lokalne Dopasowanie par (sekwencji) Dopasowanie globalne C A T W A L K C A T W A L K C O W A R D C X X O X W X A X R X D X Globalne dopasowanie Schemat punktowania (uproszczony)
Dopasowanie sekwencji (sequence alignment)
") Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
Co to jest alignment? Dopasowanie sekwencji (sequence alignment) Alignment jest sposobem dopasowania struktur pierwszorzędowych DNA, RNA lub białek do zidentyfikowanych regionów w celu określenia podobieństwa;
PRZYRÓWNANIE SEKWENCJI
 http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
http://theta.edu.pl/ Podstawy Bioinformatyki III PRZYRÓWNANIE SEKWENCJI 1 Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych), które posiadają
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (19, 26.X.2010) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Przyrównanie sekwencji. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
Przyrównanie sekwencji Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Sequence alignment - przyrównanie sekwencji Poszukiwanie ciągów znaków (zasad nukleotydowych lub reszt aminokwasowych),
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Dopasowanie sekwencji Sequence alignment. Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012)
") Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
Dopasowanie sekwencji Sequence alignment Bioinformatyka, wykłady 3 i 4 (16, 23.X.2012) krzysztof_pawlowski@sggw.pl terminologia alignment 33000 dopasowanie sekwencji 119 uliniowienie sekwencji 82 uliniowianie
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI
 PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
PODSTAWY BIOINFORMATYKI WYKŁAD 4 DOPASOWANIE SEKWENCJI DOPASOWANIE SEKWENCJI 1. Dopasowanie sekwencji - definicja 2. Wizualizacja dopasowania sekwencji 3. Miary podobieństwa sekwencji 4. Przykłady programów
Dopasowania par sekwencji DNA
 Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Dopasowania par sekwencji DNA Tworzenie uliniowień (dopasowań, tzw. alignmentów ) par sekwencji PSA Pairwise Sequence Alignment Dopasowania globalne i lokalne ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA
Bioinformatyka. (wykład monograficzny) wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM
 wykład 5. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM") Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Bioinformatyka (wykład monograficzny) wykład 5. E. Banachowicz Zakład Biofizyki Molekularnej IF UM http://www.amu.edu.pl/~ewas lgorytmy macierze punktowe (DotPlot) programowanie dynamiczne metody heurystyczne
Dopasowywanie sekwencji (ang. sequence alignment) Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania
 Metody dopasowywania sekwencji. Homologia a podobieństwo sekwencji. Rodzaje dopasowania") Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Wprowadzenie do Informatyki Biomedycznej Wykład 2: Metody dopasowywania sekwencji Wydział Informatyki PB Dopasowywanie sekwencji (ang. sequence alignment) Dopasowywanie (przyrównywanie) sekwencji polega
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek narodziła się nowa dyscyplina nauki ewolucja molekularna Ewolucja molekularna
Dopasowanie par sekwencji
 BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
BIOINFORMTYK edycja 2016 / 2017 wykład 3 Dopasowanie par sekwencji dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Idea i cele dopasowania sekwencji 2. Definicje
Porównywanie i dopasowywanie sekwencji
 Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Porównywanie i dopasowywanie sekwencji Związek bioinformatyki z ewolucją Wraz ze wzrostem dostępności sekwencji DNA i białek pojawiła się nowa możliwość śledzenia ewolucji na poziomie molekularnym Ewolucja
Przyrównywanie sekwencji
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański Przyrównywanie sekwencji 1. Porównywanie sekwencji wprowadzenie Sekwencje porównujemy po to, aby
dopasowanie sekwencji Porównywanie sekwencji Etapy dopasowywania sekwencji Homologia, podobieństwo i analogia
 Porównywanie sekwencji Homologia, podobieństwo i analogia dopasowanie sekwencji Dopasowanie/porównywanie Uliniowienie Alignment W bioinformatyce, dopasowanie sekwencji jest sposobem dopasowania struktur
Porównywanie sekwencji Homologia, podobieństwo i analogia dopasowanie sekwencji Dopasowanie/porównywanie Uliniowienie Alignment W bioinformatyce, dopasowanie sekwencji jest sposobem dopasowania struktur
Bioinformatyka. Porównywanie sekwencji
 Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Bioinformatyka Wykład 5 E. Banachowicz Zakład Biofizyki Molekularnej IF UM 1 http://www.amu.edu.pl/~ewas Porównywanie sekwencji Pierwsze pytanie biologa molekularnego, kiedy odkryje nową sekwencję: zy
Spis treści. Przedmowa... XI. Wprowadzenie i biologiczne bazy danych. 1 Wprowadzenie... 3. 2 Wprowadzenie do biologicznych baz danych...
 Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
Przedmowa... XI Część pierwsza Wprowadzenie i biologiczne bazy danych 1 Wprowadzenie... 3 Czym jest bioinformatyka?... 5 Cele... 5 Zakres zainteresowań... 6 Zastosowania... 7 Ograniczenia... 8 Przyszłe
prof. dr hab. inż. Marta Kasprzak Instytut Informatyki, Politechnika Poznańska Dopasowanie sekwencji
 Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka wykład 5: dopasowanie sekwencji prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie podobieństwa sekwencji stanowi podstawę wielu gałęzi
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Często dopasować chcemy nie dwie sekwencje ale kilkanaście lub więcej 2 Istnieją dokładne algorytmy, lecz są one niewydajne
Motywy i podobieństwo
 Motywy i podobieństwo Całość funkcja Modularna budowa białek Elementy składowe czyli miejsca wiązania, domeny 1 Motywy Motyw jest opisem określonej części trójwymiarowej struktury zawierającym charakterystyczny
Motywy i podobieństwo Całość funkcja Modularna budowa białek Elementy składowe czyli miejsca wiązania, domeny 1 Motywy Motyw jest opisem określonej części trójwymiarowej struktury zawierającym charakterystyczny
Wstęp do programowania
 Wstęp do programowania Programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. X Jesień 2013 1 / 21 Dziel i zwyciężaj przypomnienie 1 Podział problemu na 2 lub
Wstęp do programowania Programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2013 P. Daniluk(Wydział Fizyki) WP w. X Jesień 2013 1 / 21 Dziel i zwyciężaj przypomnienie 1 Podział problemu na 2 lub
Programowanie dynamiczne cz. 2
 Programowanie dynamiczne cz. 2 Wykład 7 16 kwietnia 2019 (Wykład 7) Programowanie dynamiczne cz. 2 16 kwietnia 2019 1 / 19 Outline 1 Mnożenie ciągu macierzy Konstruowanie optymalnego rozwiązania 2 Podstawy
Programowanie dynamiczne cz. 2 Wykład 7 16 kwietnia 2019 (Wykład 7) Programowanie dynamiczne cz. 2 16 kwietnia 2019 1 / 19 Outline 1 Mnożenie ciągu macierzy Konstruowanie optymalnego rozwiązania 2 Podstawy
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA
 PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
PODSTAWY BIOINFORMATYKI WYKŁAD 5 ANALIZA FILOGENETYCZNA ANALIZA FILOGENETYCZNA 1. Wstęp - filogenetyka 2. Struktura drzewa filogenetycznego 3. Metody konstrukcji drzewa 4. Etapy konstrukcji drzewa filogenetycznego
Wstęp do programowania
 Wstęp do programowania Algorytmy zachłanne, programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2014 P. Daniluk(Wydział Fizyki) WP w. IX Jesień 2014 1 / 26 Algorytmy zachłanne Strategia polegająca
Wstęp do programowania Algorytmy zachłanne, programowanie dynamiczne Paweł Daniluk Wydział Fizyki Jesień 2014 P. Daniluk(Wydział Fizyki) WP w. IX Jesień 2014 1 / 26 Algorytmy zachłanne Strategia polegająca
Dopasowanie sekwencji c.d. Sequence alignment. Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl
 krzysztof_pawlowski@sggw.pl") Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl dopasowanie - metody dopasowanie par sekwencji: Macierz punktów - dot matrix, dotplot Programowanie
Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (16.XI.2010) krzysztof_pawlowski@sggw.pl dopasowanie - metody dopasowanie par sekwencji: Macierz punktów - dot matrix, dotplot Programowanie
Wstęp do Biologii Obliczeniowej
 Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Wstęp do Biologii Obliczeniowej Zagadnienia na kolokwium Bartek Wilczyński 5. czerwca 2018 Sekwencje DNA i grafy Sekwencje w biologii, DNA, RNA, białka, alfabety, transkrypcja DNA RNA, translacja RNA białko,
Metody badania polimorfizmu/mutacji DNA. Aleksandra Sałagacka Pracownia Diagnostyki Molekularnej i Farmakogenomiki Uniwersytet Medyczny w Łodzi
 Metody badania polimorfizmu/mutacji DNA Aleksandra Sałagacka Pracownia Diagnostyki Molekularnej i Farmakogenomiki Uniwersytet Medyczny w Łodzi Mutacja Mutacja (łac. mutatio zmiana) - zmiana materialnego
Metody badania polimorfizmu/mutacji DNA Aleksandra Sałagacka Pracownia Diagnostyki Molekularnej i Farmakogenomiki Uniwersytet Medyczny w Łodzi Mutacja Mutacja (łac. mutatio zmiana) - zmiana materialnego
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
Algorytmy kombinatoryczne w bioinformatyce
 lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
lgorytmy kombinatoryczne w bioinformatyce wykład 4: dopasowanie sekwencj poszukiwanie motywów prof. dr hab. inż. Marta Kasprzak Instytut Informatyk Politechnika Poznańska Dopasowanie sekwencji Badanie
DHPLC. Denaturing high performance liquid chromatography. Wiktoria Stańczyk Zofia Kołeczko
 DHPLC Denaturing high performance liquid chromatography Wiktoria Stańczyk Zofia Kołeczko Mini-słowniczek SNP (Single Nucleotide Polymorphism) - zmienność sekwencji DNA; HET - analiza heterodupleksów; HPLC
DHPLC Denaturing high performance liquid chromatography Wiktoria Stańczyk Zofia Kołeczko Mini-słowniczek SNP (Single Nucleotide Polymorphism) - zmienność sekwencji DNA; HET - analiza heterodupleksów; HPLC
WIZUALIZACJA ALGORYTMÓW OPTYMALNEGO DOPASOWANIA SEKWENCJI NUKLEOTYDÓW I AMINOKWASÓW
 STUDIA INFORMATICA 2011 Volume 32 Number 2A (96) Adam SKOWRON, Dariusz MROZEK Politechnika Śląska, Instytut Informatyki WIZUALIZACJA ALGORYTMÓW OPTYMALNEGO DOPASOWANIA SEKWENCJI NUKLEOTYDÓW I AMINOKWASÓW
STUDIA INFORMATICA 2011 Volume 32 Number 2A (96) Adam SKOWRON, Dariusz MROZEK Politechnika Śląska, Instytut Informatyki WIZUALIZACJA ALGORYTMÓW OPTYMALNEGO DOPASOWANIA SEKWENCJI NUKLEOTYDÓW I AMINOKWASÓW
Bioinformatyka. Ocena wiarygodności dopasowania sekwencji.
 Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
Bioinformatyka Ocena wiarygodności dopasowania sekwencji www.michalbereta.pl Załóżmy, że mamy dwie sekwencje, które chcemy dopasować i dodatkowo ocenić wiarygodność tego dopasowania. Interesujące nas pytanie
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI JOANNA SZYDA MAGDALENA FRĄSZCZAK MAGDA MIELCZAREK WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Dopasowanie sekwencji c.d. Sequence alignment. Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl
 krzysztof_pawlowski@sggw.pl") Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl Dopasowanie sekwencji - znaczenie Podobieństwo porównywanych sekwencji (similarity) może świadczyć
Dopasowanie sekwencji c.d. Sequence alignment Bioinformatyka, wykład 5 (6.XI.2012) krzysztof_pawlowski@sggw.pl Dopasowanie sekwencji - znaczenie Podobieństwo porównywanych sekwencji (similarity) może świadczyć
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
FUNKCJA REKURENCYJNA. function s(n:integer):integer; begin if (n>1) then s:=n*s(n-1); else s:=1; end;
:integer; begin if (n>1) then s:=n*s(n-1); else s:=1; end;") Rekurencja Wykład: rekursja, funkcje rekurencyjne, wywołanie samej siebie, wyznaczanie poszczególnych liczb Fibonacciego, potęgowanie, algorytm Euklidesa REKURENCJA Rekurencja (z łac. recurrere), zwana
Rekurencja Wykład: rekursja, funkcje rekurencyjne, wywołanie samej siebie, wyznaczanie poszczególnych liczb Fibonacciego, potęgowanie, algorytm Euklidesa REKURENCJA Rekurencja (z łac. recurrere), zwana
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl Zasady zaliczenia przedmiotu Kolokwia (3 4 ) Ocena aktywności i przygotowania Obecność Literatura, materiały Bioinformatyka i ewolucja
Homologia, podobieństwo i analogia
 Porównywanie sekwencji Homologia, podobieństwo i analogia Homologi Ortologi homologiczne geny, których rozdzielenie nastąpiło na skutek specjacji, czyli rozdzielenia gatunków, lub rzadziej horyzontalnego
Porównywanie sekwencji Homologia, podobieństwo i analogia Homologi Ortologi homologiczne geny, których rozdzielenie nastąpiło na skutek specjacji, czyli rozdzielenia gatunków, lub rzadziej horyzontalnego
1. Nagłówek funkcji: int funkcja(void); wskazuje na to, że ta funkcja. 2. Schemat blokowy przedstawia algorytm obliczania
; wskazuje na to, że ta funkcja. 2. Schemat blokowy przedstawia algorytm obliczania") 1. Nagłówek funkcji: int funkcja(void); wskazuje na to, że ta funkcja nie ma parametru i zwraca wartość na zewnątrz. nie ma parametru i nie zwraca wartości na zewnątrz. ma parametr o nazwie void i zwraca
1. Nagłówek funkcji: int funkcja(void); wskazuje na to, że ta funkcja nie ma parametru i zwraca wartość na zewnątrz. nie ma parametru i nie zwraca wartości na zewnątrz. ma parametr o nazwie void i zwraca
D: Dopasowanie sekwencji. Programowanie dynamiczne
 D: Dopasowanie sekwencji. Programowanie dynamiczne Problem: jak porównywać sekwencje DNA? Czy te sekwencje są podobne? Jeśli są podobne, to jak mierzyć to podobieństwo? Odpowiedzi są kluczowe dla konstrukcji
D: Dopasowanie sekwencji. Programowanie dynamiczne Problem: jak porównywać sekwencje DNA? Czy te sekwencje są podobne? Jeśli są podobne, to jak mierzyć to podobieństwo? Odpowiedzi są kluczowe dla konstrukcji
Porównywanie sekwencji białkowych
 Bioinformatyka -9 Bioinformatyka Wykład 4. E. Banachowicz Zakład Biofizyki Molekularnej http://www.amu.edu.pl/~ewas Porównywanie sekwencji białkowych Wykład 4, Bioinformatyka -9 Porównywanie sekwencji
Bioinformatyka -9 Bioinformatyka Wykład 4. E. Banachowicz Zakład Biofizyki Molekularnej http://www.amu.edu.pl/~ewas Porównywanie sekwencji białkowych Wykład 4, Bioinformatyka -9 Porównywanie sekwencji
Bioinformatyka 2 (BT172) Progresywne metody wyznaczania MSA: T-coffee
 Progresywne metody wyznaczania MSA: T-coffee") Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Bioinformatyka 2 (BT172) Wykład 5 Progresywne metody wyznaczania MSA: T-coffee Krzysztof Murzyn 14.XI.2005 PLAN WYKŁADU Ostatnio : definicje, zastosowania MSA, złożoność obliczeniowa algorytmu wyznaczania
Ćwiczenie 5/6. Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST.
 Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Ćwiczenie 5/6 Informacja genetyczna i geny u różnych grup organizmów. Porównywanie sekwencji nukleotydowych w bazie NCBI z wykorzystaniem BLAST. Prof. dr hab. Roman Zieliński 1. Informacja genetyczna u
Algorytmy i str ruktury danych. Metody algorytmiczne. Bartman Jacek
 Algorytmy i str ruktury danych Metody algorytmiczne Bartman Jacek jbartman@univ.rzeszow.pl Metody algorytmiczne - wprowadzenia Znamy strukturę algorytmów Trudność tkwi natomiast w podaniu metod służących
Algorytmy i str ruktury danych Metody algorytmiczne Bartman Jacek jbartman@univ.rzeszow.pl Metody algorytmiczne - wprowadzenia Znamy strukturę algorytmów Trudność tkwi natomiast w podaniu metod służących
Programowanie dynamiczne i algorytmy zachłanne
 Programowanie dynamiczne i algorytmy zachłanne Tomasz Głowacki tglowacki@cs.put.poznan.pl Zajęcia finansowane z projektu "Rozwój i doskonalenie kształcenia na Politechnice Poznańskiej w zakresie technologii
Programowanie dynamiczne i algorytmy zachłanne Tomasz Głowacki tglowacki@cs.put.poznan.pl Zajęcia finansowane z projektu "Rozwój i doskonalenie kształcenia na Politechnice Poznańskiej w zakresie technologii
Schemat programowania dynamicznego (ang. dynamic programming)
") Schemat programowania dynamicznego (ang. dynamic programming) Jest jedną z metod rozwiązywania problemów optymalizacyjnych. Jej twórcą (1957) był amerykański matematyk Richard Ernest Bellman. Schemat ten
Schemat programowania dynamicznego (ang. dynamic programming) Jest jedną z metod rozwiązywania problemów optymalizacyjnych. Jej twórcą (1957) był amerykański matematyk Richard Ernest Bellman. Schemat ten
Struktury danych i złożoność obliczeniowa Wykład 2. Prof. dr hab. inż. Jan Magott
 Struktury danych i złożoność obliczeniowa Wykład 2. Prof. dr hab. inż. Jan Magott Metody konstrukcji algorytmów: Siłowa (ang. brute force), Dziel i zwyciężaj (ang. divide-and-conquer), Zachłanna (ang.
