(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
|
|
|
- Robert Stachowiak
- 5 lat temu
- Przeglądów:
Transkrypt
 O udzieleniu patentu europejskiego ogłoszono: 02.11.11 Europejski Biuletyn Patentowy 11/44 EP 1734971 B1 (13) (1) T3 Int.Cl. A61K 31/73 (06.01) C07K 1/00 (06.01) C07K 7/00 (06.")
1 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: (97) O udzieleniu patentu europejskiego ogłoszono: Europejski Biuletyn Patentowy 11/44 EP B1 (13) (1) T3 Int.Cl. A61K 31/73 (06.01) C07K 1/00 (06.01) C07K 7/00 (06.01) C07K 14/00 (06.01) (4) Tytuł wynalazku: Urządzenie o przedłużonym uwalnianiu na bazie polimeru () Pierwszeństwo: (43) Zgłoszenie ogłoszono: w Europejskim Biuletynie Patentowym nr 06/2 (4) O złożeniu tłumaczenia patentu ogłoszono: Wiadomości Urzędu Patentowego 12/08 (73) Uprawniony z patentu: Alkermes Pharma Ireland Limited, Athlone, IE Amylin Pharmaceuticals, Inc., San Diego, US (72) Twórca(y) wynalazku: PL/EP T3 STEVEN G. WRIGHT, Madeira, US TROY CHRISTENSON, Mason, US THEAN Y. YEOH, Foxboro, US MICHAEL E. RICKEY, Morrow, US JOYCE M. HOTZ, CINCINNATI, US RAJESH KUMAR, Marlborough, US HENRY R. COSTANTINO, Woodinville, US CHRISTINE SMITH, San Diego, US DAVID LOKENS (74) Pełnomocnik: rzecz. pat. Agnieszka Marszałek SULIMA-GRABOWSKA-SIERZPUTOWSKA BIURO PATENTÓW I ZNAKÓW TOWAROWYCH SP.J. Skr. poczt Warszawa Uwaga: W ciągu dziewięciu miesięcy od publikacji informacji o udzieleniu patentu europejskiego, każda osoba może wnieść do Europejskiego Urzędu Patentowego sprzeciw dotyczący udzielonego patentu europejskiego. Sprzeciw wnosi się w formie uzasadnionego na piśmie oświadczenia. Uważa się go za wniesiony dopiero z chwilą wniesienia opłaty za sprzeciw (Art. 99 (1) Konwencji o udzielaniu patentów europejskich).
2 SGS-06/VAL EP B1 Opis 1 2 TŁO WYNALAZKU [0001] Liczne białka i peptydy, łącznie określane tutaj jako polipeptydy, wykazują działanie biologiczne in vivo i są przydatne jako leki. Wiele chorób lub stanów wymaga podawania podtrzymywanego poziomu leku, aby zapewnić najskuteczniejsze działania profilaktyczne i/lub terapeutyczne. Podtrzymywane poziomy często osiąga się przez podawanie biologicznie czynnych polipeptydów na drodze częstych wstrzyknięć podskórnych, co często powoduje wahanie poziomów leku i słabe zdyscyplinowanie pacjenta. [0002] Alternatywnie moŝna zastosować jako układ o przedłuŝonym uwalnianiu uŝycie biodegradowalnych materiałów kapsułkujących lek, takich jak polimery. Zastosowanie biodegradowalnych polimerów, przykładowo, w postaci mikrocząstek lub mikronośników, moŝe zapewnić przedłuŝone uwalnianie leku, przez wykorzystanie naturalnej zdolności polimeru do biodegradacji do regulowania uwalniania leku, a tym samym zapewnienie bardziej zgodnego, podtrzymywanego poziomu leku i zwiększonego zdyscyplinowania pacjenta. [0003] JednakŜe takie urządzenia o przedłuŝonym uwalnianiu mogą często wykazywać wysokie początkowe gwałtowne uwalnianie leku, a potem jego uwalnianie minimalne, co powoduje, Ŝe surowicze poziomy leku znajdą się poza przedziałem terapeutycznym i/lub złą dostępność biologiczną leku. Na dodatek obecność polimeru, fizjologiczne temperatury i odpowiedź organizmu na kompozycję o przedłuŝonym uwalnianiu mogą powodować zmiany w leku (np. degradację, agregację), zakłócając tym samym Ŝądany profil uwalniania leku. [0004] Ponadto sposoby stosowane do wytwarzania kompozycji o przedłuŝonym uwalnianiu mogą powodować utratę aktywności leku z uwagi na nietrwałość leku i degradujące działania etapów przetwórstwa. Działania degradujące stanowią szczególnie problem, gdy lekiem jest polipeptyd. W WO 03/024 ujawniono preparat z małą ilością eksendyny-4 i sacharozy, zawierający ponadto bufor octanowy. [000] W związku z tym istnieje zapotrzebowanie na środki podawania biologicznie czynnych polipeptydów w sposób przedłuŝony, w przypadku których polipeptyd dostarczany jest ilości stanowiącej poziomy terapeutyczne i zachowuje aktywność i siłę działania przez Ŝądany okres uwalniania. Choć wykonano wiele prac w celu rozwiązania tego problemu, wymagane są nowe rozwiązania.
3 STRESZCZENIE WYNALAZKU [0006] Wynalazek dotyczy odkrycia, Ŝe ulepszone profile uwalniania (takie jak te charakteryzujące się stosunkiem C max do C ave około 3 lub poniŝej) moŝna osiągnąć w przypadku preparatu zawierającego niewiele składników, poprzez optymalizację stosunku oleju silikonowego do polimeru w procesie wytwarzania, osiągając tym samym małą objętość porów. Niniejszy wynalazek dotyczy kompozycji do przedłuŝonego uwalniania biologicznie czynnych polipeptydów. Kompozycje o przedłuŝonym uwalnianiu według niniejszego wynalazku zawierają biologicznie zgodny polimer, eksendynę-4 jako biologicznie czynny polipeptyd, oraz sacharozę. Według zastrzeŝenia 1 polipeptyd i cukier są zdyspergowane w polimerze. Polipeptyd i cukier moŝna dyspergować osobno lub, korzystnie, razem. Kompozycja o przedłuŝonym uwalnianiu zapewnia poŝądany i zgodny profil uwalniania charakteryzujący się stosunkiem C max do C ave około 3 lub poniŝej. [0007] Całkowita objętość porów w kompozycji wynosi około 0,1 ml/g lub poniŝej. Całkowitą objętość porów oznacza się z zastosowaniem intruzyjnej porozymetrii rtęciowej. [0008] Biologicznie zgodnym polimerem jest korzystnie polimer polilaktydo-ko-glikolid. [0009] Opisano tutaj równieŝ sposób wytwarzania kompozycji do przedłuŝonego uwalniania środków biologicznie czynnych, takich jak polipeptydy, który obejmuje wytwarzanie mieszaniny przez połączenie fazy wodnej zawierającej wodę, środek, taki jak polipeptyd rozpuszczalny w wodzie i cukier, z fazą olejową zawierającą biologicznie zgodny polimer i rozpuszczalnik polimeru; wytworzenie emulsji woda w oleju przez, przykładowo, obróbkę ultradźwiękami lub homogenizację mieszaniny; dodanie oleju silikonowego do mieszaniny z wytworzeniem mikrocząstek zarodkowych; przeniesienie mikrocząstek zarodkowych do rozpuszczalnika hartującego w celu utwardzenia mikrocząstek; zebranie utwardzonych mikrocząstek; oraz wysuszenie utwardzonych mikrocząstek. W konkretnej postaci olej silikonowy dodaje się w ilości wystarczającej do osiągnięcia stosunku oleju silikonowego do rozpuszczalnika polimeru około 1,:1. Dodatkowo lub alternatywnie polimer jest obecny w fazie olejowej w ilości około % wag./obj. lub poniŝej. [00] Eksendyna-4 jest obecna w opisanej tutaj kompozycji w stęŝeniu % wag./wag. w przeliczeniu na całkowitą masę końcowej kompozycji. Ponadto sacharoza jest obecna w stęŝeniu 2% wag./wag. końcowej masy kompozycji. [0011] Kompozycję według niniejszego wynalazku moŝna podawać człowiekowi lub innej istocie Ŝywej drogą wstrzyknięć, wszczepienia (np. podskórnie, domięśniowo, wewnątrzotrzewnowo, wewnątrzczaszkowo i śródskórnie), przez podawanie do błon śluzowych (np. donosowo, dopochwowo, dopłucnie lub za pomocą czopków) albo przez podawanie in situ (np. przez wlew lub rozpylanie aerozolu).
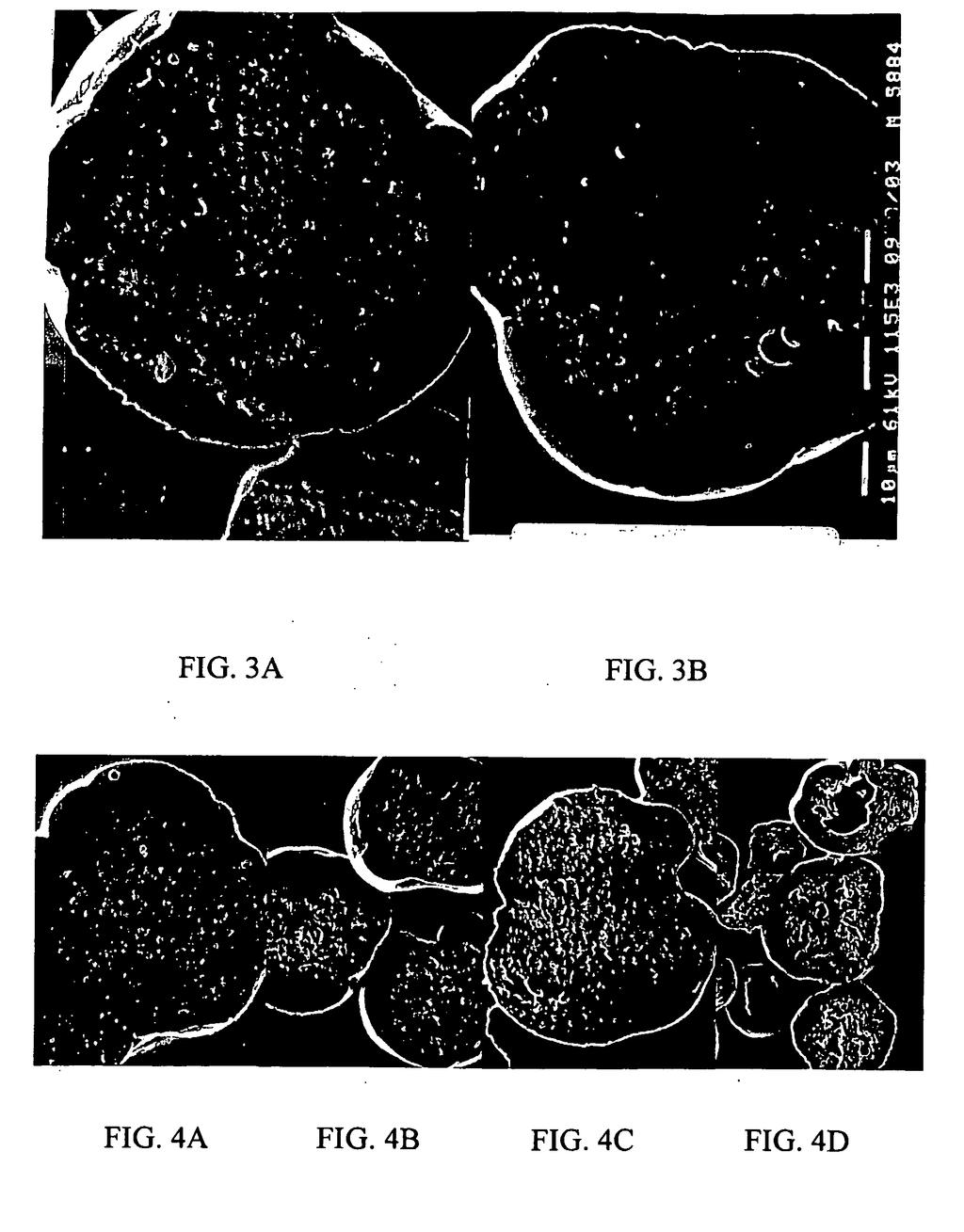
4 [0012] Kompozycję podaje się w ilości terapeutycznie skutecznej do leczenia pacjenta cierpiącego na cukrzycę, nieprawidłową tolerancję glukozy (IGT), otyłość, zaburzenie sercowo-naczyniowe (CV) lub dowolne inne zaburzenie, które moŝna leczyć eksendyną-4. [0013] Zastosowanie cukru w kompozycjach o przedłuŝonym uwalnianiu według wynalazku poprawia dostępność biologiczną wprowadzonego biologicznie czynnego polipeptydu, np. peptydów przeciwcukrzycowych i regulujących poziom glukozy i ogranicza do minimum ubytek aktywności z uwagi na nietrwałość i/lub oddziaływania chemiczne pomiędzy polipeptydem i innymi składnikami zawartymi lub stosowanymi w formułowaniu kompozycji o przedłuŝonym uwalnianiu, przy utrzymaniu doskonałego profilu uwalniania. [0014] Zalety opisanych tutaj preparatów o przedłuŝonym uwalnianiu obejmują zwiększone zdyscyplinowanie i akceptację pacjenta przez wyeliminowanie konieczności powtarzanego podawania, zwiększoną korzyść terapeutyczną przez wyeliminowanie wahań stęŝenia substancji czynnej we krwi przez zapewnienie poŝądanego profilu uwalniania oraz potencjalne zmniejszenie całkowitej ilości biologicznie czynnego polipeptydu, niezbędnej do uzyskania terapeutycznej korzyści dzięki zmniejszeniu tych wahań. KRÓTKI OPIS RYSUNKÓW [001] FIG. 1 przedstawia wykres pokazujący zaleŝność pomiędzy średnią średnicą porów i uwalnianiem in vitro dla opisanych tutaj kompozycji o przedłuŝonym uwalnianiu (A.S. = siarczan amonu). FIG. 2 przedstawia wykres pokazujący wpływ porowatości na uwalnianie in vitro eksendyny-4 z mikrocząstek oraz wpływ, jaki warunki przetwórstwa, a w szczególności stosunek oleju silikonowego do chlorku metylenu, mają na porowatość powstałych mikrocząstek. FIG. 3A-3B stanowią skany kriogenicznych SEM dla wybranych opisanych tutaj preparatów mikrocząstek. FIG. 4A-4D stanowią skany kriogenicznych SEM dla wybranych opisanych tutaj preparatów mikrocząstek. FIG. przedstawia wykres % resztkowego etanolu i chloru metylenu w funkcji Tg dla opisanych tutaj preparatów mikrocząstek. FIG. 6 przedstawia reprezentatywną krzywą farmakokinetyczną (stęŝenie, pg/ml w funkcji czasu, dni, z wstawką pokazującą stęŝenia w ciągu pierwszego dnia) dla Preparatu 2-1 (3% eksendyny-4 i 2% sacharozy), Preparatu 1 (tylko 3% eksendyny-4) i Preparatu 4 (3% eksendyny-4 i 0,% siarczanu amonu). FIG. 7 przedstawia wykres profilu uwalniania in vivo dla trzech Preparatów mikro-