Struktury danych i złożoność obliczeniowa Wykład 2. Prof. dr hab. inż. Jan Magott Metody konstrukcji algorytmów: Siłowa (ang. brute force), Dziel i zwyciężaj (ang. divide-and-conquer), Zachłanna (ang.
Dr hab.n.med. Renata Jacewicz
 GENOM CZŁOWIEKA >99 % 0,05%(100MtDNA) 65% Dr hab.n.med. Renata Jacewicz Kierownik Pracowni Genetyki Medycznej i Sądowej 3% 32% 2013 Pracownia Genetyki Medycznej i Sądowej ZMS Dojrzałe erytrocyty, trzony
GENOM CZŁOWIEKA >99 % 0,05%(100MtDNA) 65% Dr hab.n.med. Renata Jacewicz Kierownik Pracowni Genetyki Medycznej i Sądowej 3% 32% 2013 Pracownia Genetyki Medycznej i Sądowej ZMS Dojrzałe erytrocyty, trzony
Statystyczna analiza danych
 Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
Statystyczna analiza danych ukryte modele Markowa, zastosowania Anna Gambin Instytut Informatyki Uniwersytet Warszawski plan na dziś Ukryte modele Markowa w praktyce modelowania rodzin białek multiuliniowienia
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI
 ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
ANALIZA DANYCH POCHODZĄCYCH Z SEKWENCJONOWANIA NASTĘPNEJ GENERACJI Joanna Szyda Magdalena Frąszczak Magda Mielczarek WSTĘP 1. Katedra Genetyki 2. Pracownia biostatystyki 3. Projekty NGS 4. Charakterystyka
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing
 Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
Sekwencjonowanie Nowej Generacji ang. Next Generation Sequencing Wykład 7 Etapy analizy NGS Dr Wioleta Drobik-Czwarno Etapy analizy NGS Kontrola jakości surowych danych (format fastq) Jakość odczytów,
października 2013: Elementarz biologii molekularnej. Wykład nr 2 BIOINFORMATYKA rok II
 10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
10 października 2013: Elementarz biologii molekularnej www.bioalgorithms.info Wykład nr 2 BIOINFORMATYKA rok II Komórka: strukturalna i funkcjonalne jednostka organizmu żywego Jądro komórkowe: chroniona
Wykład Bioinformatyka 2012-09-24. Bioinformatyka. Wykład 7. E. Banachowicz. Zakład Biofizyki Molekularnej IF UAM. Ewolucyjne podstawy Bioinformatyki
 Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
Bioinformatyka Wykład 7 E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas 1 Plan Bioinformatyka Ewolucyjne podstawy Bioinformatyki Filogenetyka Bioinformatyczne narzędzia
Dr hab.n.med. Renata Jacewicz
 GENOM CZŁOWIEKA >99,999% 0,0005% 65% Dr hab.n.med. Renata Jacewicz Kierownik Pracowni Genetyki Medycznej i Sądowej 3% 32% 2013 Pracownia Genetyki Medycznej i Sądowej ZMS Dojrzałe erytrocyty, trzony włosów
GENOM CZŁOWIEKA >99,999% 0,0005% 65% Dr hab.n.med. Renata Jacewicz Kierownik Pracowni Genetyki Medycznej i Sądowej 3% 32% 2013 Pracownia Genetyki Medycznej i Sądowej ZMS Dojrzałe erytrocyty, trzony włosów
Bioinformatyka Laboratorium, 30h. Michał Bereta
 Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Bioinformatyka Laboratorium, 30h Michał Bereta mbereta@pk.edu.pl www.michalbereta.pl 1 Wyszukiwanie sekwencji Jak wyszukad z baz danych bioinformatycznych sekwencje podobne do sekwencji zadanej (ang. query
Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2
 ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
ALEKSANDRA ŚWIERCZ Co to jest transkryptom? A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH 2 Ekspresja genów http://genome.wellcome.ac.uk/doc_wtd020757.html A. Świercz ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
PoniŜej znajdują się pytania z egzaminów zawodowych teoretycznych. Jest to materiał poglądowy.
 PoniŜej znajdują się pytania z egzaminów zawodowych teoretycznych. Jest to materiał poglądowy. 1. Instrukcję case t of... w przedstawionym fragmencie programu moŝna zastąpić: var t : integer; write( Podaj
PoniŜej znajdują się pytania z egzaminów zawodowych teoretycznych. Jest to materiał poglądowy. 1. Instrukcję case t of... w przedstawionym fragmencie programu moŝna zastąpić: var t : integer; write( Podaj
BIOINFORMATYKA. edycja 2016 / wykład 11 RNA. dr Jacek Śmietański
 BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
BIOINFORMATYKA edycja 2016 / 2017 wykład 11 RNA dr Jacek Śmietański jacek.smietanski@ii.uj.edu.pl http://jaceksmietanski.net Plan wykładu 1. Rola i rodzaje RNA 2. Oddziaływania wewnątrzcząsteczkowe i struktury
Bioinformatyka. Rodzaje Mutacji
 Bioinformatyka (wykład monograficzny) wykład 3. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas Rodzaje Mutacji zmienność sekwencji (sequence variation) mutacje polimorfizm
Bioinformatyka (wykład monograficzny) wykład 3. E. Banachowicz Zakład Biofizyki Molekularnej IF UAM http://www.amu.edu.pl/~ewas Rodzaje Mutacji zmienność sekwencji (sequence variation) mutacje polimorfizm
Programowanie dynamiczne (optymalizacja dynamiczna).