5 cząstek 2, 2-1 i 2-2. FIG. 8 przedstawia wykres danych farmakokinetycznych dla Preparatów mikrocząstek -1, -2 i -3. FIG. 9 przedstawia wykres ilustrujący zaleŝność pomiędzy parametrami procesu i wielkością kropel emulsji wewnętrznej uzyskiwaną w procesie. SZCZEGÓŁOWY OPIS WYNALAZKU [0016] Wynalazek dostarcza kompozycję do przedłuŝonego uwalniania biologicznie czynnego polipeptydu, zawierającą biologicznie zgodny polimer z rozproszonym w nim biologicznie czynnym polipeptydem, tak Ŝe jest on obecny w ilości % (wag./wag.) w stosunku do masy kompozycji, oraz z rozproszoną w nim sacharozą, tak Ŝe jest ona obecna w ilości 2% (wag./wag.) w stosunku do masy kompozycji, przy czym biologicznie czynnym polipeptydem jest eksendyna-4; przy czym całkowita objętość porów w kompozycji, oznaczona z zastosowaniem intruzyjnej porozymetrii rtęciowej, wynosi 0,1 ml/g lub poniŝej; oraz przy czym kompozycja jest wolna od buforu i wysalających soli. [0017] Eksendyna-4 i sacharoza są rozpraszane w biologicznie zgodnym polimerze osobno lub, korzystnie, razem. W szczególności kompozycja o przedłuŝonym uwalnianiu charakteryzuje się profilem uwalniania o stosunku maksymalnego stęŝenia w surowicy (C max ) do średniego stęŝenia w surowicy (C ave ) około 3 lub poniŝej. W uŝytym tutaj znaczeniu odniesienie do liczby pojedynczej odnosi się równieŝ do liczby mnogiej. Środek [0018] Biologicznie czynnym polipeptydem jest przeciwcukrzycowy lub regulujący poziom glukozy polipeptyd eksendyna-4. [0019] Eksendyna-4 jest 39-aminokwasowym polipeptydem. Sekwencję aminokwasową eksendyny-4 moŝna znaleźć w opisie patentowym US nr wydanym dla Enga 13 czerwca 199 r. AC2993 i eksenatyd są równoznaczne z określeniem eksendyna-4. Wykazano, Ŝe eksendyna-4 stymuluje u ludzi i zwierząt wydzielanie insuliny w obecności podwyŝszonych stęŝeń glukozy w krwi, ale nie w okresach o niskim stęŝeniu glukozy w krwi (hipoglikemia). Wykazano równieŝ, Ŝe hamuje ona wydzielanie glukagonu, spowalnia opróŝnianie Ŝołądka i wpływa na przyjmowanie pokarmu oraz masę ciała, wykazując równieŝ inne działania. Jako takie eksendyna-4 oraz jej analogi i agonisty mogą być przydatne w leczeniu cukrzycy, IGT, otyłości itp. Cukier [00] Ilość sacharozy obecnej w kompozycji o przedłuŝonym uwalnianiu wynosi 2% (wag./wag.), co zapewnia doskonałe profile uwalniania.
6 1 2 3 Polimer [0021] Polimerami przydatnymi do wytwarzania kompozycji o przedłuŝonym uwalnianiu według tego wynalazku są biologicznie zgodne polimery, którymi mogą być biodegradowalne lub niebiodegradowalne polimery lub ich mieszanki albo kopolimery. Polimer jest biologicznie zgodny, jeśli polimer i jakiekolwiek produkty degradacji polimeru są nietoksyczne dla biorcy, a takŝe nie wykazują znaczących szkodliwych lub nieprzewidzianych działań na organizm biorcy, takich jak znaczący odczyn odpornościowy w miejscu wstrzyknięcia. [0022] W uŝytym tutaj znaczeniu biodegradowalna oznacza, Ŝe kompozycja będzie ulegać degradacji lub erozji in vivo tworząc mniejsze jednostki lub formy chemiczne. Degradacja moŝe być wynikiem, przykładowo, procesów enzymatycznych, chemicznych i fizycznych. Przydatne biologicznie zgodne, biodegradowalne polimery obejmują, przykładowo, poli(laktydy), poli(glikolidy), poli(laktydo-ko-glikolidy), poli(kwasy mlekowe), poli(kwasy glikolowe), poliwęglany, poliestroamidy, polibezwodniki, poli(aminokwasy), poliortoestry, poli(dioksanon)y, poli(alkilaty alkilenu), kopolimery lub glikol polietylenowy i poliortoester, biodegradowalny poliuretan, ich mieszanki i ich kopolimery. [0023] Odpowiednie biologicznie zgodne, niebiodegradowalne polimery obejmują niebiodegradowalne polimery wybrane z grupy obejmującej poliakrylany, polimery etylen-octan winylu i inne acylo-podstawione octany celulozy, niedegradowane poliuretany, polistyreny, polichlorek winylu, polifluorek winylu, poli(winyloimidazol), chlorosulfonowane poliolefiny, politlenek etylenu, ich mieszanki i ich kopolimery. [0024] Dopuszczalne wartości masy cząsteczkowej polimerów stosowanych według tego wynalazku mogą być ustalone przez przeciętnego znawcę w dziedzinie przy uwzględnieniu czynników, takich jak Ŝądana szybkość degradacji polimeru, właściwości fizyczne, takie jak wytrzymałość mechaniczna, chemizm grup końcowych i szybkość rozpuszczania polimeru w rozpuszczalniku. Typowo, dopuszczalny zakres masy cząsteczkowej wynosi około 00 daltonów do około daltonów. W korzystnej postaci polimer jest biodegradowalnym polimerem lub kopolimerem. W korzystniejszej postaci polimerem jest poli(laktydo-ko-glikolid) (poniŝej PLG ) o stosunku laktyd:glikolid około 1:1 i o masie cząsteczkowej około 000 daltonów do około daltonów. W jeszcze korzystniejszej postaci masa cząsteczkowa PLG stosowanego w niniejszym wynalazku wynosi około 000 daltonów do około daltonów, tak jak około 0000 do około daltonów. [002] PLG mogą zawierać kwasowe grupy końcowe lub zablokowane grupy końcowe, takie jak te, które moŝna uzyskać przez estryfikację kwasu. Doskonałe wyniki uzyskano w przypadku PLG z kwasową grupą końcową.
7 [0026] Polimery moŝna równieŝ wybierać w oparciu o liczbę lepkościową polimeru. Odpowiednie wartości liczby lepkościowej wynoszą około 0,06 do 1,0 dl/g, tak jak około 0,2 do 0,6 dl/g, korzystniej pomiędzy około 0,3 i 0, dl/g. Jako korzystne polimery wybiera się te, które ulegają degradacji w ciągu 3 do 4 tygodni. Odpowiednie polimery moŝna nabyć z Alkermes, Inc. pod nazwą handlową Medisorb, takie jak te sprzedawane jako 00 DL 3A lub 00 DL 4A. MoŜna równieŝ stosować PLG Resomer z Boehringer Ingelheim, takie jak Resomer RG03 i 03H. [0027] Kompozycję o przedłuŝonym uwalnianiu według tego wynalazku moŝna wytwarzać w wielu kształtach, takich jak folia, peletka, walec, krąŝek lub mikrocząstka. Mikrocząstkę, w uŝytym tutaj znaczeniu, stanowi składnik polimerowy o średnicy poniŝej około jednego milimetra, zawierający rozproszony w nim lub rozpuszczony biologicznie czynny polipeptyd. Mikrocząstka moŝe mieć kulisty, inny niŝ kulisty lub nieregularny kształt. Zazwyczaj mikrocząstka będzie miała wielkość odpowiednią do wstrzyknięcia. Typowy zakres wielkości mikrocząstek wynosi 00 mikronów lub poniŝej. W konkretnej postaci średnica mikrocząstek mieści się w zakresie od około jednego do około 180 mikronów. Dodatkowe zaróbki [0028] Choć moŝliwe jest dodawanie do preparatów według zastrzeganego wynalazku dodatkowych zaróbek, jak to jest dobrze znane w dziedzinie, nieoczekiwane odkrycie niniejszego wynalazku stanowi to, Ŝe doskonały profil uwalniania moŝna osiągnąć w przypadku opisanego tutaj prostego preparatu. Takie dodatkowe zaróbki mogą zwiększyć lub zmniejszyć szybkość uwalniania środka. Składniki, które mogą zasadniczo zwiększyć szybkość uwalniania, obejmują porofory i zaróbki ułatwiające degradację polimeru. Przykładowo, szybkość hydrolizy polimeru zwiększa się w przypadku ph innego niŝ obojętne. W związku z tym kwasową lub zasadową zaróbkę, taką jak kwas nieorganiczny lub zasada nieorganiczna, moŝna dodawać do roztworu polimeru, stosowanego do wytwarzania mikrocząstek, aby zmienić szybkość erozji polimeru. Składniki, które mogą zasadniczo zmniejszyć szybkość uwalniania, obejmują zaróbki, które zmniejszają rozpuszczalność środka w wodzie. [0029] Nieoczekiwanie odkryto, Ŝe środki buforujące, takie jak octanowy, cytrynianowy, fosforanowy lub inny biologicznie zgodny bufor, nie są niezbędne w fazie wodnej w celu uzyskania preparatu o przedłuŝonym uwalnianiu z eksendyną-4, o dobrej do doskonałej dostępności biologicznej. Ponadto nieoczekiwanie odkryto, Ŝe sole wysalające nie były niezbędne do regulowania gwałtownego uwalniania eksendyny-4. W związku z tym opisane tutaj kompozycje według wynalazku charakteryzują się zasadniczą (lub całkowitą) nieobecnością buforu i/lub wysalających soli.
8 Podawanie [00] Kompozycje według wynalazku moŝna podawać sposobami ogólnie znanymi w dziedzinie. Kompozycję według tego wynalazku moŝna podawać pacjentowi (np. człowiekowi potrzebującemu środka) lub innej istocie Ŝywej, drogą wstrzyknięcia, wszczepienia (np. podskórnie, domięśniowo, wewnątrzotrzewnowo, wewnątrzczaszkowo i śródskórnie), przez podawanie do błon śluzowych (np. donosowo, dopochwowo, dopłucnie lub za pomocą czopków) albo przez podawanie in situ (np. przez wlew lub rozpylanie aerozolu). [0031] Kompozycję o przedłuŝonym uwalnianiu moŝna podawać z zastosowaniem trybu dawkowania, który zapewnia wymagane poziomy terapeutyczne przez wymagany okres. Przykładowo, moŝna podawać kompozycję o przedłuŝonym uwalnianiu i monitorować pacjenta, aŝ poziomy podawanego leku powrócą do wartości podstawowych. Po powrocie do wartości podstawowej kompozycję o przedłuŝonym uwalnianiu moŝna podać ponownie. Alternatywnie, następne podanie kompozycji o przedłuŝonym uwalnianiu moŝe nastąpić przed osiągnięciem u pacjenta poziomów podstawowych. [0032] Przykładowo, kompozycję podaje się w terapeutycznie skutecznej ilości w celu leczenia pacjenta chorego na cukrzycę, cukrzycę typu II, IGT, otyłość, zaburzenie sercowo-naczyniowe (CV) lub dowolne inne zaburzenie, które moŝna leczyć eksendyną-4. [0033] Kompozycję o przedłuŝonym uwalnianiu według niniejszego wynalazku moŝna podawać wspólnie z kortykosteroidem. Wspólne podawanie kompozycji o przedłuŝonym uwalnianiu według wynalazku z kortykosteroidem moŝe jeszcze bardziej zwiększyć dostępność biologiczną biologicznie czynnego polipeptydu w kompozycji o przedłuŝonym uwalnianiu. Wspólne podawanie kortykosteroidu w połączeniu z kompozycjami o przedłu- Ŝonym uwalnianiu jest opisane szczegółowo w zgłoszeniu patentowym US 60/4194 zatytułowanym, Method of Modifying the Release Profile of Sustained Release Compositions, Dasch i in. [0034] W uŝytym tutaj znaczeniu określenie kortykosteroidy dotyczy steroidowych środków przeciwzapalnych, określanych równieŝ jako glukokortykoidy. [003] Odpowiednie kortykosteroidy obejmują, ale nie wyłącznie, 21-acetoksypregnenolon, alklometazon, algeston, amcynonid, beklometazon, betametazon, budezonid, chloroprednizon, klobetazol, klobetazon, klokortolon, kloprednol, kortykosteron, kortyzon, kortywazol, deflazakort, dezonid, dezoksymetazon, deksametazon, diflorazon, diflukortolon, difluprednat, enoksolon, fluazakort, flukloronid, flumetazon, flunizolid, acetonid flucynolonu, fluocynonid, ester butylowy fluokortyny, flukortolon, fluorometolon, octan fluperolonu, octan fluprednidenu, fluprednizolon, flurandrenolid, propionian flutikazonu, formokortal, halcynonid, propionian halobetazolu, halometazon, octan halopredonu, hy-
9 drokortamat, hydrokortyzon, etabonian loteprednolu, mazypredon, medryzon, meprednizon, metyloprednizolon, furoinian mometazonu, parametazon, prednikarbat, prednizolon, 2-dietylolaminooctan prednizolonu, sól sodowa fosforanu prednizolonu, prednizon, predniwal, prednyliden, rimeksolon, tiksokortol, triamcynolon (wszystkie postacie), przykładowo, acetonid triamcynolonu, acetonid estru metylowego kwasu triamcynolon-21-owego, benetonid triamcynolonu, heksacetonid triamcynolonu, dioctan triamcynolonu, ich farmaceutycznie dopuszczalne mieszaniny i ich sole oraz dowolne inne ich pochodne i analogi. [0036] Kortykosteroid moŝna wprowadzać jednocześnie do kompozycji o przedłuŝonym uwalnianiu, zawierającej biologicznie zgodny polimer z wprowadzonym do niego środkiem w postaci biologicznie czynnego polipeptydu. [0037] Kortykosteroid moŝna wprowadzać osobno do drugiego biologicznie zgodnego polimeru. Drugi biologicznie zgodny polimer moŝe być taki sam lub inny niŝ pierwszy biologicznie zgodny polimer z wprowadzonym do niego środkiem w postaci biologicznie czynnego polipeptydu. [0038] Kortykosteroid moŝe być obecny w postaci niekapsułkowanej, ale wymieszany z kompozycją o przedłuŝonym uwalnianiu. Przykładowo, kortykosteroid moŝe być rozpuszczony w nośniku stosowanym do dostarczania kompozycji o przedłuŝonym uwalnianiu. Alternatywnie, kortykosteroid moŝe być obecny w postaci substancji stałej w zawiesinie w odpowiednim nośniku. Ponadto, kortykosteroid moŝe być obecny w postaci proszku wymieszanego z kompozycją o przedłuŝonym uwalnianiu. [0039] Zrozumiałe jest, Ŝe kortykosteroid jest obecny w ilości wystarczającej do zwiększenia dostępności biologicznej biologicznie czynnego polipeptydu z kompozycji o przedłu- Ŝonym uwalnianiu. Zwiększona dostępność biologiczna dotyczy wzrostu dostępności biologicznej biologicznie czynnego polipeptydu z kompozycji o przedłuŝonym uwalnianiu przy jednoczesnym podawaniu z kortykosteroidem w porównaniu z podawaniem bez kortykosteroidu w okresie rozpoczynającym się dwa dni po podaniu i kończącym się pod koniec cyklu uwalniania dla konkretnego preparatu. [0040] W uŝytym tutaj znaczeniu pacjent oznacza człowieka, takiego jak człowiek potrzebujący środka lub terapii, profilaktyki albo metody diagnostycznej. [0041] W uŝytym tutaj znaczeniu przedłuŝone uwalnianie biologicznie czynnego polipeptydu stanowi uwalnianie polipeptydu z kompozycji o przedłuŝonym uwalnianiu według wynalazku, które występuje w okresie dłuŝszym niŝ okres, w którym biologiczne znacząca ilość polipeptydu powinna być dostępna po bezpośrednim podaniu roztworu polipeptydu. Korzystnie przedłuŝone uwalnianie stanowi uwalnianie, które występuje w okresie co najmniej około jednego tygodnia, taki jak co najmniej około dwóch, co najmniej około trzech
10 tygodni lub co najmniej około czterech tygodni. PrzedłuŜone uwalnianie moŝe być uwalnianiem ciągłym lub nieciągłym, ze stosunkowo stałymi lub zmiennymi szybkościami uwalniania. Na ciągłość uwalniania i poziom uwalniania moŝe mieć wpływ typ uŝytej kompozycji polimeru (np. stosunki monomerów, masa cząsteczkowa, skład bloków i zmienianie połączeń polimerów), obciąŝenie polipeptydem i/lub dobór zaróbek w celu osiągnięcia Ŝądanego działania. [0042] W uŝytym tutaj znaczeniu terapeutycznie skuteczna ilość, profilaktycznie skuteczna ilość lub diagnostycznie skuteczna ilość stanowi ilość kompozycji o przedłuŝonym uwalnianiu wymagana do uzyskania Ŝądanej odpowiedzi biologicznej po podaniu. [0043] C max w uŝytym tutaj znaczeniu stanowi maksymalne stęŝenie leku w surowicy, które występuje w trakcie okresu uwalniania, który jest monitorowany. [0044] C ave w uŝytym tutaj znaczeniu oznacza średnie stęŝenie leku w surowicy, uzyskane przez podzielenie pola powierzchni pod krzywą (AUC) dla profilu uwalniania przez czas trwania uwalniania. [004] Korzystnie stosunek C max do C ave wynosi około 3 lub poniŝej. Taki profil jest szczególnie poŝądany w przypadku polipeptydów przeciwcukrzycowych lub regulujących poziom glukozy, takich jak te opisane powyŝej. Stosunek około 3 lub poniŝej moŝe zapewnić C ave w przedziale terapeutycznym z uniknięciem niekorzystnych skutków ubocznych leku, które mogą wynikać z wyŝszych stosunków. [0046] Dostępność biologiczna, jako uŝyte tutaj określenie, odnosi się do ilości leku, która dociera do układu krąŝenia. Dostępność biologiczną moŝna zdefiniować jako obliczone pole powierzchni pod krzywą (AUC) dla profilu uwalniania konkretnego polipeptydu w okresie rozpoczynającym się od momentu po podaniu i kończącym się w ustalonym punkcie czasowym. Jak to jest zrozumiałe w dziedzinie, profil uwalniania generuje się przez sporządzenie wykresu poziomów surowiczych biologicznie czynnego środka u osobnika (na osi Y) w ustalonych punktach czasowych (na osi X). Dostępność biologiczną często podaje się jako % dostępności biologicznej, który stanowi dostępność biologiczną osiągniętą dla konkretnego polipeptydu po podaniu kompozycji o przedłuŝonym uwalnianiu, podzieloną przez dostępność biologiczną osiąganą dla konkretnego polipeptydu po doŝylnym podaniu takiej samej dawki leku, pomnoŝoną przez 0. [0047] Modyfikację profilu uwalniania moŝna potwierdzić odpowiednim farmakokinetycznym monitorowaniem obecności w surowicy pacjenta środka w postaci biologicznie czynnego polipeptydu. Przykładowo, odpowiednie badanie oparte na przeciwciałach (np. ELISA i IRMA), jak to jest dobrze znane w dziedzinie, moŝna zastosować do oznaczania stęŝenia pewnych biologicznie czynnych środków polipeptydowych w surowicy pacjenta.
11 1 2 3 Przykład takiego badania opisano tutaj dla eksendyny-4. [0048] Farmakodynamiczne monitorowanie pacjenta w celu monitorowania działania terapeutycznego środka u pacjenta moŝna zastosować do potwierdzenia zachowania aktywności biologicznej uwolnionego środka. Sposoby monitorowania działań farmakodynamicznych moŝna wybrać w oparciu o podawany biologicznie czynny środek polipeptydowy, z zastosowaniem powszechnie dostępnych technik. Wytwarzanie [0049] Znanych jest wiele sposobów, jakimi moŝna wytwarzać kompozycje o przedłuŝonym uwalnianiu (matryce z polimeru/biologicznie czynnego polipeptydu) według wynalazku, w szczególności opisane tutaj kompozycje o małej porowatości. Szczegółowe procedury dla pewnych sposobów wytwarzania mikrocząstek podano poniŝej w Przykładach Wykonania. W korzystnej postaci sposób według wynalazku wytwarzania kompozycji do przedłuŝonego uwalniania biologicznie czynnego polipeptydu obejmuje wytwarzanie mieszaniny przez połączenie fazy wodnej zawierającej wodę, środek, taki jak polipeptyd rozpuszczalny w wodzie i cukier z fazą olejową zawierającą biologicznie zgodny polimer i rozpuszczalnik polimeru; wytworzenie emulsji woda w oleju; dodanie do mieszaniny środka powodującego koacerwację, przykładowo oleju silikonowego, oleju roślinnego lub oleju mineralnego, z wytworzeniem mikrocząstek zarodkowych; przeniesienie mikrocząstek zarodkowych do rozpuszczalnika hartującego w celu utwardzenia mikrocząstek; zebranie utwardzonych mikrocząstek; oraz wysuszenie utwardzonych mikrocząstek. Sposób ten jest tutaj ogólnie określany jako sposób woda-olej-olej (W/O/O). [000] Korzystnie, polimer moŝe być obecny w fazie olejowej w stęŝeniu w zakresie od około 3% wag./wag. do około 2% wag./wag., korzystnie od około 4% wag./wag. do około 1% wag./wag., tak jak od około % wag./wag. do około % wag./wag. Doskonałe wyniki uzyskano tutaj stosując stęŝenie 6% wag./wag. PLG w fazie olejowej. [001] Polimer zazwyczaj łączy się z rozpuszczalnikiem polimeru. Gdy polimerem jest PLG, taki jak ten korzystnie stosowany tutaj, polimer dodaje się do rozpuszczalnika PLG. Takie rozpuszczalniki są dobrze znane w dziedzinie. Korzystnym rozpuszczalnikiem jest chlorek metylenu. [002] Środek i cukier dodaje się w fazie wodnej, korzystnie w tej samej fazie wodnej. StęŜenie środka korzystnie wynosi do 0 mg/g, korzystnie pomiędzy 0 do 0 mg/g. StęŜenie cukru korzystnie wynosi do 0 mg/g i do 0 mg/g. [003] Dwie fazy miesza się następnie w celu wytworzenia emulsji. Korzystnie emulsję wytwarza się tak, Ŝe wielkość kropel emulsji wewnętrznej wynosi poniŝej około 1 mikrona, korzystnie poniŝej około 0,7 mikrona, korzystniej poniŝej około 0, mikrona, tak jak