.") Programowanie dynamiczne (optymalizacja dynamiczna). W wielu przypadkach zadania, których złożoność wynikająca z pełnego przeglądu jest duża (zwykle wyk ładnicza) można rozwiązać w czasie wielomianowym
Programowanie dynamiczne (optymalizacja dynamiczna). W wielu przypadkach zadania, których złożoność wynikająca z pełnego przeglądu jest duża (zwykle wyk ładnicza) można rozwiązać w czasie wielomianowym
Algorytmy i struktury danych. Co dziś? Tytułem przypomnienia metoda dziel i zwyciężaj. Wykład VIII Elementarne techniki algorytmiczne
 Algorytmy i struktury danych Wykład VIII Elementarne techniki algorytmiczne Co dziś? Algorytmy zachłanne (greedyalgorithms) 2 Tytułem przypomnienia metoda dziel i zwyciężaj. Problem można podzielić na
Algorytmy i struktury danych Wykład VIII Elementarne techniki algorytmiczne Co dziś? Algorytmy zachłanne (greedyalgorithms) 2 Tytułem przypomnienia metoda dziel i zwyciężaj. Problem można podzielić na
MODEL ODPOWIEDZI I SCHEMAT PUNKTOWANIA ZADAŃ ETAP SZKOLNY KONKURSU GEOGRAFICZNEGO
 MODEL ODPOWIEDZI I SCHEMAT PUNKTOWANIA ZADAŃ ETAP SZKOLNY KONKURSU GEOGRAFICZNEGO Nr zadania 1. 2. Przewidywana odpowiedź Punktacja Zasady oceniania Skala mapy Ali: C. 1:50 000 Skala mapy Izy: H. 1:200
MODEL ODPOWIEDZI I SCHEMAT PUNKTOWANIA ZADAŃ ETAP SZKOLNY KONKURSU GEOGRAFICZNEGO Nr zadania 1. 2. Przewidywana odpowiedź Punktacja Zasady oceniania Skala mapy Ali: C. 1:50 000 Skala mapy Izy: H. 1:200
GENOMIKA. MAPOWANIE GENOMÓW MAPY GENOMICZNE
 GENOMIKA. MAPOWANIE GENOMÓW MAPY GENOMICZNE Bioinformatyka, wykład 3 (21.X.2008) krzysztof_pawlowski@sggw.waw.pl tydzień temu Gen??? Biologiczne bazy danych historia Biologiczne bazy danych najważniejsze
GENOMIKA. MAPOWANIE GENOMÓW MAPY GENOMICZNE Bioinformatyka, wykład 3 (21.X.2008) krzysztof_pawlowski@sggw.waw.pl tydzień temu Gen??? Biologiczne bazy danych historia Biologiczne bazy danych najważniejsze
operacje porównania, a jeśli jest to konieczne ze względu na złe uporządkowanie porównywanych liczb zmieniamy ich kolejność, czyli przestawiamy je.
 Problem porządkowania zwanego również sortowaniem jest jednym z najważniejszych i najpopularniejszych zagadnień informatycznych. Dane: Liczba naturalna n i ciąg n liczb x 1, x 2,, x n. Wynik: Uporządkowanie
Problem porządkowania zwanego również sortowaniem jest jednym z najważniejszych i najpopularniejszych zagadnień informatycznych. Dane: Liczba naturalna n i ciąg n liczb x 1, x 2,, x n. Wynik: Uporządkowanie
Przykładowe B+ drzewo
 Przykładowe B+ drzewo 3 8 1 3 7 8 12 Jak obliczyć rząd indeksu p Dane: rozmiar klucza V, rozmiar wskaźnika do bloku P, rozmiar bloku B, liczba rekordów w indeksowanym pliku danych r i liczba bloków pliku
Przykładowe B+ drzewo 3 8 1 3 7 8 12 Jak obliczyć rząd indeksu p Dane: rozmiar klucza V, rozmiar wskaźnika do bloku P, rozmiar bloku B, liczba rekordów w indeksowanym pliku danych r i liczba bloków pliku
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW
 PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
PODSTAWY BIOINFORMATYKI 8 DOPASOWYWANIE SEKWENCJI AMINOKWASÓW DOPASOWYWANIE SEKWENCJI 1. Miary podobieństwa sekwencji aminokwasów 2. Zastosowanie programów: CLUSTAL OMEGA BLAST Copyright 2013, Joanna Szyda
TEORETYCZNE PODSTAWY INFORMATYKI
 1 TEORETYCZNE PODSTAWY INFORMATYKI WFAiS UJ, Informatyka Stosowana I rok studiów, I stopień Wykład 2 2 Problemy algorytmiczne Klasy problemów algorytmicznych Liczby Fibonacciego Przeszukiwanie tablic Największy
1 TEORETYCZNE PODSTAWY INFORMATYKI WFAiS UJ, Informatyka Stosowana I rok studiów, I stopień Wykład 2 2 Problemy algorytmiczne Klasy problemów algorytmicznych Liczby Fibonacciego Przeszukiwanie tablic Największy
Na czym skończyliśmy BLACK BOX. Sekwencjonowanie polega na odczytaniu sekwencji liter DNA/RNA badanego fragmentu genomu
 ALEKSANDRA ŚWIERCZ Na czym skończyliśmy BLACK BOX AAATGCCTGCCCTGAAGGCCTGCGTA GTTTTGGGAGAAGACCCACGGATA AAGGTGTAGCCCCGTAGC GGGGGGTATTATTTATTTTATACCCAC.. ACAGGAUCGUUGGAUGGTGGGA. Sekwencjonowanie polega na
ALEKSANDRA ŚWIERCZ Na czym skończyliśmy BLACK BOX AAATGCCTGCCCTGAAGGCCTGCGTA GTTTTGGGAGAAGACCCACGGATA AAGGTGTAGCCCCGTAGC GGGGGGTATTATTTATTTTATACCCAC.. ACAGGAUCGUUGGAUGGTGGGA. Sekwencjonowanie polega na
Składnia funkcji i Rekurencja w języku Haskell