12 około 0,4 mikrona. Do wytwarzania takiej emulsji moŝna stosować sonikatory i homogenizatory. [004] W uŝytym tutaj znaczeniu środek powodujący koacerwację stanowi dowolny olej, w którym roztwór polimeru (polimer i rozpuszczalnik) nie jest łatwo rozpuszczalny i w związku z tym tworzy odrębną fazę w stosunku do roztworu polimeru. Odpowiednie środki powodujące koacerwację do stosowania w niniejszym wynalazku obejmują, ale nie wyłącznie, olej silikonowy, olej roślinny i olej mineralny. W konkretnej postaci środkiem powodującym koacerwację jest olej silikonowy i jest on dodawany w ilości wystarczającej do uzyskania stosunku oleju silikonowego do rozpuszczalnika polimeru od około 0,7:1 do około 2:1. W konkretnej postaci stosunek oleju silikonowego do polimeru wynosi od około 1:1 do około 1,:1. W korzystnej postaci stosunek oleju silikonowego do polimeru wynosi około 1,:1. [00] Uzyskaną mieszaninę dodaje się do rozpuszczalnika hartującego, który stanowi nierozpuszczalnik polimeru. Nierozpuszczalniki polimeru są ogólnie dobrze znane w dziedzinie. Szczególnie korzystny rozpuszczalnik hartujący stanowi układ rozpuszczalników heptan/etanol. [006] Stały lek moŝna równieŝ kapsułkować z zastosowaniem zmodyfikowanej wersji opisanego powyŝej sposobu. Ten zmodyfikowany sposób moŝna określić jako substancja stała/olej/olej (S/O/O). [007] Przykładowo, stałą eksendynę-4 przeprowadza się w zawiesinę w chlorku metylenu zawierającym 6% PLG i poddaje się na lodzie obróbce ultradźwiękowej przez około cztery minuty. Następną obróbkę przeprowadza się w sposób analogiczny jak w sposobie W/O/O. [008] Wynalazek zostanie obecnie dokładniej i konkretnie opisany w poniŝszych przykładach. PRZYKŁADY WYTWARZANIE MIKROCZĄSTEK I [009] Opisane tutaj kompozycje o przedłuŝonym uwalnianiu otrzymywano sposobem rozdzielania faz. Ogólny sposób opisano poniŝej dla mikrocząstek zawierających eksendynę-4 i sacharozę, dla partii o wielkości 1 kg. A. Wytwarzanie emulsji wewnętrznej woda w oleju [0060] Emulsję woda w oleju wytworzono z zastosowaniem homogenizatora. Odpowiednie homogenizatory obejmują pracujący w linii homogenizator Megatron MT-V 3-6 F/FF/FF, Kinematica AG, Szwajcaria. Fazę wodną emulsji przygotowano przez rozpuszczenie eksendyny-4 i zaróbek, takich jak sacharoza, w wodzie. StęŜenie leku w uzyskanym roztworze moŝe wynosić od około 0 mg/g do około 0 mg/g. Przykładowo, gdy lekiem
13 jest eksendyna-4, stęŝenie leku w roztworze moŝe wynosić od około g do około 60 g na 600 g wody. W konkretnej postaci 0 g eksendyny-4 i g sacharozy rozpuszczono w 600 g wody do irygacji (WFI). Konkretne ilości podane powyŝej stanowią nominalne obciąŝenie bez nastawiania w celu skompensowania uwzględniającego konkretną moc zawartego peptydu w uŝytej partii eksendyny-4. Fazę olejową emulsji przygotowano przez rozpuszczenie polimeru PLGA (np. 9 g oczyszczonego 0:0 PLGA DL4A (Alkermes, Inc.) w chlorku metylenu (14,6 kg lub 6% wag./wag.). [0061] Fazę wodną dodano następnie do fazy olejowej w celu uzyskania zgrubnej emulsji z uŝyciem mieszadła z górnym napędem przez około trzy minuty. Następnie zgrubną emulsję homogenizowano przy około 000 obrotów/minutę w temperaturze otoczenia. Uzyskano w ten sposób wielkość kropel emulsji wewnętrznej poniŝej 1 mikrona. Zrozumiałe jest, Ŝe tworzenie emulsji wewnętrznej moŝna osiągnąć z uŝyciem dowolnych odpowiednich środków. Odpowiednie środki do wytwarzania emulsji obejmują, ale nie wyłącznie, homogenizację w sposób opisany powyŝej oraz obróbkę ultradźwiękami. B. Wytwarzanie koacerwatu [0062] Następnie przeprowadzono etap koacerwacji przez dodanie oleju silikonowego (21,8 kg Dimethicone, NF, cs) w ciągu około pięciu minut do emulsji wewnętrznej. Odpowiada to stosunkowo 1,:1, oleju silikonowego do chlorku metylenu. Chlorek metylenu z roztworu polimeru oddziela się do oleju silikonowego i polimer zaczyna wytrącać się wokół fazy wodnej zawierającej eksendynę-4, co prowadzi do mikrokapsułkowania. Powstałe w ten sposób mikrosfery zarodkowe są miękkie i wymagają utwardzenia. Często mikrosfery zarodkowe odstawia się na krótki czas, przykładowo od około 1 minuty do około minut przed skierowaniem do etapu utwardzania mikrosfer. C. Utwardzanie i płukanie mikrosfer [0063] Następnie mikrosfery zarodkowe natychmiast przeniesiono do mieszaniny rozpuszczalników heptan/etanol. Niezbędną objętość mieszaniny heptan/etanol moŝna ustalić na podstawie wielkości partii mikrosfer, przy czym stosunek chlorku metylenu do rozpuszczalnika heptan/etanol wynosi zazwyczaj 16:1. W niniejszym przykładzie zastosowano około 2 kg heptanu i 23 kg etanolu w ochłodzonym do 3 C zbiorniku z mieszadłem. Taka mieszanina rozpuszczalników utwardza mikrosfery przez wyekstrahowanie dodatkowej ilości chlorku metylenu z mikrosfer. Ten etap utwardzania moŝna równieŝ określić jako hartowanie. Po hartowaniu przez 1 godzinę w 3 C mieszaninę rozpuszczalników dekantuje się i dodaje się świeŝy heptan (13 kg) w 3 C i przetrzymuje się przez 1 godzinę w celu wypłukania resztkowego oleju silikonowego, etanolu i chlorku metylenu obecnych na powierzchni mikrosfer, albo pompuje się bezpośrednio do etapu zbierania.
14 D. Suszenie i zbieranie mikrosfer [0064] Po zakończeniu etapu hartowania lub dekantacji/przemywania mikrosfery przeniesiono i zebrano z zastosowaniem urządzenia 12-calowy filtr/suszarka Sweco Pharmasep Model PH12Y6. W filtrze/suszarce stosuje się mikronowe wielowarstwowe sito zbierające, połączone z silnikiem, który wprawia w drgania sito podczas zbierania i suszenia. Końcowe przemywanie heptanem (6 kg w 3 C) przeprowadzono w celu zapewnienia maksymalnej przepustowości linii i usunięcia jakiegokolwiek nadmiaru oleju silikonowego. Mikrosfery wysuszono następnie pod próŝnią ze stałym przedmuchem gazowym azotem, z kontrolowaną szybkością, zgodnie z następującym harmonogramem: 6 godzin w 3 C; 6 godzin ze wzrostem do 41 C; i 84 godziny w 41 C. [006] Po zakończeniu suszenia mikrosfery wyładowano do zbiornika gromadzącego, przesiano przez sito µm i przechowywano w około - C do czasu napełniania. [0066] W przypadku wszystkich wytworzonych tutaj preparatów mikrocząstek ilość polipeptydu, przykładowo eksendyny-4, oraz zaróbek obecnych w wytworzonych preparatach, wyraŝa się w % (wag./wag.) w przeliczeniu na końcową masę kompozycji o przedłuŝonym uwalnianiu. % (wag./wag.) stanowi procent nominalny, z wyjątkiem zaznaczonych przypadków. WYTWARZANIE MIKROCZĄSTEK II A. Wytwarzanie emulsji wewnętrznej woda w oleju [0067] Emulsję woda w oleju wytworzono z zastosowaniem sonikatora. Odpowiednie sonikatory obejmują Vibracell VCX 70 z głowicą sondującą model CV33, Sonics and Materials Inc., Newtown, CT. Fazę wodną emulsji przygotowano przez rozpuszczenie eksendyny-4 i zaróbek, takich jak sacharoza, w wodzie. StęŜenie leku w uzyskanym roztworze moŝe wynosić od około 0 mg/ml do około 0 mg/ml. Przykładowo, gdy lekiem jest eksendyna-4, stęŝenie leku w roztworze moŝe wynosić od około 3,28 g do około 6, g na 6, g wody. W konkretnej postaci,46 g eksendyny-4 i 2,18 g sacharozy rozpuszczono w 6, g wody do irygacji czyli WFI. Konkretne ilości podane powyŝej reprezentują 4% nadmiar w stosunku do obciąŝenia docelowego w celu skompensowania strat związanych z filtracyjną sterylizacją składników. Fazę olejową emulsji przygotowano przez rozpuszczenie polimeru PLGA (np. 97,7 g oczyszczonego 0:0 PLGA DL4A (Alkermes, Inc.)) w chlorku metylenu (139 g lub 6% wag./obj.). [0068] Fazę wodną dodano następnie do fazy olejowej w ciągu około trzech minut z obróbką ultradźwiękami przy 0% amplitudzie w temperaturze pokojowej. Fazę wodną pompowano przez ¼-calową rurę ze stali nierdzewnej z 1-calową końcówką rury do HPLC (Śr. wewn. = /00 cali) pod nadciśnieniem funtów/cal 2, wprowadzając ją poniŝej
15 sondy do obróbki ultradźwiękami wewnątrz strefy obróbki ultradźwiękami. Zawartość reaktora mieszano następnie z szybkością 1400 do 1600 obrotów/minutę, z dodatkową obróbką ultradźwiękami przy 0% amplitudzie przez 2 minuty, po czym następowało sekundowe zatrzymanie, a następnie jeszcze 1 minutowa obróbka ultradźwiękami. Uzyskano w ten sposób wielkość kropel emulsji wewnętrznej poniŝej 0, mikrona. Zrozumiałe jest, Ŝe tworzenie emulsji wewnętrznej moŝna osiągnąć z uŝyciem dowolnych odpowiednich środków. Odpowiednie środki do wytwarzania emulsji obejmują, ale nie wyłącznie, opisaną powyŝej obróbkę ultradźwiękami oraz homogenizację. B. Wytwarzanie koacerwatu [0069] Następnie przeprowadzono etap koacerwacji przez dodanie oleju silikonowego (2294 g Dimethicone, NF, cs) w ciągu około trzech do pięciu minut do emulsji wewnętrznej. Odpowiada to stosunkowi 1,:1, oleju silikonowego do chlorku metylenu. Chlorek metylenu z roztworu polimeru oddziela się do oleju silikonowego i polimer zaczyna wytrącać się wokół fazy wodnej zawierającej eksendynę-4, co prowadzi do mikrokapsułkowania. Powstałe w ten sposób mikrosfery zarodkowe są miękkie i wymagają utwardzenia. Często mikrosfery zarodkowe odstawia się na krótki czas, przykładowo od około 1 minuty do około minut przed skierowaniem do etapu utwardzania mikrosfer. C. Utwardzanie i płukanie mikrosfer [0070] Następnie mikrosfery zarodkowe natychmiast przeniesiono do mieszaniny rozpuszczalników heptan/etanol. Niezbędną objętość mieszaniny heptan/etanol moŝna ustalić na podstawie wielkości partii mikrosfer. W niniejszym przykładzie zastosowano około 22 kg heptanu i 2448 g etanolu w ochłodzonym do 3 C zbiorniku z mieszaniem ( do 40 obrotów/minutę). Taka mieszanina rozpuszczalników utwardza mikrosfery przez wyekstrahowanie dodatkowej ilości chlorku metylenu z mikrosfer. Ten etap utwardzania moŝna równieŝ określić jako hartowanie. Po hartowaniu przez 1 godzinę w 3 C mieszaninę rozpuszczalników zdekantowano i świeŝy heptan (13 kg) dodano w 3 C i przetrzymywano przez 1 godzinę w celu wypłukania resztkowego oleju silikonowego, etanolu i chlorku metylenu obecnych na powierzchni mikrosfer. D. Suszenie i zbieranie mikrosfer [0071] Po zakończeniu etapu płukania mikrosfery przeniesiono i zebrano na mikronowym wielowarstwowym sicie o średnicy 6 cali wewnątrz komory suszącej w kształcie stoŝka, który działał jako filtr końcowy. Końcowe płukanie heptanem (6 kg w 4 C) przeprowadzono w celu zapewnienia maksymalnej przepustowości linii. Mikrosfery wysuszono następnie ze stałym przedmuchem gazowym azotem, z kontrolowaną szybkością, zgodnie z następującym harmonogramem: 18 godzin w 3 C; 24 godziny w 2 C; 6 godzin w
16 C; i 42 godziny w 38 C. [0072] Po zakończeniu suszenia mikrosfery wyładowano do wysterylizowanego zbiornika gromadzącego z teflonu/stali nierdzewnej, przymocowanego do stoŝka suszącego. Zbiornik gromadzący zamyka się szczelnie, odłącza od stoŝka suszącego i przechowuje w - ± C do czasu napełniania. Materiał pozostający w stoŝku po demontaŝu w celu oczyszczenia pobiera się do analizy zawartości leku. Wydajność wyniosła około 0 gramów mikrosfer. [0073] W przypadku wszystkich wytworzonych tutaj preparatów mikrocząstek ilość polipeptydu, przykładowo eksendyny-4, oraz zaróbek obecnych w wytworzonych preparatach, wyraŝa się w % (wag./wag.) w przeliczeniu na końcową masę kompozycji o przedłuŝonym uwalnianiu. % (wag./wag.) stanowi procent nominalny, z wyjątkiem zaznaczonych przypadków. POLIMER: [0074] Przykłady przydatnych do stosowania konkretnych polimerów PLG zestawiono poniŝej. Wszystkie polimery stosowane w poniŝszych przykładach zostały umieszczone na liście i wszystkie zestawione polimery otrzymano z Alkermes, Inc. Cincinnati, OH i moŝna je opisać następująco: Polimer 2A: Poli(laktydo-ko-glikolid); stosunek laktyd:glikolid 0:0; Masa cząst. 12,3 kd; IV=0,1 (dl/g). Polimer 4A: Poli(laktydo-ko-glikolid); stosunek laktyd:glikolid 0:0; Masa cząst kd; IV=0,4-0,47 (dl/g). [007] OCZYSZCZANIE PLG: Wiadomo (patrz, przykładowo, Peptide Acylation by Poly(α-Hydroxy Esters), Lucke i in., Pharmaceutical Research, tom 19, nr 2, str , luty 02), Ŝe białka i peptydy, które są wprowadzane do matryc PLG, mogą ulegać niepo- Ŝądanym zmianom (np. degradacji lub modyfikacji chemicznej) w wyniku oddziaływania z produktami degradacji PLG lub zanieczyszczeniami pozostającymi po wytworzeniu polimeru. W związku z tym polimery PLG stosowane do wytwarzania większości opisanych tutaj preparatów mikrocząstek oczyszczano przed wytwarzaniem kompozycji o przedłuŝonym uwalnianiu, z zastosowaniem znanych w dziedzinie sposobów oczyszczania. METODY CHARAKTERYZOWANIA: [0076] Ustalono, Ŝe następujące metody charakteryzowania są przydatne do identyfikacji mikrocząstek, które będą zapewniać poŝądany profil uwalniania substancji czynnej. SEM [0077] SEM stosowano do oceny wielkości cząstek, kształtu i cech powierzchniowych mikrocząstek. Obrazowanie SEM wykonywano z zastosowaniem układu Personal SEM (ASPEX, LLC). Wszystkie próbki osadzano za pomocą szpachelki na standardowych
17 16 1 stolikach SEM pokrytych dwustronną taśmą węglową. Próbki powlekano przez rozpylanie Au przez około 90 sekund przy prądzie emisji 18 ma, z zastosowaniem powlekarki Mini Sputter Coater Model SC 76 (Energy Beam Sciences). Wszystkie próby obrazowania SEM wykonywano z uŝyciem wiązki elektronów o KeV w zakresie powiększenia około do 0X. KRIOGENICZNA SEM [0078] Przekrój mikrocząstek badano z zastosowaniem kriogenicznej SEM. Próbkę mikrocząstek zmieszano z roztworem HISTO PREP (Fischer) i przetrzymywano przez noc w kriostacie w - C. Utwardzone mikrocząstki zamocowano na szkiełku nakrywkowym, a następnie pocięto z uŝyciem metalowego noŝa. Pocięte cząstki zamocowano na stolikach aluminiowych, powleczono przez rozpylanie platyną i palladem, i obserwowano pod skaningowym mikroskopem elektronowym (Phillips 2M). Obserwacja sekcji wzrokiem dostarcza metodę określania stopnia porowatości mikrocząstek. POMIAR POROWATOŚCI INTRUZJA RTĘCI [0079] Rozkład objętości porów w mikrocząstkach oznaczano z zastosowaniem porozymetru Moden Mercury Intrusion Porosimeter model SutoPor IV 900 (Mikromeritics, Norcross, GA). W skrócie, rtęć wtłaczano w znaną ilość mikrocząstek w penetrometrze przez stopniowe przykładanie ciśnienia aŝ do osiągnięcia maksymalnego ciśnienia funtów/cal 2. Mierzono objętość rtęci intrudowanej w pory przy róŝnych wartościach ciśnienia. Sposób ten pozwala na ilościowe oznaczenie rozkładu porów w mikrocząstkach. Jest faktem, Ŝe wielkość poddanych intruzji porów jest odwrotnie proporcjonalna do przyłoŝonego ciśnienia. Równowagę pomiędzy siłami wewnętrznymi i zewnętrznymi w układzie cieczciało stałe-para moŝna opisać równaniem Washburna. ZaleŜność pomiędzy przyłoŝonym ciśnieniem i wielkością porów, w które wtłaczana jest rtęć, jest określona jako: 2 gdzie: D = średnica pora γ = napięcie powierzchniowe (stałe) θ = kąt zwilŝania (stały) P= ciśnienie W związku z tym, wielkość pora, do którego rtęć będzie intrudowana, jest odwrotnie proporcjonalna do przyłoŝonego ciśnienia. Przy załoŝeniu, Ŝe wszystkie pory są ciasnymi walcami, średnią średnicę pora (D=4V/A) moŝna obliczyć przez podzielenie objętości pora (V=πD2h/4) przez powierzchnię pora (A=πDh). RESZTKOWE ROZPUSZCZALNIKI
18 17 [0080] Jedną metodę zastosowano do ilościowego oznaczania heptanu, etanolu i chlorku metylenu. Aparat stanowił chromatograf gazowy HP 890 Series 2 z kolumną Rtx 11, cm x 0,3 mm. Około 1 mg mikrocząstek rozpuszczano w ml N,Ndimetyloformamidu. Octan propylu stosowano jako wzorzec wewnętrzny. Preparat próbki nastawiano tak, aby moŝna było oznaczyć ilościowo stęŝenie chlorku metylenu wynoszące zaledwie 0,03%. WYTWARZANIE MIKROCZĄSTEK [0081] Partie mikrocząstek podane w Tabeli 1 wytwarzano w sposób opisany powyŝej w skali 0 gramów stosując polimer 4A przy stosunku oleju silikonowego do chlorku metylenu 1,:1 lub 1:1, przy czym lepkość oleju silikonowego wynosiła cs. Ilości eksendyny-4 i zaróbek uŝyte w preparacie podano równieŝ w Tabeli 1. TABELA 1 Partia # Preparat Gwałtowne uwalnianie in vitro (%) Uwagi (#1)** 0% Sacharozy, 0% AS 0,40 Olej Si:MeCl 2 1,: (#2)** 2% Sacharozy (F16) 0,40 Olej Si:MeCl 2 1,: (#2-1)** (#2-2)** (#2-3)** (#2-4)** (#2-)** 2% Sacharozy (F16) 0,44 Olej Si:MeCl 2 1,:1 2% Sacharozy (F16) 0,4 Olej Si:MeCl 2 1,:1 2% Sacharozy (F16) 0,80 Olej Si:MeCl 2 1:1 2% Sacharozy (F16) 1,0 Olej Si:MeCl 2 1:1 2% Sacharozy (F16) 1,1 Olej Si:MeCl 2 1: (#3-1)** (#3-2)** 2% Sacharozy, 0,% AS (F14) 2% Sacharozy, 0,% AS (F14) 1,3 Hartowanie 0:0 2,2 Olej Si:MeCl 2 1,: (#4)** 0% Sacharozy, 0,% AS 3,8 Olej Si:MeCl 2 1,: A (#3-3)** 2% Sacharozy, 0,% AS (F14) 12,7 Hartowanie 0% heptanu
19 18 Partia # Preparat Gwałtowne uwalnianie in vitro (%) Uwagi (#) (% obciąŝenie lekiem) 2% Sacharozy (F17) 0, % obciąŝenie lekiem, Olej Si:MeCl 2 1,: (#3-4)** (#3-)** (#3-6)** (#3-7)** 2% Sacharozy, 0,% AS (F14) 2% Sacharozy, 0,% AS (F14) 2% Sacharozy, 0,% AS (F14) 2% Sacharozy, 0,% AS (F14) 0, Olej Si:MeCl 2 1,:1 1, Olej Si:MeCl 2 1:1 2,70 Olej Si:MeCl 2 1:1 6,70 Olej Si:MeCl 2 1:1 *OBCIĄśENIE LEKIEM WE WSZYSTKICH PREPARATACH WYNOSIŁO 3% Z WYJĄTKIEM POROWATOŚCI # ** Przykład porównawczy [0082] Całkowitą objętość intruzji uzyskaną metodą intruzyjnej porozymetrii rtęciowej i obliczone średnie średnice porów podano w TABELI 2. ZaleŜność pomiędzy średnią średnicą porów i uwalnianiem in vitro pokazano na FIG. 1 TABELA 2 Partia # Całkowita objętość porów (ml/g) Gwałtowne uwalnianie in vitro (%) Średnia średnica porów (µm) (#1)* 0,033 0,40 0, (#2)* 0,03 0,40 0, (#2-1)* 0,037 0,44 0, (#2-2)* 0,03 0,4 0, (#2-3)* 0,036 0,80 0, (#2-4)* 0,038 1,0 0, (#2-)* 0,039 1,1 0, (#3-1)* 0,041 1,3 0, (#3-2)* 0,039 2,2 0, (#4)* 0,067 3,8 0,012
20 19 Partia # Całkowita objętość porów (ml/g) Gwałtowne uwalnianie in vitro (%) Średnia średnica porów (µm) A(#3-3)* 0,13 12,7 0, (#3-4)* 0,0 0, 0, (#3-)* 0,060 1, 0, (#3-6)* 0,060 2,70 0, (#3-7)* 0,180 6,70 0,0170 *Przykład porównawczy 1 2 [0083] Na FIG. 1 pokazano wpływ siarczanu amonu na początkowe uwalnianie in vitro. Dane wskazują, Ŝe początkowe uwalnianie in vitro jest skorelowane ze średnicą porów mikrocząstek. Preparaty wykonane z siarczanem amonu wykazują zmienne poziomy uwalniania in vitro i zmienną porowatość w przeciwieństwie do preparatów bez siarczanu amonu, które wykazują stałą porowatość i uwalnianie. Przy wytwarzaniu mikrocząstek obecność siarczanu amonu w fazie wodnej moŝe powodować wysalanie substancji leczniczej podczas wytwarzania emulsji wewnętrznej. RóŜnice w mikrośrodowisku wytrąconych osadów mogą przyczyniać się do róŝnicy w porowatości i w związku z tym do zmian w początkowym uwalnianiu. Działania takiego nie zaobserwowano w przypadku preparatów wytwarzanych bez siarczanu amonu. Preparaty z sacharozą i eksendyną-4 wykazują bardziej poŝądany i stały poziom początkowego uwalniania w porównaniu z preparatami zawierającymi eksendynę-4, sacharozę i siarczan amonu. [0084] FIG. 2 dodatkowo pokazuje wpływ porowatości na uwalnianie in vitro oraz wpływ, jaki warunki obróbki, a konkretnie stosunek oleju silikonowego do chlorku metylenu, mają na porowatość wytworzonych mikrocząstek. W skrócie, preparaty mikrocząstek wytworzonych przy zastosowaniu stosunku oleju silikonowego do chlorku metylenu 1:1 (Preparaty 2-4 i 2- w Tabeli 1) wykazują wyŝsze początkowe uwalnianie niŝ takie same preparaty wytworzone przy zastosowaniu stosunku oleju silikonowego do chlorku metylenu 1,:1 (Preparaty 2, 2-1 i 2-2 z Tabeli 1). FIG. 2 sugeruje, Ŝe wyŝszy stosunek oleju silikonowego do chlorku metylenu powoduje mniejszą porowatość, co prowadzi do mniejszego początkowego uwalniania. KRIOGENICZNA SEM [008] Analizę metodą kriogenicznej SEM przeprowadzono w sposób opisany powyŝej dla Preparatów Typów 2, 3 i z Tabeli 1. FIG. 3A-3B przedstawiają skany mikrofotografii dla wybranych preparatów Typu 2 (Preparat 2-2, FIG. 3A) i Typu (% eksendyny-4, 2%