 Składnia funkcji i w języku Haskell Tomasz Ostrowski, Adrian Niechciał, Michał Workiewicz, Marcin Wilk 26 marca 2015 Składnia funkcji i w języku Haskell Spis treści Składnia funkcji Tomasz Ostrowski Adrian
Składnia funkcji i w języku Haskell Tomasz Ostrowski, Adrian Niechciał, Michał Workiewicz, Marcin Wilk 26 marca 2015 Składnia funkcji i w języku Haskell Spis treści Składnia funkcji Tomasz Ostrowski Adrian
Generator testów Bioinformatyka wer / 0 Strona: 1
 Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Przedmiot: Nazwa przedmiotu Nazwa testu: Bioinformatyka wer. 1.0.6 Nr testu 0 Klasa: V zaoczne WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Analiza porównawcza białek zwykle zaczyna się na badaniach
Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online
 Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
Techniki molekularne ćw. 5 1 z 13 Ćwiczenia nr 5. Wykorzystanie baz danych i narzędzi analitycznych dostępnych online I. Zasoby NCBI Strona: http://www.ncbi.nlm.nih.gov/ stanowi punkt startowy dla eksploracji
Konstruowanie drzew filogenetycznych. Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu
 Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Konstruowanie drzew filogenetycznych Magda Mielczarek Katedra Genetyki Uniwersytet Przyrodniczy we Wrocławiu Drzewa filogenetyczne ukorzenione i nieukorzenione binarność konstrukcji topologia (sposób rozgałęziana
Złożoność obliczeniowa zadania, zestaw 2
 Złożoność obliczeniowa zadania, zestaw 2 Określanie złożoności obliczeniowej algorytmów, obliczanie pesymistycznej i oczekiwanej złożoności obliczeniowej 1. Dana jest tablica jednowymiarowa A o rozmiarze
Złożoność obliczeniowa zadania, zestaw 2 Określanie złożoności obliczeniowej algorytmów, obliczanie pesymistycznej i oczekiwanej złożoności obliczeniowej 1. Dana jest tablica jednowymiarowa A o rozmiarze
MARKERY MIKROSATELITARNE
 MARKERY MIKROSATELITARNE Badania laboratoryjne prowadzone w Katedrze Genetyki i Ogólnej Hodowli Zwierząt SGGW w ramach monitoringu genetycznego wykorzystują analizę genetyczną markerów mikrosatelitarnych.
MARKERY MIKROSATELITARNE Badania laboratoryjne prowadzone w Katedrze Genetyki i Ogólnej Hodowli Zwierząt SGGW w ramach monitoringu genetycznego wykorzystują analizę genetyczną markerów mikrosatelitarnych.
Bioinformatyczne bazy danych - część 2. -przeszukiwanie baz danych -pobieranie danych
 Bioinformatyczne bazy danych - część 2 -przeszukiwanie baz danych -pobieranie danych Numery dostępowe baz danych (accession number) to ciąg liter i cyfr służących jako etykieta identyfikująca sekwencję
Bioinformatyczne bazy danych - część 2 -przeszukiwanie baz danych -pobieranie danych Numery dostępowe baz danych (accession number) to ciąg liter i cyfr służących jako etykieta identyfikująca sekwencję
Programowanie w VB Proste algorytmy sortowania
 Programowanie w VB Proste algorytmy sortowania Sortowanie bąbelkowe Algorytm sortowania bąbelkowego polega na porównywaniu par elementów leżących obok siebie i, jeśli jest to potrzebne, zmienianiu ich
Programowanie w VB Proste algorytmy sortowania Sortowanie bąbelkowe Algorytm sortowania bąbelkowego polega na porównywaniu par elementów leżących obok siebie i, jeśli jest to potrzebne, zmienianiu ich
Bioinformatyka wykład 10
 Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Bioinformatyka wykład 10 21.XII.2010 białkowa bioinformatyka strukturalna, c.d. krzysztof_pawlowski@sggw.pl 2011-01-17 1 Regiony nieuporządkowane disordered regions trudna definicja trudne do przewidzenia
Wprowadzenie do genetyki medycznej i sądowej
 Genetyka medyczno-sądowa Wprowadzenie do genetyki medycznej i sądowej Kierownik Pracowni Genetyki Medycznej i Sądowej Ustalanie tożsamości zwłok Identyfikacja sprawców przestępstw Identyfikacja śladów
Genetyka medyczno-sądowa Wprowadzenie do genetyki medycznej i sądowej Kierownik Pracowni Genetyki Medycznej i Sądowej Ustalanie tożsamości zwłok Identyfikacja sprawców przestępstw Identyfikacja śladów
Algorytmy przeszukiwania
 Algorytmy przeszukiwania Przeszukiwanie liniowe Algorytm stosowany do poszukiwania elementu w zbiorze, o którym nic nie wiemy. Aby mieć pewność, że nie pominęliśmy żadnego elementu zbioru przeszukujemy
Algorytmy przeszukiwania Przeszukiwanie liniowe Algorytm stosowany do poszukiwania elementu w zbiorze, o którym nic nie wiemy. Aby mieć pewność, że nie pominęliśmy żadnego elementu zbioru przeszukujemy
DNA musi współdziałać z białkami!
 DNA musi współdziałać z białkami! Specyficzność oddziaływań między DNA a białkami wiążącymi DNA zależy od: zmian konformacyjnych wzdłuż cząsteczki DNA zróżnicowania struktury DNA wynikającego z sekwencji
DNA musi współdziałać z białkami! Specyficzność oddziaływań między DNA a białkami wiążącymi DNA zależy od: zmian konformacyjnych wzdłuż cząsteczki DNA zróżnicowania struktury DNA wynikającego z sekwencji
W kierunku równoległej implementacji pakietu T-Coffee
 W kierunku równoległej implementacji pakietu T-Coffee Adrian Rospondek 1 1 Wydział Inżynierii Mechanicznej i Informatyki Kierunek Informatyka, Rok V a.rospondek@poczta.fm Streszczenie Artykuł ten prezentuje
W kierunku równoległej implementacji pakietu T-Coffee Adrian Rospondek 1 1 Wydział Inżynierii Mechanicznej i Informatyki Kierunek Informatyka, Rok V a.rospondek@poczta.fm Streszczenie Artykuł ten prezentuje
Klasa 2 INFORMATYKA. dla szkół ponadgimnazjalnych zakres rozszerzony. Założone osiągnięcia ucznia wymagania edukacyjne na. poszczególne oceny
 Klasa 2 INFORMATYKA dla szkół ponadgimnazjalnych zakres rozszerzony Założone osiągnięcia ucznia wymagania edukacyjne na poszczególne oceny Algorytmy 2 3 4 5 6 Wie, co to jest algorytm. Wymienia przykłady
Klasa 2 INFORMATYKA dla szkół ponadgimnazjalnych zakres rozszerzony Założone osiągnięcia ucznia wymagania edukacyjne na poszczególne oceny Algorytmy 2 3 4 5 6 Wie, co to jest algorytm. Wymienia przykłady
Algorytmy i struktury danych. Wykład 6 Tablice rozproszone cz. 2
 Algorytmy i struktury danych Wykład 6 Tablice rozproszone cz. 2 Na poprzednim wykładzie Wiele problemów wymaga dynamicznych zbiorów danych, na których można wykonywać operacje: wstawiania (Insert) szukania
Algorytmy i struktury danych Wykład 6 Tablice rozproszone cz. 2 Na poprzednim wykładzie Wiele problemów wymaga dynamicznych zbiorów danych, na których można wykonywać operacje: wstawiania (Insert) szukania
 Ś Ą Ś Ą Ś Ą Ą Ś Ą Ą ŚĆ Ą Ą Ś Ś ć ź ź Ń Ś Ą ć Ź Ą Ą Ś ć Ą Ą Ą Ś Ą ć Ą Ą ć Ą ć ć Ć Ź ć Ś Ź Ź ć Ź Ź ć Ź ź Ź Ś ź Ź ć ć Ń ź ć ć Ń Ć ź ć ć Ś ć ć ć Ź Ń ć Ź ć ć ź Ą Ś Ć Ź ź ź Ź ć ć Ś ź Ń ć ć ć ź Ą Ś Ń Ś ć ć Ź
Ś Ą Ś Ą Ś Ą Ą Ś Ą Ą ŚĆ Ą Ą Ś Ś ć ź ź Ń Ś Ą ć Ź Ą Ą Ś ć Ą Ą Ą Ś Ą ć Ą Ą ć Ą ć ć Ć Ź ć Ś Ź Ź ć Ź Ź ć Ź ź Ź Ś ź Ź ć ć Ń ź ć ć Ń Ć ź ć ć Ś ć ć ć Ź Ń ć Ź ć ć ź Ą Ś Ć Ź ź ź Ź ć ć Ś ź Ń ć ć ć ź Ą Ś Ń Ś ć ć Ź
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii
 Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Transgeneza - genetycznie zmodyfikowane oraganizmy 2. Medycyna i ochrona zdrowia 3. Genomika poznawanie genomów Przełom XX i
Możliwości współczesnej inżynierii genetycznej w obszarze biotechnologii 1. Transgeneza - genetycznie zmodyfikowane oraganizmy 2. Medycyna i ochrona zdrowia 3. Genomika poznawanie genomów Przełom XX i
Jeśli czas działania algorytmu zależy nie tylko od rozmiaru danych wejściowych i przyjmuje różne wartości dla różnych danych o tym samym rozmiarze,
 Oznaczenia: Jeśli czas działania algorytmu zależy nie tylko od rozmiaru danych wejściowych i przyjmuje różne wartości dla różnych danych o tym samym rozmiarze, to interesuje nas złożoność obliczeniowa
Oznaczenia: Jeśli czas działania algorytmu zależy nie tylko od rozmiaru danych wejściowych i przyjmuje różne wartości dla różnych danych o tym samym rozmiarze, to interesuje nas złożoność obliczeniowa
Algorytm. a programowanie -
 Algorytm a programowanie - Program komputerowy: Program komputerowy można rozumieć jako: kod źródłowy - program komputerowy zapisany w pewnym języku programowania, zestaw poszczególnych instrukcji, plik
Algorytm a programowanie - Program komputerowy: Program komputerowy można rozumieć jako: kod źródłowy - program komputerowy zapisany w pewnym języku programowania, zestaw poszczególnych instrukcji, plik
Bioinformatyka wykład 8, 27.XI.2012
 Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
Bioinformatyka wykład 8, 27.XI.2012 białkowa bioinformatyka strukturalna c.d. krzysztof_pawlowski@sggw.pl 2013-01-21 1 Plan wykładu regiony nieuporządkowane sposoby przedstawienia struktur białkowych powierzchnia
PROGRAMOWANIE W PYTHONIE OD PIERWSZYCH KROKÓW
 PROGRAMOWANIE W PYTHONIE OD PIERWSZYCH KROKÓW http://metodycy.torun.pl/ m.informatyka@metodycy.torun.pl 1. Wprowadzenie do Pythona podstawowe informacje Python to język programowania wysokiego poziomu,
PROGRAMOWANIE W PYTHONIE OD PIERWSZYCH KROKÓW http://metodycy.torun.pl/ m.informatyka@metodycy.torun.pl 1. Wprowadzenie do Pythona podstawowe informacje Python to język programowania wysokiego poziomu,
POPULARNE POLECENIA SKRYPTY. Pracownia Informatyczna 2
 SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
SKRYPTY Pracownia Informatyczna 2 PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK PRACOWNIA INFORMATYCZNA 2017/2018 MAGDA MIELCZAREK 2 cal wyświetlenie kalendarza Składnia: cal 2017, cal Polecenie cal
MSA i analizy filogenetyczne
 Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
Instytut Informatyki i Matematyki Komputerowej UJ, opracowanie: mgr Ewa Matczyńska, dr Jacek Śmietański MSA i analizy filogenetyczne 1. Dopasowania wielosekwencyjne - wprowadzenie Dopasowanie wielosekwencyjne
Lokalizacja genów DNA/RNA. Nukleotydy i ich łańcuchy 11/21/2013. Genom ludzki. Struktura genomu. Pirymidyny i Puryny
 Genom ludzki Lokalizacja genów Cała informacja genetyczna wdna. Zawiera sekwencje kodujące i niekodujące Rozpoczęty w 1989 Pierwsza wersja w 2000 Koszt $3 mld 3*10 9 bp(base pairs) 20,000 genów 1 Struktura
Genom ludzki Lokalizacja genów Cała informacja genetyczna wdna. Zawiera sekwencje kodujące i niekodujące Rozpoczęty w 1989 Pierwsza wersja w 2000 Koszt $3 mld 3*10 9 bp(base pairs) 20,000 genów 1 Struktura
Uwaga: Funkcja zamień(a[j],a[j+s]) zamienia miejscami wartości A[j] oraz A[j+s].