21 1 2 3 sacharozy, FIG. 3B). FIG. 4A-D przedstawiają skany mikrofotografii dla Preparatów odpowiednio 3-4, 3-, 3-6 i 3-7, z Tabeli 1. Podobnie, zmiany porowatości osiągane przy stosowaniu siarczanu amonu, które mogą przyczyniać się do zmienności początkowego uwalniania, moŝna zaobserwować na kriogenicznej SEM przekrojów na FIG. 4A-D. POZIOMY RESZTKOWYCH ROZPUSZCZALNIKÓW [0086] Poziomy resztkowych rozpuszczalników w danym preparacie mogą wpływać na Tg preparatu. Poziomy resztkowych rozpuszczalników oznaczono dla preparatów mikrocząstek Typu 2 i z Tabeli 1. Jedną metodę zastosowano do ilościowego oznaczania heptanu, etanolu i chlorku metylenu. Aparat stanowił chromatograf gazowy HP 890 Series 2 z kolumną Rtx 11, m x 0,3 mm. Około 1 mg mikrocząstek rozpuszczano w ml N,N-dimetyloformamidu. Octan propylu stosowano jako wzorzec wewnętrzny. Preparat próbki nastawiano tak, aby moŝna było oznaczyć ilościowo stęŝenie chlorku metylenu wynoszące zaledwie 0,03%. [0087] FIG. stanowi wykres % resztkowego etanolu i chlorku metylenu w preparatach Typu 2 i z Tabeli 1 (3 lub % eksendyny-4, 2% sacharozy). FIG. wykazuje, Ŝe Tg obniŝa się, gdy ilość resztkowego rozpuszczalnika zwiększa się. WYTWARZANIE MIKROCZĄSTEK ZAWIERAJĄCYCH 3% EKSENDYNY-4 I 2% SACHAROZY [0088] Z uwagi na zmienność porowatości uzyskiwanej dzięki obecności siarczanu amonu w preparatach mikrocząstek oraz zidentyfikowanie porowatości jako cechy, która znacząco wpływa na początkowe uwalnianie, w dalszych badaniach nie zajmowano się siarczanem amonu. WPŁYW WIELKOŚCI KROPEL EMULSJI WEWNĘTRZNEJ [0089] PoniŜsze badanie wykonano w celu ustalenia wpływu parametrów procesu na tworzenie się emulsji wewnętrznej, a takŝe trwałość uzyskanej emulsji i osiągane 24-godzinne uwalnianie in vitro z mikrosfer wytworzonych przy zastosowaniu róŝnych parametrów procesu. Emulsje wewnętrzne z fazy wodnej i fazy rozpuszczalnika wytwarzano na drodze obróbki ultradźwiękami, jak to opisano powyŝej, w skali 0 g, albo przez homogenizację z zastosowaniem homogenizatora MT000 z generatorem 36/4 (Kinematica AG, Szwajcaria) przy małej szybkości (800 obrotów/minutę) lub duŝej szybkości (210 obrotów/minutę). Po wytworzeniu emulsji wewnętrznej róŝnymi technikami, emulsje przetrzymywano w reaktorze w warunkach łagodnego mieszania mieszadłem z górnym napędem przez, 1 lub 60 minut przed usunięciem próbki. Po ustalonym czasie przetrzymywania emulsję wewnętrzną poddawano w sposób opisany powyŝej dalszej przeróbce do mikrocząstek, po czym oznaczano 24-godzinne uwalnianie in vitro dla kaŝdej partii, w
22 sposób opisany poniŝej. Charakterystykę wielkości kropel emulsji wewnętrznej moŝna ustalić z zastosowaniem analizatora wielkości cząstek Horiba [0090] Próbkę emulsji wewnętrznej pobierano z reaktora za pomocą szklanej pipety. Z uŝyciem pipety do przenoszenia ~ kropel emulsji wewnętrznej dodawano do ~ ml 6% roztworu 0:0 Medisorb :polimer PLG 4A PLG w zakręcanej fiolce scyntylacyjnej o pojemności cm 3, po czym całość mieszano. 6% roztwór 0:0 Medisorb :polimer PLG 4A PLG słuŝył równieŝ jako roztwór odniesienia, stanowiący ślepą próbę. Próbkę około 9 ml tej rozcieńczonej emulsji przenoszono następnie do czystego ml pojemnika Horiba na próbki. Pokrywę umieszczano na pojemniku na próbki, aby zapobiec szybkiemu odparowaniu rozpuszczalnika polimeru. Przygotowana próbka mieściła się w dopuszczalnym zakresie % odczytu przepuszczalności, 0,6% - 0,90% na błękitny pasek (Lampa). Nastawę względnego współczynnika załamania 0,94-0,00i wybrano przy nastawianiu programu. Następnie wykonywano pomiary wielkości kropel w próbce w analizatorze wielkości cząstek Horiba, takim jak model LA 9. Dane dotyczące korelacji parametrów procesu i osiąganej wielkości kropel emulsji wewnętrznej dla czasów przetrzymywania, 1 i 60 minut, a takŝe wyniki 24- godzinnego testu uwalniania in vitro (w nawiasach) przedstawiono na Figurze 9. CHARAKTERYZOWANIE MIKROSFER [0091] Mikrosfery eksendyny-4 rutynowo charakteryzowano w odniesieniu do zawartości leku, wielkości cząstek, resztkowych rozpuszczalników, początkowego uwalniania in vitro oraz charakterystyki PK u szczurów. Dla wybranych partii po kapsułkowaniu lek poddano ekstrakcji w celu dokonania wstępnej oceny czystości eksendyny-4. POCZĄTKOWE UWALNIANIE IN VITRO [0092] Początkowe uwalnianie eksendyny-4 oznaczano mierząc stęŝenie eksendyny-4 po 1 godzinie w buforze uwalniania ( mm HEPES, 0 mm NaCl, ph 7,4). ± mg mikrosfer umieszczano w,0 ml buforu z mm HEPES, 0 mm NaCl, ph 7,4, w temperaturze pokojowej, worteksowano przez około sekund w celu przeprowadzenia roztworu w zawiesinę, a następnie umieszczano w komorze powietrznej w 37 C na 1 godzinę. Po 1 godzinie próbki wyjmowano z komory i odwracano kilka razy w celu wymieszania, po czym odwirowywano przy 0 obrotach/minutę przez minut. Supernatant usuwano i analizowano natychmiast metodą HPLC w następujących warunkach: Kolumna: TSK-GEL, 7,8 mm x cm, m (TSOH BIOSEP PART #0840); Temperatura pieca kolumny: otoczenia; Temperatura autosamplera: 6 C; NatęŜenie przepływu: 0,8 ml/minutę; Wykrywanie: 280 nm; Wstrzykiwana objętość: l; Faza ruchoma: 3% Acetonitryl/6% woda z 0,1% TFA/litr (obj.); Czas trwania próby: około minut. Jako wzorzec stosowano substancję
23 leczniczą w masie, eksendynę-4, 0,2 mg/ml, przygotowaną w mm buforze octanowym, ph 4,. BADANIA NA ZWIERZĘTACH [0093] Wszystkie opisane tutaj badania farmakokinetyczne (PK) prowadzono na dorosłych samcach szczura Sprague-Dawley waŝących około 00±0 g. [0094] Do charakteryzowania PK preparatów mikrocząstek, kaŝdemu zwierzęciu wykonywano wstrzyknięcie podskórne mikrocząstek w zawiesinie w rozcieńczalniku (3% karboksymetyloceluloza, 0,9% NaCl, 0,1% Tween ) do obszaru międzyłopatkowego. Zazwyczaj dawka wynosiła 1,0 mg eksendyny-4 na szczura, wstrzykiwana w objętości 0,7 ml. Próbki krwi zbierano z bocznej Ŝyły ogonowej w 0,, 2, 4, 6,, 24 godziny oraz 2, 4, 7,, 14, 17, 21, 24 i 28 dni po podaniu. Próbki krwi natychmiast umieszczano w probówkach MICROTAINER zawierających EDTA i odwirowywano przy około X g przez około dwie minuty. Osocze przenoszono następnie do probówek MICROTAINER bez dodatku i przechowywano w -70 C do czasu wykonania testu. IRMA zastosowano do oznaczania stęŝeń eksendyny w osoczu. UWALNIANIE IN VIVO - IRMA [009] Jako metodę ilościowego oznaczania eksendyny-4 w osoczu zastosowano kanapkowy test immunologiczny, z analitem wychwytywanym przez monoklonalne przeciwciało EXE4:2-8.4 na fazie stałej i wykrywaniem przez promieniotwórczo jodowane monoklonalne przeciwciało GLP-1:3-3. Związane impulsy oznaczano ilościowo z kalibracyjnej krzywej wzorcowej. Jest to test specyficzny dla eksendyny-4 pełnej długości czyli nienaruszonej i nie wykrywa eksendyny-4 (3-39). Typowy zakres krzywej wzorcowej wynosi od pg/ml do 00 pg/ml, w zaleŝności od wieku przeciwciała znacznikowego. UWALNIANIE IN VITRO I IN VIVO [0096] Początkowe uwalnianie in vitro preparatów 2, 2-1 i 2-2 (3% eksendyny-4 i 2% sacharozy) zbadano w sposób opisany powyŝej. Uwalnianie in vitro wynosiło odpowiednio 0,4%, 0,4% i 0,%. Wszystkie trzy partie wykazywały równieŝ stosunkowo powolne uwalnianie in vivo w zakresie 114 do 1 pg/ml dla C max w dniu 0-1. FIG. 6 przedstawia reprezentatywną krzywą farmakokinetyczną dla preparatów zawierających 3% eksendyny- 4 i 2% sacharozy (2-1), a takŝe tylko 3% eksendyny-4 (1) oraz 3% eksendyny-4 i 0,% siarczanu amonu (4). Zastosowano olej silikonowy do chlorku metylenu w stosunku 1,:1, a lepkość oleju silikonowego wynosiła cs. [0097] Z FIG. 6 moŝna stwierdzić, Ŝe preparaty nie zawierające siarczanu amonu wykazują mniejsze początkowe uwalnianie. Choć preparat zawierający samą eksendynę-4 wykazywał odpowiednie początkowe uwalnianie, czystość po kapsułkowaniu leku była zmniej-
24 szona w porównaniu z preparatem zawierającym eksendynę-4 w połączeniu z sacharozą. Dodatek cukru do preparatów zmniejsza degradację środka. [0098] Profil uwalniania in vivo dla trzech preparatów 2, 2-1 i 2-2 porównywanych powy- Ŝej, przedstawiono na FIG. 7. Wszystkie trzy partie wykazywały stosunkowo niskie początkowe uwalnianie, po którym następowało uwalnianie minimalne (niskie poziomy w surowicy pomiędzy około dniem 4 i dniem 17), a następnie przedłuŝone uwalnianie od około dnia 21 do dnia 28. Niskie początkowe uwalnianie i kształt profilu uwalniania dla trzech preparatów były zgodne. PREPARAT PRZY ZASTOSOWANIU OLEJU SILIKONOWEGO DO CHLORKU ME- TYLENU W STOSUNKU 1:1 [0099] Preparaty 2-3, 2-4 i 2- z Tabeli 1 (3% eksendyny-4, 2% sacharozy) otrzymano przy zastosowaniu oleju silikonowego i chlorku metylenu w stosunku 1:1. Uwalnianie wyjściowe w przypadku tych preparatów było większe niŝ w przypadku preparatów 2, 2-1 i 2-2 z Tabeli 1 (3% eksendyny-4, 2% sacharozy przy stosunku silikonu do chlorku metylenu 1,:1). W szczególności preparaty przy stosunku 1,:1 zapewniały średnie początkowe uwalnianie około 0,4%, podczas gdy preparaty przy stosunku 1:1 zapewniały średnie początkowe uwalnianie około 1,0%. Tę samą tendencję obserwowano in vivo, przy czym C max w dniu 0-1 u szczurów wynosiło 2288 ± pg/ml dla stosunku 1:1, podczas gdy C max w dniu 0-1 u szczurów wynosiło 10±221 pg/ml dla stosunku 1,:1. ZWIĘKSZONE OBCIĄśENIE LEKIEM [00] Zwiększenie obciąŝenia eksendyną-4 do 4% przy utrzymaniu 2% sacharozy powodowało początkowe uwalnianie in vitro i in vivo w tym samym zakresie jak w przypadku obciąŝenia 3%. [01] Wykonano trzy preparaty Typu według Tabeli 1 (% obciąŝenia lekiem, 2% sacharozy, stosunek oleju silikonowego do chlorku metylenu 1,:1). Wszystkie trzy partie, - 1, -2 i -3, wykazywały niskie początkowe uwalnianie in vitro w zakresie od 0,2 do 0,%. Podobnie in vivo C max preparatów było zgodnie niskie w zakresie 467 pg/ml do 1267 pg/ml. Na FIG. 8 pokazano wykres z danych farmakokinetycznych dla tych trzech zbadanych partii. W porównaniu z zachowaniem preparatu z 3% eksendyny-4 zawierającym 2% sacharozy, preparaty z % wykazywały wyŝsze poziomy leku w surowicy w około dniu 1 i dniu 2. Pozostała część profilu dla preparatów z % była podobna jak dla preparatów z 3% wykazujących minimalne uwalnianie, po którym następowało uwalnianie leku głównie od dnia 21 do dnia 28.
25 24 ZastrzeŜenia patentowe Kompozycja do przedłuŝonego uwalniania biologicznie czynnego polipeptydu, zawierająca biologicznie zgodny polimer z rozproszonym w nim biologicznie czynnym polipeptydem, tak Ŝe jest on obecny w ilości % (wag./wag.) w stosunku do masy kompozycji, oraz rozproszoną w nim sacharozą, tak Ŝe jest ona obecna w ilości 2% (wag./wag.) w stosunku do masy kompozycji, przy czym biologicznie czynnym polipeptydem jest eksendyna-4; przy czym całkowita objętość porów w kompozycji, oznaczana metodą intruzyjnej porozymetrii rtęciowej wynosi 0,1 ml/g lub poniŝej; oraz przy czym kompozycja jest wolna od buforu i wysalających soli. 2. Kompozycja o przedłuŝonym uwalnianiu według zastrzeŝenia 1, w której biologicznie zgodny polimer jest wybrany z grupy obejmującej poli(laktydy), poli(glikolidy), poli(laktydo-ko-glikolidy), poli(kwasy mlekowe), poli(kwasy glikolowe), poliwęglany, poliestroamidy, polibezwodniki, poli(aminokwasy), poliortoestry, poli(dioksanony), biodegradowalne poliuretany, ich mieszanki i ich kopolimery. 3. Kompozycja o przedłuŝonym uwalnianiu według zastrzeŝenia 1, w której biologicznie zgodny polimer jest wybrany spośród poli(laktydów), poli(glikolidów), poli(laktydo-ko-glikolidów), poli(kwasów mlekowych), poli(kwasów glikolowych) oraz ich mieszanek i kopolimerów. 4. Kompozycja o przedłuŝonym uwalnianiu według zastrzeŝenia 1, w której biologicznie zgodny polimer obejmuje poli(laktydo-ko-glikolid).. Kompozycja o przedłuŝonym uwalnianiu według zastrzeŝenia 1, w której biologicznie zgodny polimer stanowi oczyszczony poli(laktydo-ko-glikolid) 0:0. 6. Kompozycja o przedłuŝonym uwalnianiu według któregokolwiek z zastrzeŝeń 1 do, która jest w postaci mikrocząstek. 7. Kompozycja o przedłuŝonym uwalnianiu według któregokolwiek z zastrzeŝeń 1 do 6, do stosowania w leczeniu cukrzycy typu 2. Uprawnieni: Alkermes Pharma Ireland Limited Amylin Pharmaceuticals, Inc. Pełnomocnik: mgr inŝ. Agnieszka Marszałek Rzecznik patentowy
26 2
27 26
28 27
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2190940 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.09.2008 08802024.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2480370 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 08.09.2010 10773557.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1968711 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 05.01.2007 07712641.5
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1711158 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.11.2004 04806793.8
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2528702 (96) Data i numer zgłoszenia patentu europejskiego: 03.12.2010 10796315.9 (13) (51) T3 Int.Cl. B21D 53/36 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1854925 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2005 05826699.0 (13) (51) T3 Int.Cl. E03D 1/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1773451 (96) Data i numer zgłoszenia patentu europejskiego: 08.06.2005 05761294.7 (13) (51) T3 Int.Cl. A61K 31/4745 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1732433 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.01.2005 05702820.1
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1477128 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 14.05.2004 04076445.8 (51) Int. Cl. A61D1/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2086467 (96) Data i numer zgłoszenia patentu europejskiego: 26.11.2007 07824706.1 (13) (51) T3 Int.Cl. A61F 2/16 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1754519 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2006 06016676.6 (51) Int. Cl. A62C13/66 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1754519 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2006 06016676.6 (51) Int. Cl. A62C13/66 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1663252 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 10.09.2004 04786930.0
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1663252 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 10.09.2004 04786930.0
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609. (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1879609 (96) Data i numer zgłoszenia patentu europejskiego: 04.05.2006 06742792.2 (13) (51) T3 Int.Cl. A61K 38/17 (2006.01)
(86) Data i numer zgłoszenia międzynarodowego: , PCT/AT01/ (87) Data i numer publikacji zgłoszenia międzynarodowego:
 Data i numer zgłoszenia międzynarodowego: , PCT/AT01/ (87) Data i numer publikacji zgłoszenia międzynarodowego:") RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 206658 (21) Numer zgłoszenia: 355294 (22) Data zgłoszenia: 05.10.2001 (86) Data i numer zgłoszenia międzynarodowego:
RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 206658 (21) Numer zgłoszenia: 355294 (22) Data zgłoszenia: 05.10.2001 (86) Data i numer zgłoszenia międzynarodowego:
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1586320 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.02.2005 05472001.6 (51) Int. Cl. A61K31/435 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1586320 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.02.2005 05472001.6 (51) Int. Cl. A61K31/435 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 71811 (96) Data i numer zgłoszenia patentu europejskiego: 29.09.06 06791167.7 (13) (1) T3 Int.Cl. H04Q 11/00 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1999308 (96) Data i numer zgłoszenia patentu europejskiego: 28.03.2007 07727422.3 (13) (51) T3 Int.Cl. D06F 35/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1999308 (96) Data i numer zgłoszenia patentu europejskiego: 28.03.2007 07727422.3 (13) (51) T3 Int.Cl. D06F 35/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680966 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791390.0 (13) T3 (51) Int. Cl. A23L1/172 A23P1/08
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680966 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791390.0 (13) T3 (51) Int. Cl. A23L1/172 A23P1/08
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2998028 (96) Data i numer zgłoszenia patentu europejskiego: 31.07.2015 15002280.4 (13) (51) T3 Int.Cl. B04B 11/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2998028 (96) Data i numer zgłoszenia patentu europejskiego: 31.07.2015 15002280.4 (13) (51) T3 Int.Cl. B04B 11/00 (2006.01)
PL B1 (12) O P I S P A T E N T O W Y (19) P L (11) (13) B 1 A61K 9/20. (22) Data zgłoszenia:
 O P I S P A T E N T O W Y (19) P L (11) (13) B 1 A61K 9/20. (22) Data zgłoszenia:") R Z E C Z PO SPO L IT A PO LSK A (12) O P I S P A T E N T O W Y (19) P L (11) 1 7 7 6 0 7 (21) Numer zgłoszenia: 316196 (13) B 1 Urząd Patentowy Rzeczypospolitej Polskiej (22) Data zgłoszenia: 13.03.1995
R Z E C Z PO SPO L IT A PO LSK A (12) O P I S P A T E N T O W Y (19) P L (11) 1 7 7 6 0 7 (21) Numer zgłoszenia: 316196 (13) B 1 Urząd Patentowy Rzeczypospolitej Polskiej (22) Data zgłoszenia: 13.03.1995
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690923 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 1.02.0 0460002.8 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690923 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 1.02.0 0460002.8 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2179743 (96) Data i numer zgłoszenia patentu europejskiego: 13.07.2009 09460028.5 (13) (51) T3 Int.Cl. A61K 38/18 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 197092 (96) Data i numer zgłoszenia patentu europejskiego: 22.11.06 06824279.1 (13) (1) T3 Int.Cl. A61K 3/36 (06.01) A61P
PL B1. POLWAX SPÓŁKA AKCYJNA, Jasło, PL BUP 21/12. IZABELA ROBAK, Chorzów, PL GRZEGORZ KUBOSZ, Czechowice-Dziedzice, PL
 PL 214177 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 214177 (13) B1 (21) Numer zgłoszenia: 394360 (51) Int.Cl. B22C 1/02 (2006.01) C08L 91/08 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
PL 214177 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 214177 (13) B1 (21) Numer zgłoszenia: 394360 (51) Int.Cl. B22C 1/02 (2006.01) C08L 91/08 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618. (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618. (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618 (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.0 (13) (1) T3 Int.Cl. A41B 11/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2120618 (96) Data i numer zgłoszenia patentu europejskiego: 28.02.2008 08719309.0 (13) (1) T3 Int.Cl. A41B 11/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1886669 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 02.08.2007 07113670.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2321564 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2008 08785479.0 (13) (51) T3 Int.Cl. F16L 21/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2321564 (96) Data i numer zgłoszenia patentu europejskiego: 10.08.2008 08785479.0 (13) (51) T3 Int.Cl. F16L 21/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 191441 (96) Data i numer zgłoszenia patentu europejskiego: 16.08.06 0680183.3 (13) (1) T3 Int.Cl. C12N 9/00 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 191441 (96) Data i numer zgłoszenia patentu europejskiego: 16.08.06 0680183.3 (13) (1) T3 Int.Cl. C12N 9/00 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1680075 (13) T3 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 11.10.2004
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2162456 (96) Data i numer zgłoszenia patentu europejskiego: 29.05.2008 08748372.3 (13) (51) T3 Int.Cl. C07D 475/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2162456 (96) Data i numer zgłoszenia patentu europejskiego: 29.05.2008 08748372.3 (13) (51) T3 Int.Cl. C07D 475/04 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1661542 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 12.08.2004 04762070.3 (51) Int. Cl. A61G7/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1661542 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 12.08.2004 04762070.3 (51) Int. Cl. A61G7/00 (2006.01)
PL B1. INSTYTUT BIOPOLIMERÓW I WŁÓKIEN CHEMICZNYCH, Łódź, PL
 PL 214380 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 214380 (13) B1 (21) Numer zgłoszenia: 385032 (51) Int.Cl. C08B 37/08 (2006.01) D01D 5/40 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
PL 214380 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 214380 (13) B1 (21) Numer zgłoszenia: 385032 (51) Int.Cl. C08B 37/08 (2006.01) D01D 5/40 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 221611 (96) Data i numer zgłoszenia patentu europejskiego: 19.01. 000481.1 (13) (1) T3 Int.Cl. B28C /42 (06.01) B60P 3/16
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 221611 (96) Data i numer zgłoszenia patentu europejskiego: 19.01. 000481.1 (13) (1) T3 Int.Cl. B28C /42 (06.01) B60P 3/16
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2210706 (96) Data i numer zgłoszenia patentu europejskiego: 21.01.2010 10000580.0 (13) (51) T3 Int.Cl. B24B 21/20 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2210706 (96) Data i numer zgłoszenia patentu europejskiego: 21.01.2010 10000580.0 (13) (51) T3 Int.Cl. B24B 21/20 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787644 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.11.2006 06123574.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787644 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.11.2006 06123574.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2946811 (96) Data i numer zgłoszenia patentu europejskiego: 21.04.2015 15164439.0 (13) (51) T3 Int.Cl. A62C 2/12 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2946811 (96) Data i numer zgłoszenia patentu europejskiego: 21.04.2015 15164439.0 (13) (51) T3 Int.Cl. A62C 2/12 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1802536 (96) Data i numer zgłoszenia patentu europejskiego: 20.09.2004 04774954.4 (13) T3 (51) Int. Cl. B65D77/20 B65D85/72
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1802536 (96) Data i numer zgłoszenia patentu europejskiego: 20.09.2004 04774954.4 (13) T3 (51) Int. Cl. B65D77/20 B65D85/72
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1748241 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 26.07.200 0106841.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1748241 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 26.07.200 0106841.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1708988 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 13.01.200 0706914.8
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1708988 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 13.01.200 0706914.8
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1740398 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 1.03.200 071703.9 (1) Int. Cl. B60C1/06 (2006.01) (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1740398 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 1.03.200 071703.9 (1) Int. Cl. B60C1/06 (2006.01) (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2224595 (96) Data i numer zgłoszenia patentu europejskiego: 10.02.2010 10001353.1 (13) (51) T3 Int.Cl. H03K 17/96 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2224595 (96) Data i numer zgłoszenia patentu europejskiego: 10.02.2010 10001353.1 (13) (51) T3 Int.Cl. H03K 17/96 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 187318 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 06.04.06 06731279.3
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 187318 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 06.04.06 06731279.3
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2657547 (96) Data i numer zgłoszenia patentu europejskiego: 24.04.2012 12165334.9 (13) (51) T3 Int.Cl. F16B 25/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2657547 (96) Data i numer zgłoszenia patentu europejskiego: 24.04.2012 12165334.9 (13) (51) T3 Int.Cl. F16B 25/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1747298 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 02.05.2005 05747547.7 (51) Int. Cl. C22C14/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1799953 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.08.2005 05770398.5
(96) Data i numer zgłoszenia patentu europejskiego:
 Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 10232 (96) Data i numer zgłoszenia patentu europejskiego: 17.06.2004 04102787.1 (13) T3 (1) Int. Cl. E0C9/00 (2006.01) E0C9/02
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 10232 (96) Data i numer zgłoszenia patentu europejskiego: 17.06.2004 04102787.1 (13) T3 (1) Int. Cl. E0C9/00 (2006.01) E0C9/02
PL B1. Preparat o właściwościach przeciwutleniających oraz sposób otrzymywania tego preparatu. POLITECHNIKA ŁÓDZKA, Łódź, PL
 PL 217050 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 217050 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 388203 (22) Data zgłoszenia: 08.06.2009 (51) Int.Cl.
PL 217050 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 217050 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 388203 (22) Data zgłoszenia: 08.06.2009 (51) Int.Cl.
PL B1. Sposób wytwarzania dodatku o właściwościach przewodzących do kompozytów cementowych
 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 229764 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 408318 (22) Data zgłoszenia: 26.05.2014 (51) Int.Cl. C04B 22/02 (2006.01)
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 229764 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 408318 (22) Data zgłoszenia: 26.05.2014 (51) Int.Cl. C04B 22/02 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2328822 (96) Data i numer zgłoszenia patentu europejskiego: 02.09.2009 09782487.4 (13) (51) T3 Int.Cl. B65G 15/38 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2328822 (96) Data i numer zgłoszenia patentu europejskiego: 02.09.2009 09782487.4 (13) (51) T3 Int.Cl. B65G 15/38 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.") PL/EP 1809944 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1809944 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.4 (51) Int. Cl.
PL/EP 1809944 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1809944 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 27.10.2004 04791425.4 (51) Int. Cl.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2398779 (96) Data i numer zgłoszenia patentu europejskiego: 22.02.2010 10711860.6 (13) (1) T3 Int.Cl. C07D 239/7 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2398779 (96) Data i numer zgłoszenia patentu europejskiego: 22.02.2010 10711860.6 (13) (1) T3 Int.Cl. C07D 239/7 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2555663 (96) Data i numer zgłoszenia patentu europejskiego: 06.04.2011 11730434.5 (13) (51) T3 Int.Cl. A47L 15/42 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2555663 (96) Data i numer zgłoszenia patentu europejskiego: 06.04.2011 11730434.5 (13) (51) T3 Int.Cl. A47L 15/42 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 232147 (96) Data i numer zgłoszenia patentu europejskiego: 19.06.2009 097974.4 (13) (1) T3 Int.Cl. D06F 7/18 (2006.01) F04B
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 232147 (96) Data i numer zgłoszenia patentu europejskiego: 19.06.2009 097974.4 (13) (1) T3 Int.Cl. D06F 7/18 (2006.01) F04B
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 244831 (96) Data i numer zgłoszenia patentu europejskiego: 17.06.20 72697.6 (13) (1) T3 Int.Cl. C01B 7/04 (2006.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 244831 (96) Data i numer zgłoszenia patentu europejskiego: 17.06.20 72697.6 (13) (1) T3 Int.Cl. C01B 7/04 (2006.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2122 (96) Data i numer zgłoszenia patentu europejskiego: 2..07 07866441.4 (13) (1) T3 Int.Cl. D21H 19/06 (06.01) Urząd Patentowy
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2122 (96) Data i numer zgłoszenia patentu europejskiego: 2..07 07866441.4 (13) (1) T3 Int.Cl. D21H 19/06 (06.01) Urząd Patentowy
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 187481 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 17.02.06 0673321. (1) Int. Cl. C08G61/ (06.01) (97) O
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 187481 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 17.02.06 0673321. (1) Int. Cl. C08G61/ (06.01) (97) O
(96) Data i numer zgłoszenia patentu europejskiego:
 Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1690978 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2005 05101042.9 (13) T3 (51) Int. Cl. D06F81/08 (2006.01)
(12) OPIS PATENTOWY (19) PL (11)
 OPIS PATENTOWY (19) PL (11)") RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 172296 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 302820 (22) Data zgłoszenia: 28.03.1994 (51) IntCl6: C08L 33/26 C08F
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 172296 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 302820 (22) Data zgłoszenia: 28.03.1994 (51) IntCl6: C08L 33/26 C08F
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2337642 (96) Data i numer zgłoszenia patentu europejskiego: 08.09.09 0978272.1 (13) (1) T3 Int.Cl. B21B 4/08 (06.01) B08B
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2337642 (96) Data i numer zgłoszenia patentu europejskiego: 08.09.09 0978272.1 (13) (1) T3 Int.Cl. B21B 4/08 (06.01) B08B
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1810954 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.2006 06025226.9 (13) (51) T3 Int.Cl. C03B 9/41 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2131847. (96) Data i numer zgłoszenia patentu europejskiego: 27.02.2008 08716068.