![Uwaga: Funkcja zamień(a[j],a[j+s]) zamienia miejscami wartości A[j] oraz A[j+s].](/thumbs/61/46207972.jpg "Uwaga: Funkcja zamień(a[j],a[j+s]) zamienia miejscami wartości A[j] oraz A[j+s].") Zadanie 1. Wiązka zadań Od szczegółu do ogółu Rozważmy następujący algorytm: Dane: Algorytm 1: k liczba naturalna, A[1...2 k ] tablica liczb całkowitych. n 1 dla i=1,2,,k wykonuj n 2n s 1 dopóki s
Zadanie 1. Wiązka zadań Od szczegółu do ogółu Rozważmy następujący algorytm: Dane: Algorytm 1: k liczba naturalna, A[1...2 k ] tablica liczb całkowitych. n 1 dla i=1,2,,k wykonuj n 2n s 1 dopóki s
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19
 WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
WSTĘP DO BIOINFORMATYKI Konspekt wykładu - wiosna 2018/19 Witold Dyrka 14 marca 2019 1 Wprowadzenie 1.1 Definicje bioinformatyki Według polskiej Wikipedii [1], Bioinformatyka interdyscyplinarna dziedzina
Wyrażenie include(sciezka_do_pliku) pozwala na załadowanie (wnętrza) pliku do skryptu php. Plik ten może zawierać wszystko, co może się znaleźć w
 pozwala na załadowanie (wnętrza) pliku do skryptu php. Plik ten może zawierać wszystko, co może się znaleźć w") Wyrażenie include(sciezka_do_pliku) pozwala na załadowanie (wnętrza) pliku do skryptu php. Plik ten może zawierać wszystko, co może się znaleźć w obrębie skryptu. Wyrażenia include() i require() są niemal
Wyrażenie include(sciezka_do_pliku) pozwala na załadowanie (wnętrza) pliku do skryptu php. Plik ten może zawierać wszystko, co może się znaleźć w obrębie skryptu. Wyrażenia include() i require() są niemal
Sortowanie danych. Jolanta Bachan. Podstawy programowania
 Sortowanie danych Podstawy programowania 2013-06-06 Sortowanie przez wybieranie 9 9 9 9 9 9 10 7 7 7 7 7 10 9 1 3 3 4 10 7 7 10 10 10 10 4 4 4 4 4 4 3 3 3 3 2 2 2 2 2 2 2 3 1 1 1 1 1 1 Gurbiel et al. 2000
Sortowanie danych Podstawy programowania 2013-06-06 Sortowanie przez wybieranie 9 9 9 9 9 9 10 7 7 7 7 7 10 9 1 3 3 4 10 7 7 10 10 10 10 4 4 4 4 4 4 3 3 3 3 2 2 2 2 2 2 2 3 1 1 1 1 1 1 Gurbiel et al. 2000
Generator testów 1.3.1 Bioinformatyka_zdalne wer. 1.0.13 / 0 Strona: 1
 Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Przedmiot: Bioinformatyka Nazwa testu: Bioinformatyka_zdalne wer. 1.0.13 Nr testu 0 Klasa: WNB UZ Odpowiedzi zaznaczamy TYLKO w tabeli! 1. Model Markowa substytucji aminokwasów w mutagenezie białek zakłada...
Metody Kompilacji Wykład 7 Analiza Syntaktyczna
 Metody Kompilacji Wykład 7 Analiza Syntaktyczna Parsowanie Parsowanie jest to proces określenia jak ciąg terminali może być generowany przez gramatykę. Włodzimierz Bielecki WI ZUT 2/57 Parsowanie Dla każdej
Metody Kompilacji Wykład 7 Analiza Syntaktyczna Parsowanie Parsowanie jest to proces określenia jak ciąg terminali może być generowany przez gramatykę. Włodzimierz Bielecki WI ZUT 2/57 Parsowanie Dla każdej
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: dodany punkt na temat testów do sprawozdania. Biologia, bioinformatyka:
 Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,
Wersja pliku: v.10, 13 kwietnia 2019 zmiany: - 13.04 dodany punkt na temat testów do sprawozdania Biologia, bioinformatyka: 1. DNA kwas deoksyrybonukleinowy. Zbudowany z 4 rodzajów nukleotydów: adeniny,