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2131847. (96) Data i numer zgłoszenia patentu europejskiego: 27.02.2008 08716068.") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2131847 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.02.2008 08716068.5
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2131847 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.02.2008 08716068.5
PL B1. PRZEDSIĘBIORSTWO PRODUKCJI FARMACEUTYCZNEJ HASCO-LEK SPÓŁKA AKCYJNA, Wrocław, PL BUP 09/13
 PL 222738 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 222738 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 396706 (22) Data zgłoszenia: 19.10.2011 (51) Int.Cl.
PL 222738 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 222738 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 396706 (22) Data zgłoszenia: 19.10.2011 (51) Int.Cl.
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1838670 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 0.12.0 08876.7 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1838670 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 0.12.0 08876.7 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 184620 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.0.2007 077439.7 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 184620 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 03.0.2007 077439.7 (97)
Granulowany Węgiel Aktywny z łupin orzechów kokosowych BT bitumiczny AT antracytowy
 Granulowany Węgiel Aktywny z łupin orzechów kokosowych BT bitumiczny AT antracytowy Granulowany Węgiel Aktywny GAC (GAC ang. Granular Activated Carbon) jest wysoce wydajnym medium filtracyjnym. Węgiel
Granulowany Węgiel Aktywny z łupin orzechów kokosowych BT bitumiczny AT antracytowy Granulowany Węgiel Aktywny GAC (GAC ang. Granular Activated Carbon) jest wysoce wydajnym medium filtracyjnym. Węgiel
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 28647 (96) Data i numer zgłoszenia patentu europejskiego: 30.03.09 091662.2 (13) (1) T3 Int.Cl. C07D 333/28 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 28647 (96) Data i numer zgłoszenia patentu europejskiego: 30.03.09 091662.2 (13) (1) T3 Int.Cl. C07D 333/28 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 21737 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2010 10790844.4 (13) (1) T3 Int.Cl. A47L 1/42 (2006.01) A47L
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 21737 (96) Data i numer zgłoszenia patentu europejskiego: 16.12.2010 10790844.4 (13) (1) T3 Int.Cl. A47L 1/42 (2006.01) A47L
PL B1. Ośrodek Badawczo-Rozwojowy Izotopów POLATOM,Świerk,PL BUP 12/05
 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 201238 (13) B1 (21) Numer zgłoszenia: 363932 (51) Int.Cl. G21G 4/08 (2006.01) C01F 17/00 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej (22)
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 201238 (13) B1 (21) Numer zgłoszenia: 363932 (51) Int.Cl. G21G 4/08 (2006.01) C01F 17/00 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej (22)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 213136 (96) Data i numer zgłoszenia patentu europejskiego: 14.03.2008 08723469.6 (13) (1) T3 Int.Cl. F24D 19/ (2006.01) Urząd
PL 213904 B1. Elektrolityczna, nanostrukturalna powłoka kompozytowa o małym współczynniku tarcia, zużyciu ściernym i korozji
 PL 213904 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 213904 (13) B1 (21) Numer zgłoszenia: 390004 (51) Int.Cl. C25D 3/12 (2006.01) C25D 15/00 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
PL 213904 B1 RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 213904 (13) B1 (21) Numer zgłoszenia: 390004 (51) Int.Cl. C25D 3/12 (2006.01) C25D 15/00 (2006.01) Urząd Patentowy Rzeczypospolitej Polskiej
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 223771 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.08 0886773.1 (13) (1) T3 Int.Cl. A47L 1/42 (06.01) Urząd
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 223771 (96) Data i numer zgłoszenia patentu europejskiego: 06.12.08 0886773.1 (13) (1) T3 Int.Cl. A47L 1/42 (06.01) Urząd
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 161679 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego: 24.06.0 064.7 (1) Int. Cl. B60R21/01 (06.01) (97) O udzieleniu
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787658 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 10.11.2005 05077559.2
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1787658 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 10.11.2005 05077559.2
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2259949 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2009 09727379.1 (13) (51) T3 Int.Cl. B60L 11/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2259949 (96) Data i numer zgłoszenia patentu europejskiego: 11.02.2009 09727379.1 (13) (51) T3 Int.Cl. B60L 11/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2452138 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.06.2010 10723151.6
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2452138 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 16.06.2010 10723151.6
(19) PL (11) (13)B1
 PL (11) (13)B1") RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (21) Numer zgłoszenia: 324710 (22) Data zgłoszenia: 05.02.1998 (19) PL (11)189348 (13)B1 (51) IntCl7 C08L 23/06 C08J
RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (21) Numer zgłoszenia: 324710 (22) Data zgłoszenia: 05.02.1998 (19) PL (11)189348 (13)B1 (51) IntCl7 C08L 23/06 C08J
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2253685 (96) Data i numer zgłoszenia patentu europejskiego: 11.06.2010 10165746.8 (13) (51) T3 Int.Cl. C09K 5/04 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2253685 (96) Data i numer zgłoszenia patentu europejskiego: 11.06.2010 10165746.8 (13) (51) T3 Int.Cl. C09K 5/04 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1508941 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.08.2004 04018799.9
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1508941 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 07.08.2004 04018799.9
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1588845 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2004 04405247.0
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1588845 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 22.04.2004 04405247.0
Mikrokapsułki CS. Prof. dr hab. Stanisław Ignatowicz Konsultacje Entomologiczne Warszawa
 Mikrokapsułki CS Prof. dr hab. Stanisław Ignatowicz Konsultacje Entomologiczne Warszawa Kapsułkowanie 2 Kapsułkowanie jest techniką, za pomocą której jeden materiał lub mieszanina materiałów jest powlekana
Mikrokapsułki CS Prof. dr hab. Stanisław Ignatowicz Konsultacje Entomologiczne Warszawa Kapsułkowanie 2 Kapsułkowanie jest techniką, za pomocą której jeden materiał lub mieszanina materiałów jest powlekana
PL 178509 B1 (13) B1. (51) IntCl6: C23C 8/26. (54) Sposób obróbki cieplno-chemicznej części ze stali nierdzewnej
 B1. (51) IntCl6: C23C 8/26. (54) Sposób obróbki cieplno-chemicznej części ze stali nierdzewnej") RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 178509 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 305287 (22) Data zgłoszenia: 03.10.1994 (51) IntCl6: C23C 8/26 (54)
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 178509 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 305287 (22) Data zgłoszenia: 03.10.1994 (51) IntCl6: C23C 8/26 (54)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1876175 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.04.2006 06745731.7
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1876175 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 27.04.2006 06745731.7
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2445326 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 24.10.2011 11186353.6
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2828428 (96) Data i numer zgłoszenia patentu europejskiego: 12.03.13 13731877.0 (13) (1) T3 Int.Cl. D0B 19/12 (06.01) D0B
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2828428 (96) Data i numer zgłoszenia patentu europejskiego: 12.03.13 13731877.0 (13) (1) T3 Int.Cl. D0B 19/12 (06.01) D0B
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 182634 (96) Data i numer zgłoszenia patentu europejskiego: 19.04.07 070963.1 (13) T3 (1) Int. Cl. F16H/17 F16H7/04 (06.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 182634 (96) Data i numer zgłoszenia patentu europejskiego: 19.04.07 070963.1 (13) T3 (1) Int. Cl. F16H/17 F16H7/04 (06.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1950498 (96) Data i numer zgłoszenia patentu europejskiego: 23.01.2007 07001358.6 (13) (51) T3 Int.Cl. F24C 7/08 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1950498 (96) Data i numer zgłoszenia patentu europejskiego: 23.01.2007 07001358.6 (13) (51) T3 Int.Cl. F24C 7/08 (2006.01)
(12) OPIS PATENTOWY (19) PL (11) (13) B1
 OPIS PATENTOWY (19) PL (11) (13) B1") RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 178871 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 307881 (22) Data zgłoszenia: 24.03.1995 (51) IntCl7: A61L 15/22 (54)
RZECZPOSPOLITA POLSKA (12) OPIS PATENTOWY (19) PL (11) 178871 (13) B1 Urząd Patentowy Rzeczypospolitej Polskiej (21) Numer zgłoszenia: 307881 (22) Data zgłoszenia: 24.03.1995 (51) IntCl7: A61L 15/22 (54)
Biopaliwo do silników z zapłonem samoczynnym i sposób otrzymywania biopaliwa do silników z zapłonem samoczynnym. (74) Pełnomocnik:
 Pełnomocnik:") RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 197375 (21) Numer zgłoszenia: 356573 (22) Data zgłoszenia: 10.10.2002 (13) B1 (51) Int.Cl. C10L 1/14 (2006.01)
RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 197375 (21) Numer zgłoszenia: 356573 (22) Data zgłoszenia: 10.10.2002 (13) B1 (51) Int.Cl. C10L 1/14 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 240040 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 09.07. 007077.0 (97)
PL B1. Sposób wytwarzania produktu mlecznego, zawierającego żelatynę, mleko odtłuszczone i śmietanę
 PL 212118 B1 RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 212118 (21) Numer zgłoszenia: 365023 (22) Data zgłoszenia: 22.01.2001 (86) Data i numer zgłoszenia
PL 212118 B1 RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 212118 (21) Numer zgłoszenia: 365023 (22) Data zgłoszenia: 22.01.2001 (86) Data i numer zgłoszenia
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18761 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 31.03.06 06726163.6 (97)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18761 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 31.03.06 06726163.6 (97)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") PL/EP 1699990 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1699990 (96) Data i numer zgłoszenia patentu europejskiego: 09.11.2004 04800186.1 (13) (51) T3 Int.Cl. E04G
PL/EP 1699990 T3 RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1699990 (96) Data i numer zgłoszenia patentu europejskiego: 09.11.2004 04800186.1 (13) (51) T3 Int.Cl. E04G
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2258256 (96) Data i numer zgłoszenia patentu europejskiego: 01.06.2010 10164555.4 (13) (51) T3 Int.Cl. A61B 3/103 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2258256 (96) Data i numer zgłoszenia patentu europejskiego: 01.06.2010 10164555.4 (13) (51) T3 Int.Cl. A61B 3/103 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2057877 (96) Data i numer zgłoszenia patentu europejskiego: 03.11.2008 08019246.1 (13) (51) T3 Int.Cl. A01C 23/00 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2057877 (96) Data i numer zgłoszenia patentu europejskiego: 03.11.2008 08019246.1 (13) (51) T3 Int.Cl. A01C 23/00 (2006.01)
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890558 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.05.2006 06755505.2
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1890558 Urząd Patentowy Rzeczypospolitej Polskiej (96) Data i numer zgłoszenia patentu europejskiego: 18.05.2006 06755505.2
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2127498 (96) Data i numer zgłoszenia patentu europejskiego: 14.02.2008 08716843.1 (13) (51) T3 Int.Cl. H05B 41/288 (2006.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 2127498 (96) Data i numer zgłoszenia patentu europejskiego: 14.02.2008 08716843.1 (13) (51) T3 Int.Cl. H05B 41/288 (2006.01)
(86) Data i numer zgłoszenia międzynarodowego: , PCT/JP02/ (87) Data i numer publikacji zgłoszenia międzynarodowego:
 Data i numer zgłoszenia międzynarodowego: , PCT/JP02/ (87) Data i numer publikacji zgłoszenia międzynarodowego:") RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 205828 (21) Numer zgłoszenia: 370226 (22) Data zgłoszenia: 20.06.2002 (86) Data i numer zgłoszenia międzynarodowego:
RZECZPOSPOLITA POLSKA Urząd Patentowy Rzeczypospolitej Polskiej (12) OPIS PATENTOWY (19) PL (11) 205828 (21) Numer zgłoszenia: 370226 (22) Data zgłoszenia: 20.06.2002 (86) Data i numer zgłoszenia międzynarodowego:
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1816307 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:.07.06 060114.3 (1) Int. Cl. E06B9/68 (06.01) (97) O udzieleniu
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1816307 (13) T3 (96) Data i numer zgłoszenia patentu europejskiego:.07.06 060114.3 (1) Int. Cl. E06B9/68 (06.01) (97) O udzieleniu
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1786660 (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9 (13) T3 (51) Int. Cl. B62D25/08 B60G15/06
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 1786660 (96) Data i numer zgłoszenia patentu europejskiego: 06.09.2005 05788867.9 (13) T3 (51) Int. Cl. B62D25/08 B60G15/06
(12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:
 TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) (96) Data i numer zgłoszenia patentu europejskiego:") RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18611 (96) Data i numer zgłoszenia patentu europejskiego:.03.06 06726236.0 (13) T3 (1) Int. Cl. E03C1/32 E03C1/22 (06.01)
RZECZPOSPOLITA POLSKA (12) TŁUMACZENIE PATENTU EUROPEJSKIEGO (19) PL (11) PL/EP 18611 (96) Data i numer zgłoszenia patentu europejskiego:.03.06 06726236.0 (13) T3 (1) Int. Cl. E03C1/32 E03C1/22 (06.01)